
Genomic Insights into Cyanide Biodegradation in the Pseudomonas Genus
 , , , and
, , , and
Abstract
1. Introduction
2. Results
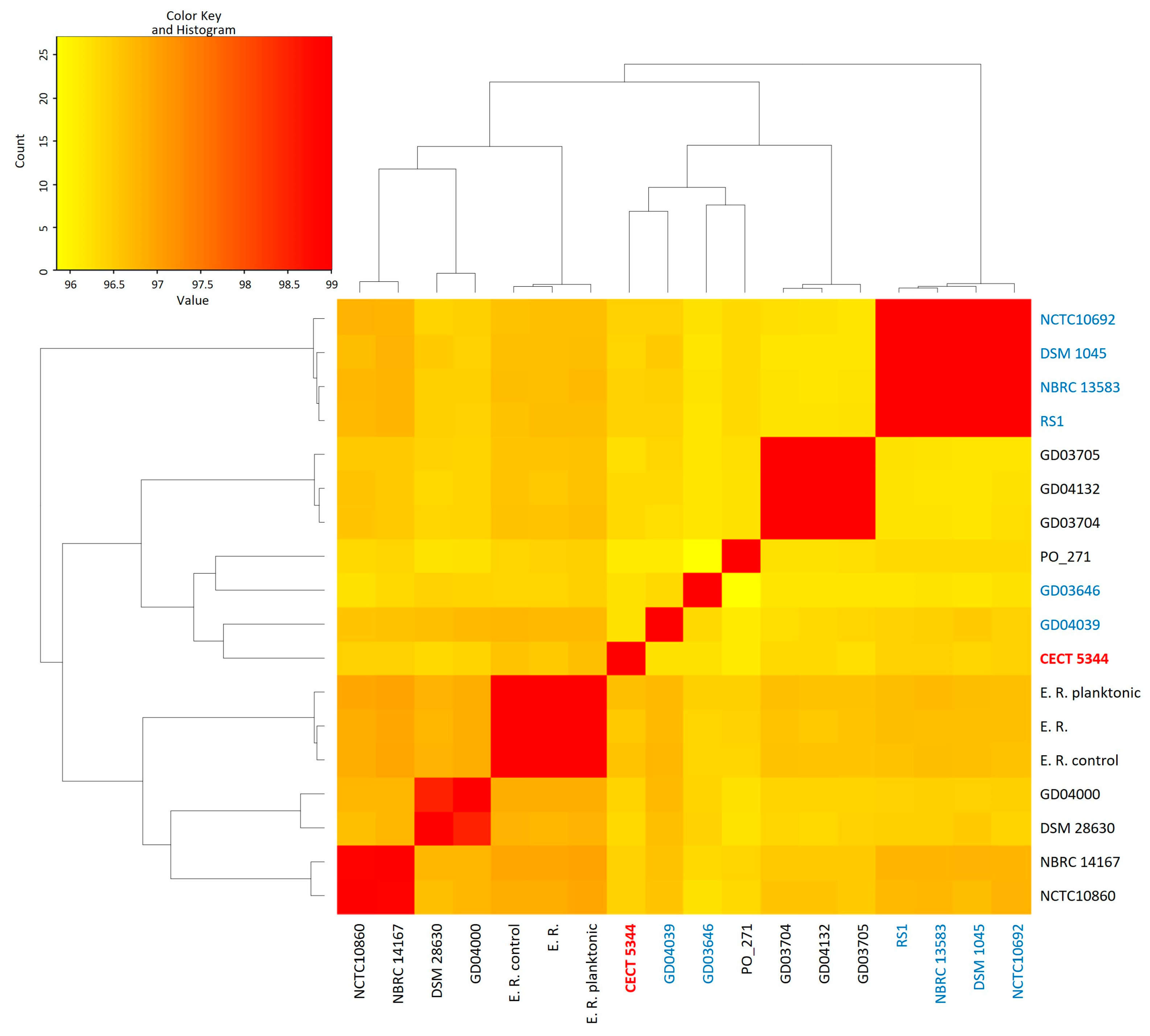
2.1. General Features of Pseudomonas oleovorans Genomes
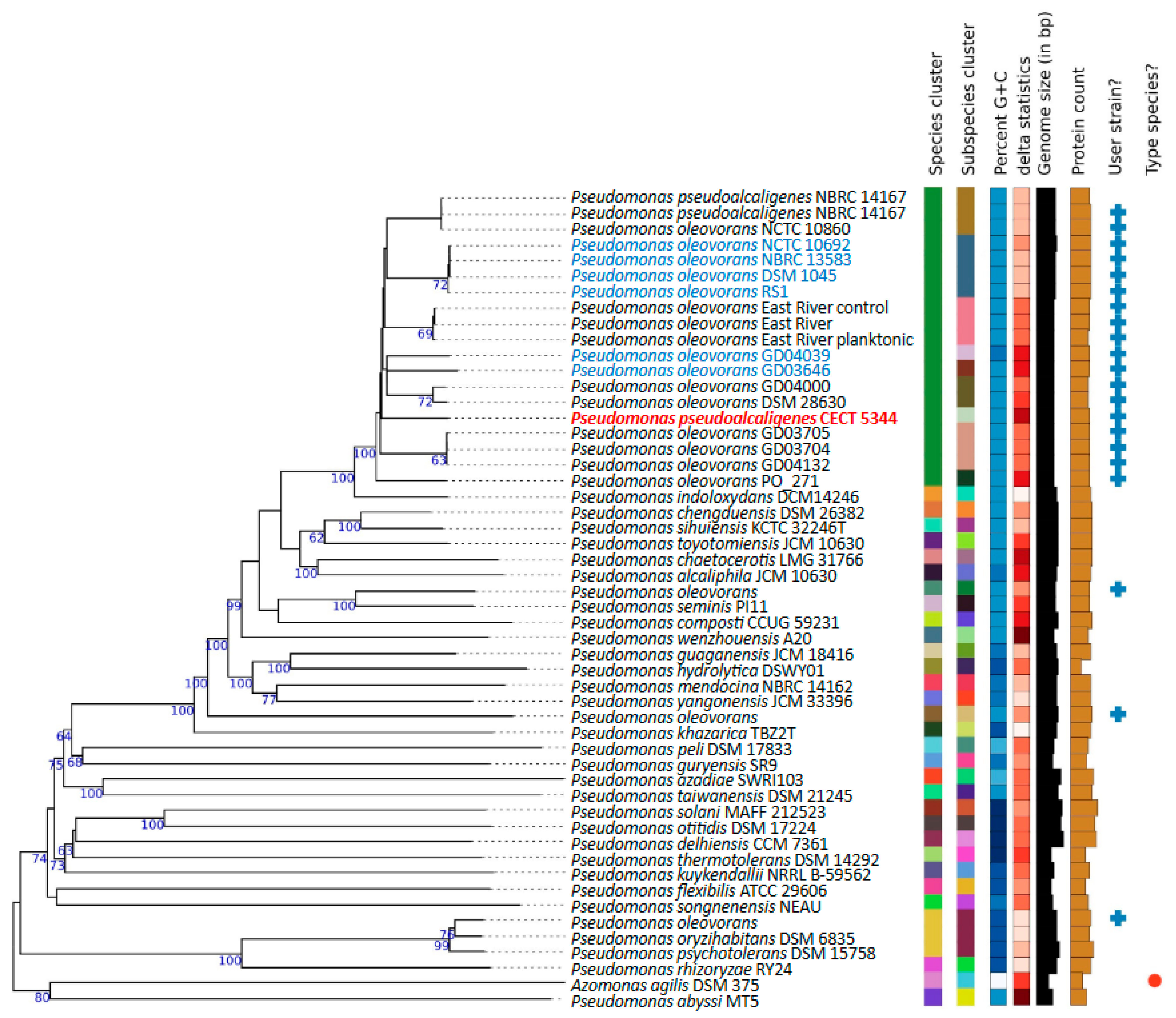
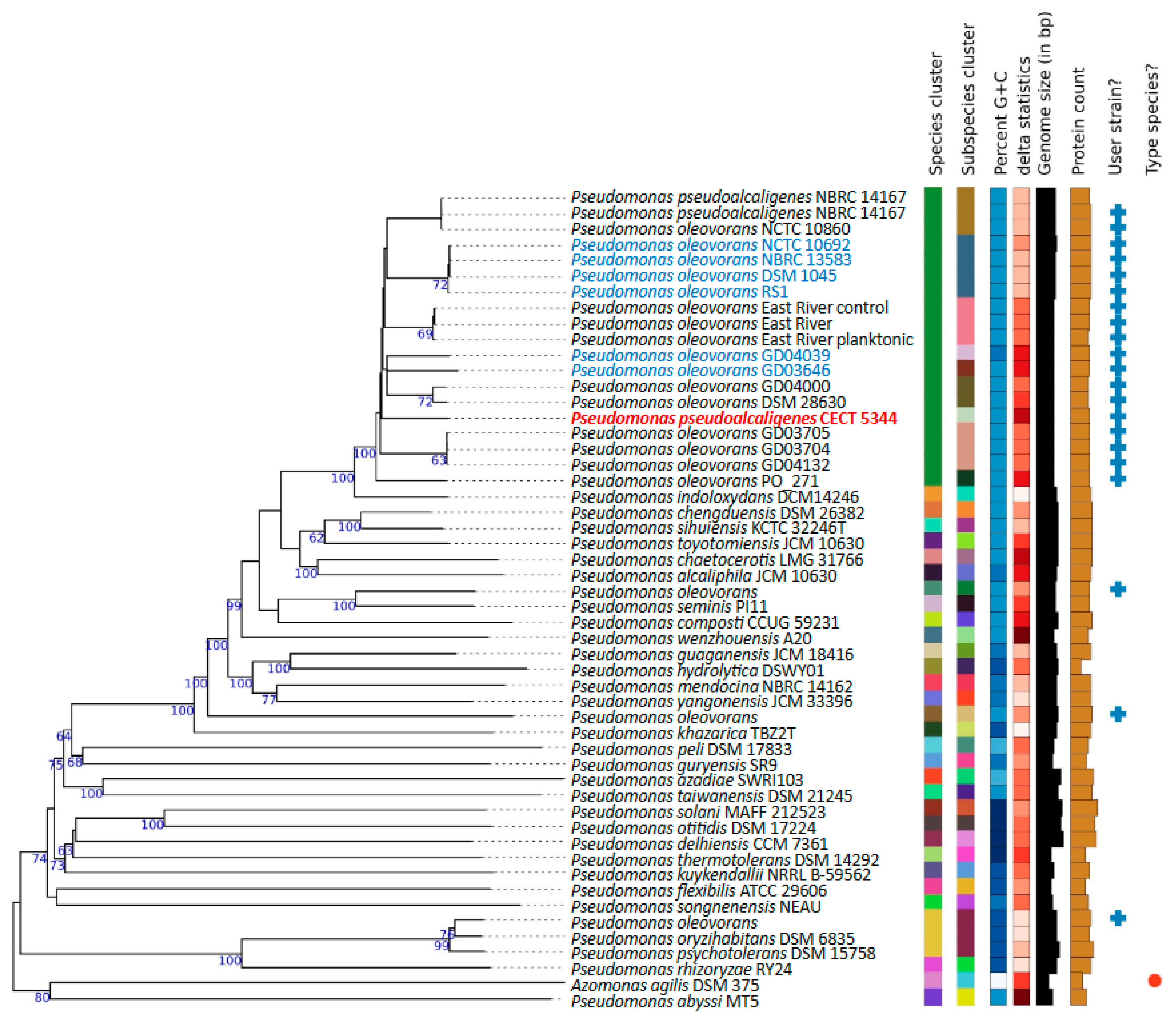
2.2. Phylogenomics of P. oleovorans Species
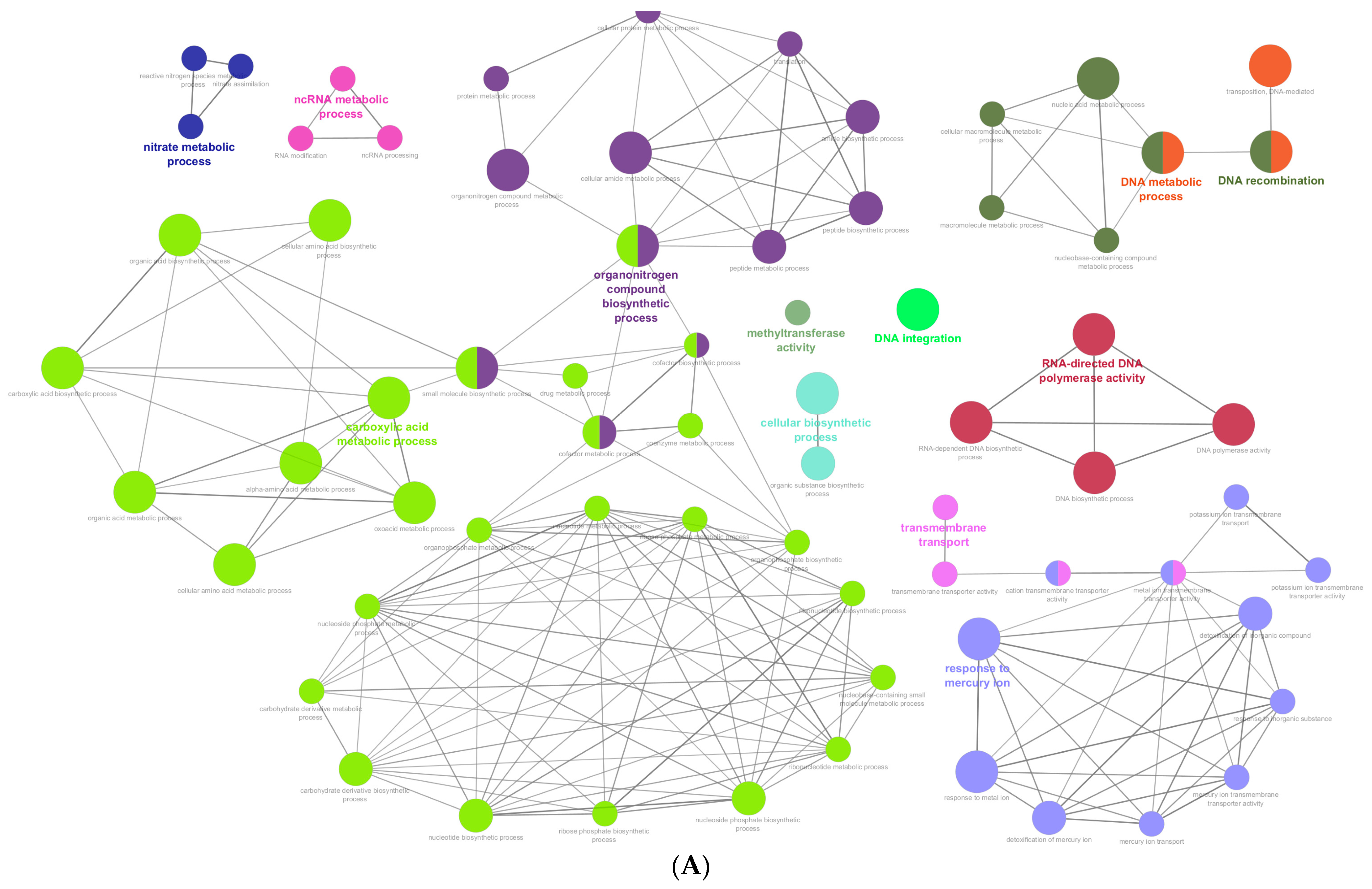
2.3. Determination and Characterization of the P. oleovorans Pan-Genome
2.4. Importance of Hydrolytic Pathways for Cyanide Biodegradation in Bacteria
2.5. Distribution of Genes Involved in Cyanide Resistance and Cyanide or Cyanate Assimilation in the Pseudomonas Genus
2.5.1. Cyanide Resistance Genes
2.5.2. Assimilation of Cyanide
2.5.3. Assimilation of Cyanate
2.5.4. Searching for Novel Genes Involved in the Biodegradation of Cyanide
3. Discussion
3.1. Genomics Supports Taxonomy Position of the Cyanotrophic Strain CECT 5344 as a Pseudomonas oleovorans Species
3.2. Defining the Pan-Genome of Pseudomonas oleovorans
3.3. The Nitrilase NitC Essential for the Cyanotrophic Phenotype Is Broadly Extended in Bacteria and It Is Encoded in the Accessory Genome of the Pseudomonas Genus
3.4. Genetic Factors Conferring Cyanide Resistance Are More Variable and Extended than Those for Cyanide Assimilation in the Pseudomonas Genus
3.5. The Presence of Genes Coding for Other Nitrilase or Cyanase Enzymes Is Not Associated to the Assimilation of Cyanide
3.6. Identification of Novel Genes with a Potential Role in Cyanide Biodegradation
4. Materials and Methods
4.1. Genomic Datasets
4.2. Average Nucleotide Identity Analysis
4.3. Phylogenomic Analysis
4.4. Pan-Genome Analysis and Comparative Genomics
4.5. Functional Analysis
4.6. Search for Sequence Homologs
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Diamond, M.L.; de Wit, C.A.; Molander, S.; Scheringer, M.; Backhaus, T.; Lohmann, R.; Arvidsson, R.; Bergman, A.; Hauschild, M.; Holoubek, I.; et al. Exploring the planetary boundary for chemical pollution. Environ. Int. 2015, 78, 8–15. [Google Scholar] [CrossRef]
- Naidu, R.; Biswas, B.; Willett, I.R.; Cribb, J.; Singh, B.K.; Paul Nathanail, C.; Coulon, F.; Semple, K.T.; Jones, K.C.; Barclay, A.; et al. Chemical pollution: A growing peril and potential catastrophic risk to humanity. Environ. Int. 2021, 156, 106616. [Google Scholar] [CrossRef]
