

Vascular Heparan Sulfate and Amyloid-β in Alzheimer’s Disease Patients

Abstract
1. Introduction
2. Results
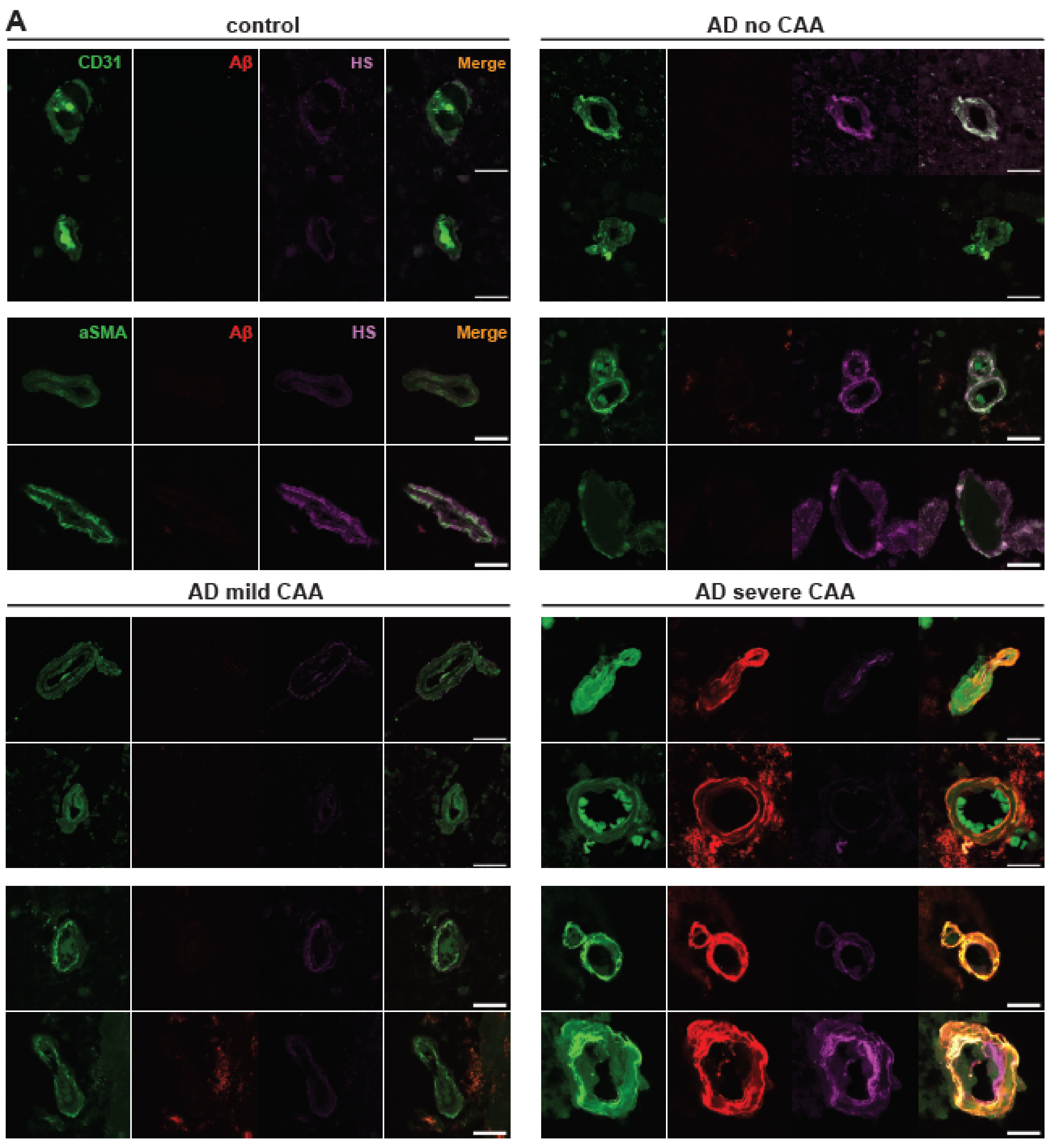
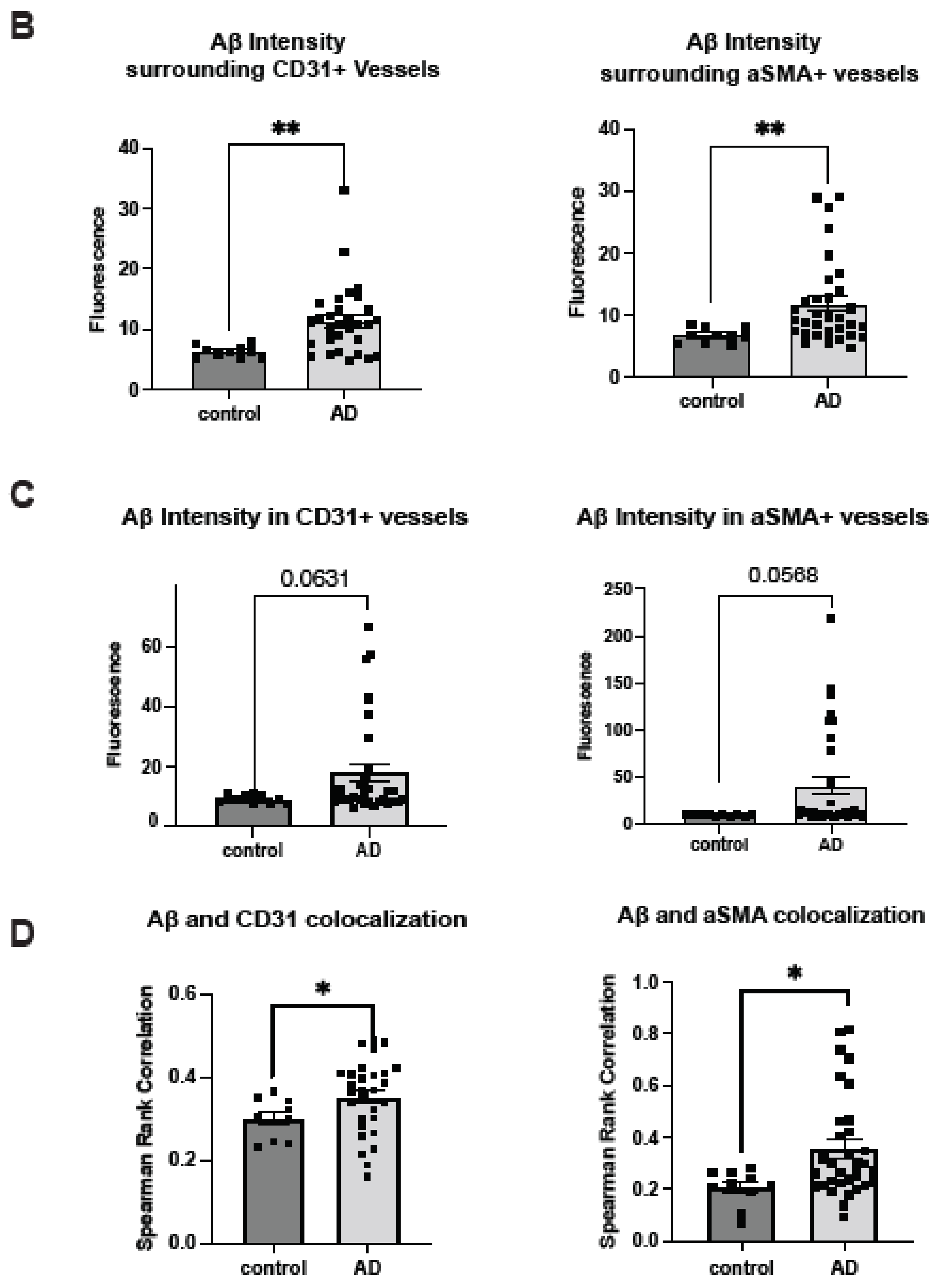