- MacLeod, M.; Arp, H.P.H.; Tekman, M.B.; Jahnke, A. The global threat from plastic pollution. Science 2021, 373, 61–65. [Google Scholar] [CrossRef]
- Alasfar, R.H.; Isaifan, R.J. Aluminum environmental pollution: The silent killer. Environ. Sci. Pollut. Res. 2021, 28, 44587–44597. [Google Scholar] [CrossRef]
- Chrzanowski, L.; Lawniczak, L. Biodegradation of conventional and emerging pollutants. Molecules 2020, 25, 1186. [Google Scholar] [CrossRef]
- Sana, B. Bioresources for control of environmental pollution. Adv. Biochem. Eng. Biotechnol. 2015, 147, 137–183. [Google Scholar]
- Liu, L.; Bilal, M.; Duan, X.; Iqbal, H.M.N. Mitigation of environmental pollution by genetically engineered bacteria -current challenges and future perspectives. Sci. Total Environ. 2019, 667, 444–454. [Google Scholar] [CrossRef]
- Sharma, N.; Thakur, N.; Raj, T.; Savitri; Bhalla, T.C. Mining of microbial genomes for the novel sources of nitrilases. Biomed Res. Int. 2017, 2017, 7039245. [Google Scholar] [CrossRef]
- Oyewusi, H.A.; Wahab, R.A.; Huyop, F. Whole genome strategies and bioremediation insight into dehalogenase-producing bacteria. Mol. Biol. Rep. 2021, 48, 2687–2701. [Google Scholar] [CrossRef]
- Caputo, A.; Fournier, P.-E.; Raoult, D. Genome and pan-genome analysis to classify emerging bacteria. Biol. Direct 2019, 14, 5. [Google Scholar] [CrossRef]
- Bobby, P.; Raj, K.K.; Murali, T.S.; Satyamoorthy, K. Species-specific genomic sequences for classification of bacteria. Comput. Biol. Med. 2020, 123, 103874. [Google Scholar]
- Jaszczak, E.; Polkowska, Z.; Narkowicz, S.; Namiesnik, J. Cyanides in the environment-analysis-problems and challenges. Environ. Sci. Pollut. Res. 2017, 24, 15929–15948. [Google Scholar] [CrossRef]
- Quesada, A.; Guijo, M.I.; Merchán, F.; Blázquez, B.; Igeño, M.I.; Blasco, R. Essential role of the cytochrome bd-related oxidase in cyanide resistance of Pseudomonas pseudoalcaligenes CECT5344. Appl. Environ. Microbiol. 2007, 73, 5118–5124. [Google Scholar] [CrossRef]
- Luque-Almagro, V.M.; Merchán, F.; Blasco, R.; Igeño, M.I.; Martínez-Luque, M.; Moreno-Vivián, C.; Castillo, F.; Roldán, M.D. Cyanide degradation by Pseudomonas pseudoalcaligenes CECT5344 involves a malate: Quinone oxidoreductase and an associated cyanide-insensitive electro transfer chain. Microbiology 2011, 157, 739–746. [Google Scholar] [CrossRef]
- Cabello, P.; Luque-Almagro, V.M.; Olaya-Abril, A.; Sáez, L.P.; Moreno-Vivián, C.; Roldán, M.D. Assimilation of cyanide and cyano-derivatives by Pseudomonas pseudoalcaligenes CECT5344: From omic approaches to biotechnological applications. FEMS Microbiol. Lett. 2018, 1, fny032. [Google Scholar] [CrossRef]
- Roldán, M.D.; Olaya-Abril, A.; Sáez, L.P.; Cabello, P.; Luque-Almagro, V.M.; Moreno-Vivián, C. Bioremediation of cyanide-containing wastes. The potential of systems and synthetic biology for cleaning up the toxic leftovers from mining. EMBO Rep. 2021, 22, e53720. [Google Scholar] [CrossRef]
- Olaya-Abril, A.; Biełło, K.; Rodríguez-Caballero, G.; Cabello, P.; Sáez, L.P.; Moreno-Vivián, C.; Luque-Almagro, V.M.; Roldán, M.D. Bacterial tolerance and detoxification of cyanide, arsenic and heavy metals: Holistic approaches applied to bioremediation of industrial complex wastes. Microb. Biotechnol. 2024, 17, e14399. [Google Scholar] [CrossRef]
- Estepa, J.; Luque-Almagro, V.M.; Manso, I.; Escribano, M.P.; Martínez-Luque, M.; Castillo, F.; Moreno-Vivián, C.; Roldán, M.D. The nit1C gene cluster of Pseudomonas pseudoalcaligenes CECT5344 involved in assimilation of nitriles is essential for growth on cyanide. Environ. Microbiol. Rep. 2012, 4, 326–334. [Google Scholar] [CrossRef]
- Jones, L.B.; Ghosh, P.; Lee, J.H.; Chou, C.N.; Kunz, D.A. Linkage of the Nit1C gene cluster to bacterial cyanide assimilation as a nitrogen source. Microbiology 2018, 164, 956–968. [Google Scholar] [CrossRef]
- Podar, M.; Eads, J.R.; Richardson, R.H. Evolution of a microbial nitrilase gene family: A comparative and environmental genomics study. BMC Evol. Biol. 2005, 5, 42. [Google Scholar] [CrossRef]
- Panwar, P.; Williams, T.J.; Allen, M.A.; Cavicchioli, R. Population structure of an Antarctic aquatic cyanobacterium. Microbiome 2022, 10, 207. [Google Scholar] [CrossRef] [PubMed]
- Pérez, M.D.; Olaya-Abril, A.; Cabello, P.; Sáez, L.P.; Roldán, M.D.; Moreno-Vivián, C.; Luque-Almagro, V.M. Alternative pathway for 3-cyanoalanine assimilation in Pseudomonas pseudoalcaligenes CECT5344 under noncyanotrophic conditions. Microbiol. Spectr. 2021, 9, e00777-21. [Google Scholar] [CrossRef] [PubMed]
- Sáez, L.P.; Cabello, P.; Ibáñez, M.I.; Luque-Almagro, V.M.; Roldán, M.D.; Moreno-Vivián, C. Cyanate assimilation by the alkaliphilic cyanide-degrading bacterium Pseudomonas pseudoalcaligenes CECT5344: Mutational analysis of the cyn gene cluster. Int. J. Mol. Sci. 2019, 20, 3008. [Google Scholar] [CrossRef] [PubMed]
- Ciufo, S.; Kannan, S.; Sharma, S.; Badretdin, A.; Clark, K.; Turner, S.; Brover, S.; Schoch, C.L.; Kimchi, A.; DiCuccio, M. Using average nucleotide identity to improve taxonomic assignments in prokaryotic genomes at the NCBI. Int. J. Syst. Evol. Microbiol. 2018, 68, 2386–2392. [Google Scholar] [CrossRef] [PubMed]
- Luque-Almagro, V.M.; Huertas, M.J.; Martínez-Luque, M.; Moreno-Vivián, C.; Roldán, M.D.; García-Gil, L.J.; Castillo, F.; Blasco, R. Bacterial degradation of cyanide and its metal complexes under alkaline conditions. Appl. Environ. Microbiol. 2005, 71, 940–947. [Google Scholar] [CrossRef]
- Konstantinidis, K.T.; Tiedje, J.M. Genomic insights that advance the species definition for prokaryotes. Proc. Natl. Acad. Sci. USA 2005, 102, 2567–2572. [Google Scholar] [CrossRef] [PubMed]
- Richter, M.; Rosselló-Móra, R. Shifting the genomic gold standard for the prokaryotic species definition. Proc. Natl. Acad. Sci. USA 2009, 106, 19126–19131. [Google Scholar] [CrossRef]
- Bodilis, J.; Nsigue-Mejlo, S.; Besaury, L.; Quillet, L. Variable copy number, intra-genomic heterogeneities and lateral transfers of the 16S rRNA gene in Pseudomonas. PLoS ONE 2012, 7, e35647. [Google Scholar] [CrossRef]
- Watanabe, A.; Yano, K.; Ikebukuro, K.; Karube, I. Cyanide hydrolysis in a cyanide-degrading bacterium, Pseudomonas stutzeri AK61, by cyanidase. Microbiology 1998, 144, 1677–1682. [Google Scholar] [CrossRef]
- Kunz, D.A.; Nagappan, O. Cyanase-mediated utilization of cyanate in Pseudomonas fluorescens NCIB 11764. Appl. Environ. Microbiol. 1989, 55, 256–258. [Google Scholar] [CrossRef]
- Luque-Almagro, V.M.; Huertas, M.J.; Sáez, L.P.; Luque-Romero, M.M.; Moreno-Vivián, C.; Castillo, F.; Roldán, M.D.; Blasco, R. Characterization of the Pseudomonas pseudoalcaligenes CECT5344 cyanase, an enzyme that is not essential for cyanide assimilation. Appl. Environ. Microbiol. 2008, 74, 6280–6288. [Google Scholar] [CrossRef] [PubMed]
- Luque-Almagro, V.M.; Acera, F.; Igeño, M.I.; Wibberg, D.; Roldán, M.D.; Sáez, L.P.; Hennig, M.; Quesada, A.; Huertas, M.J.; Blom, J.; et al. Draft whole genome sequence of the cyanide-degrading bacterium Pseudomonas pseudoalcaligenes CECT5344. Environ. Microbiol. 2013, 15, 253–270. [Google Scholar] [CrossRef] [PubMed]
- Lalucat, J.; Gomila, M.; Mulet, M.; Zaruma, A.; García-Valdés, E. Past, present and future of the boundaries of the Pseudomonas genus: Proposal of Stutzerimonas gen. Nov. Syst. Appl. Microbiol. 2022, 45, 126289. [Google Scholar] [CrossRef] [PubMed]
- Gomila, M.; Mulet, M.; García-Valdés, E.; Lalucat, J. Genome-based taxonomy of the genus Stutzerimonas and proposal of S. frequens sp. nov. and S. degradans sp. nov. and emended descriptions of S. perfectomarina and S. chloritidismutans. Microorganisms 2022, 10, 1363. [Google Scholar] [CrossRef] [PubMed]
- Stanier, R.Y.; Palleroni, N.J.; Doudoroff, M. The aerobic pseudomonads: A taxonomic study. J. Gen. Microbiol. 1966, 43, 159–271. [Google Scholar] [CrossRef]
- Yamamoto, S.; Kasai, H.; Arnold, D.L.; Jackson, R.W.; Vivian, A.; Harayama, S. Phylogeny of the genus Pseudomonas: Intrageneric structure reconstructed from the nucleotide sequences of gyrB and rpoD genes. Microbiology 2000, 146, 2385–2394. [Google Scholar] [CrossRef]
- Mulet, M.; Lalucat, J.; García-Valdés, E. DNA sequence-based analysis of the Pseudomonas species. Environ. Microbiol. 2010, 12, 1513–1530. [Google Scholar] [CrossRef] [PubMed]
- Saha, R.; Spröer, C.; Beck, B.; Bagley, S. Pseudomonas oleovorans subsp. lubricantis subsp. nov., and reclassification of Pseudomonas pseudoalcaligenes ATCC 17440T as later synonym of Pseudomonas oleovorans ATCC 8062 T. Curr. Microbiol. 2010, 60, 294–300. [Google Scholar] [CrossRef]
- Gomila, M.; Peña, A.; Mulet, M.; Lalucat, J.; García-Valdés, E. Phylogenomics and systematics in Pseudomonas. Front. Microbiol. 2015, 18, 214. [Google Scholar] [CrossRef]
- Silby, M.W.; Winstanley, C.; Godfrey, S.A.; Levy, S.B.; Jackson, R.W. Pseudomonas genomes: Diverse and adaptable. FEMS Microbiol. Rev. 2011, 35, 652–680. [Google Scholar] [CrossRef]
- Tettelin, H.; Riley, D.; Cattuto, C.; Medini, D. Comparative genomics: The bacterial pan-genome. Curr. Opin. Microbiol. 2008, 11, 472–477. [Google Scholar] [CrossRef]
- Udaondo, Z.; Molina, L.; Segura, A.; Duque, E.; Ramos, J.L. Analysis of the core genome and pangenome of Pseudomonas putida. Environ. Microbiol. 2016, 18, 3268–3283. [Google Scholar] [CrossRef]
- Kumari, K.; Rawat, V.; Shadan, A.; Sharma, P.K.; Deb, S.; Singh, R.P. In-depth genome and pan-genome analysis of a metal-resistant bacterium Pseudomonas parafulva OS-1. Front. Microbiol. 2023, 14, 1140249. [Google Scholar] [CrossRef]
- Poehlein, A.; Daniel, R.; Thürmer, A.; Bollinger, A.; Thies, S.; Katzke, N.; Jaeger, K.E. First insights into the genome sequence of Pseudomonas oleovorans DSM 1045. Genome Announc. 2017, 32, e00774-17. [Google Scholar] [CrossRef] [PubMed]
- Brockhurst, M.A.; Harrison, E.; Hall, J.P.J.; Richards, T.; McNally, A.; MacLean, C. The ecology and evolution of pangenomes. Curr. Biol. 2019, 29, R1094–R1103. [Google Scholar] [CrossRef] [PubMed]
- Cummins, E.A.; Hall, R.J.; McInerney, J.O.; McNally, A. Prokaryote pangenomes are dynamic entities. Curr. Opin. Microbiol. 2022, 66, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Parra-Sánchez, Á.; Antequera-Zambrano, L.; Martínez-Navarrete, G.; Zorrilla-Muñoz, V.; Paz, J.L.; Alvarado, Y.J.; González-Paz, L.; Fernández, E. Comparative analysis of CRISPR-Cas systems in Pseudomonas genomes. Genes 2023, 14, 1337. [Google Scholar] [CrossRef]
- Wheatley, R.M.; MacLean, R.C. CRISPR-Cas systems restrict horizontal gene transfer in Pseudomonas aeruginosa. ISME J. 2021, 15, 1420–1433. [Google Scholar] [CrossRef]
- Luque-Almagro, V.M.; Escribano, M.P.; Manso, I.; Sáez, L.P.; Cabello, P.; Moreno-Vivián, C.; Roldán, M.D. DNA microarray analysis of the cyanotroph Pseudomonas pseudoalcaligenes CECT5344 in response to nitrogen starvation, cyanide and a jewelry wastewater. J. Biotechnol. 2015, 214, 171–181. [Google Scholar] [CrossRef] [PubMed]
- Sewell, B.T.; Berman, M.N.; Meyers, P.R.; Jandhyala, D.; Benedik, M.J. The cyanide degrading nitrilase from Pseudomonas stutzeri AK61 is a two-fold symmetric, 14-subunit spiral. Structure 2003, 11, 1413–1422. [Google Scholar] [CrossRef]
- Jones, L.B.; Kunz, D.A. Draft genome sequence of the cyanotroph Pseudomonas monteilii BCN3. Microbiol. Resour. Announc. 2019, 8, e01322-18. [Google Scholar] [CrossRef] [PubMed]
- Doré, H.; Guyet, U.; Leconte, J.; Farrant, G.K.; Alric, B.; Ratin, M.; Ostrowski, M.; Ferrieux, M.; Brillet-Guéguen, L.; Hoebeke, M.; et al. Differential global distribution of marine picocyanobacteria gene clusters reveals distinct niche-related adaptive strategies. ISME J. 2023, 17, 720–732. [Google Scholar] [CrossRef] [PubMed]
- Barbaccia, P.; Gaglio, R.; Dazzi, C.; Miceli, C.; Bella, P.; Lo Papa, G.; Settanni, L. Plant growth-promoting activities of bacteria isolated from an anthropogenic soil located in Agrigento province. Microorganisms 2022, 10, 2167. [Google Scholar] [CrossRef]
- Meier-Kolthoff, J.P.; Göker, M. TYGS is an automated high-throughput platform for state-of-the-art genome-based taxonomy. Nat. Commun. 2019, 10, 2182. [Google Scholar] [CrossRef]
- Ondov, B.D.; Treangen, T.J.; Melsted, P.; Mallonee, A.B.; Bergman, N.H.; Koren, S.; Phillippy, A.M. Mash: Fast genome and metagenome distance estimation using MinHash. Genome Biol. 2016, 17, 1–14. [Google Scholar] [CrossRef]
- Lagesen, K.; Hallin, P. RNAmmer: Consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 2007, 35, 3100–3108. [Google Scholar] [CrossRef]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [PubMed]
- Meier-Kolthoff, J.P.; Auch, A.F.; Klenk, H.-P.; Göker, M. Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinform. 2013, 14, 60. [Google Scholar] [CrossRef] [PubMed]
- Meier-Kolthoff, J.P.; Carbasse, J.S.; Peinado-Olarte, R.L.; Göker, M. TYGS and LPSN: A database tandem for fast and reliable genome-based classification and nomenclature of prokaryotes. Nucleic Acid Res. 2022, 50, D801–D807. [Google Scholar] [CrossRef]
- Lefort, V.; Desper, R.; Gascuel, O. FastME 2.0: A comprehensive, accurate, and fast distance-based phylogeny inference program. Mol. Biol. Evol. 2015, 32, 2798–2800. [Google Scholar] [CrossRef]
- Farris, J.S. Estimating phylogenetic trees from distance matrices. Am. Nat. 1972, 106, 645–667. [Google Scholar] [CrossRef]
- Kreft, L.; Botzki, A.; Coppens, F.; Vandepoele, K.; Van Bel, M. PhyD3: A phylogenetic tree viewer with extended phyloXML support for functional genomics data visualization. Bioinformatics 2017, 33, 2946–2947. [Google Scholar] [CrossRef] [PubMed]
- Meier-Kolthoff, J.P.; Hahnke, R.L.; Petersen, J.; Scheuner, C.; Michael, V.; Fiebig, A.; Rohde, C.; Rohde, M.; Fartmann, B.; Goodwin, L.A.; et al. Complete genome sequence of DSM 30083T, the type strain (U5/41T) of Escherichia coli, and a proposal for delineating subspecies in microbial taxonomy. Stand. Genomic. Sci. 2014, 9, 2. [Google Scholar] [CrossRef] [PubMed]
- Dereeper, A.; Summo, M.; Meyer, D.F. PanExplorer: A web-based tool for exploratory analysis and visualization of bacterial pan-genomes. Bioinformatics 2022, 38, 4412–4414. [Google Scholar] [CrossRef] [PubMed]
- Perrin, A.; Rocha, E.P.C. PanACoTA: A modular tool for massive microbial comparative genomics. NAR Genom. Bioinform. 2021, 3, lqaa106. [Google Scholar]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef] [PubMed]
- Jong, A.; Kuipers, O.P.; Kok, J. FUNAGE-Pro: Comprehensive web server for gene set enrichment analysis of prokaryotes. Nucleic Acids Res. 2022, 50, W330–W336. [Google Scholar] [PubMed]
- Ge, S.X.; Jung, D.; Yao, R. ShinyGO: A graphical gene-set enrichment tool for animals and plants. Bioinformatics 2020, 36, 2628–2629. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
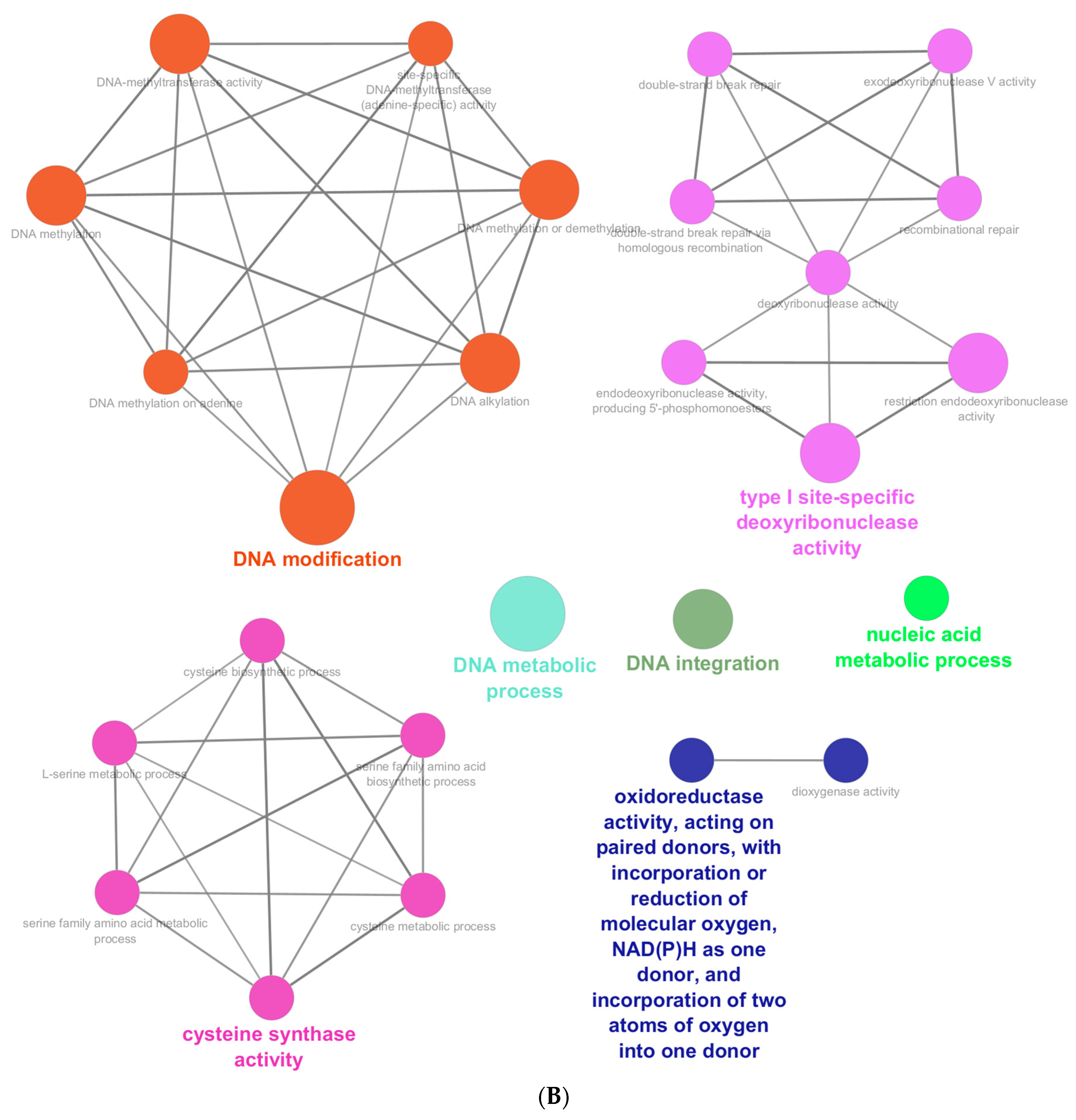{kind=link}
{kind=link}
| Species | Number of Strains with nit1C Cluster | Number of Sequenced Genomes | Relative Abundance (%) |
|---|---|---|---|
| P. abietaniphila | 1 | 3 | 33.3 |
| P. abyssi | 1 | 1 | 100.0 |
| P. arsenicoxydans | 1 | 3 | 33.3 |
| P. avellanae | 1 | 18 | 5.6 |
| P. bohemica | 1 | 1 | 100.0 |
| P. carbonaria | 1 | 1 | 100.0 |
| P. caspiana | 1 | 2 | 50.0 |
| P. daroniae | 1 | 4 | 25.0 |
| P. fluorescens | 2 | 299 | 0.7 |
| P. folii | 1 | 1 | 100.0 |
| P. gingeri | 1 | 30 | 3.3 |
| P. indoloxydans | 1 | 2 | 50.0 |
| P. kuykendallii | 2 | 7 | 28.6 |
| P. mandelii | 2 | 13 | 15.4 |
| P. migulae | 2 | 6 | 33.3 |
| P. mohnii | 1 | 3 | 33.3 |
| P. monteilii | 1 | 65 | 1.5 |
| P. moorei | 2 | 5 | 40.0 |
| P. oleovorans | 7 | 36 | 19.4 |
| P. quasicaspiana | 1 | 4 | 25.0 |
| P. reinekei | 4 | 7 | 57.1 |
| P. serbica | 1 | 1 | 100.0 |
| P. typographi | 1 | 3 | 33.3 |
| P. viridiflava | 11 | 1564 | 0.7 |
| P. wenzhouensis | 1 | 2 | 50.0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sáez, L.P.; Rodríguez-Caballero, G.; Olaya-Abril, A.; Cabello, P.; Moreno-Vivián, C.; Roldán, M.D.; Luque-Almagro, V.M. Genomic Insights into Cyanide Biodegradation in the Pseudomonas Genus. Int. J. Mol. Sci. 2024, 25, 4456. https://doi.org/10.3390/ijms25084456
Sáez LP, Rodríguez-Caballero G, Olaya-Abril A, Cabello P, Moreno-Vivián C, Roldán MD, Luque-Almagro VM. Genomic Insights into Cyanide Biodegradation in the Pseudomonas Genus. International Journal of Molecular Sciences. 2024; 25(8):4456. https://doi.org/10.3390/ijms25084456
Chicago/Turabian StyleSáez, Lara P., Gema Rodríguez-Caballero, Alfonso Olaya-Abril, Purificación Cabello, Conrado Moreno-Vivián, María Dolores Roldán, and Víctor M. Luque-Almagro. 2024. "Genomic Insights into Cyanide Biodegradation in the Pseudomonas Genus" International Journal of Molecular Sciences 25, no. 8: 4456. https://doi.org/10.3390/ijms25084456
APA StyleSáez, L. P., Rodríguez-Caballero, G., Olaya-Abril, A., Cabello, P., Moreno-Vivián, C., Roldán, M. D., & Luque-Almagro, V. M. (2024). Genomic Insights into Cyanide Biodegradation in the Pseudomonas Genus. International Journal of Molecular Sciences, 25(8), 4456. https://doi.org/10.3390/ijms25084456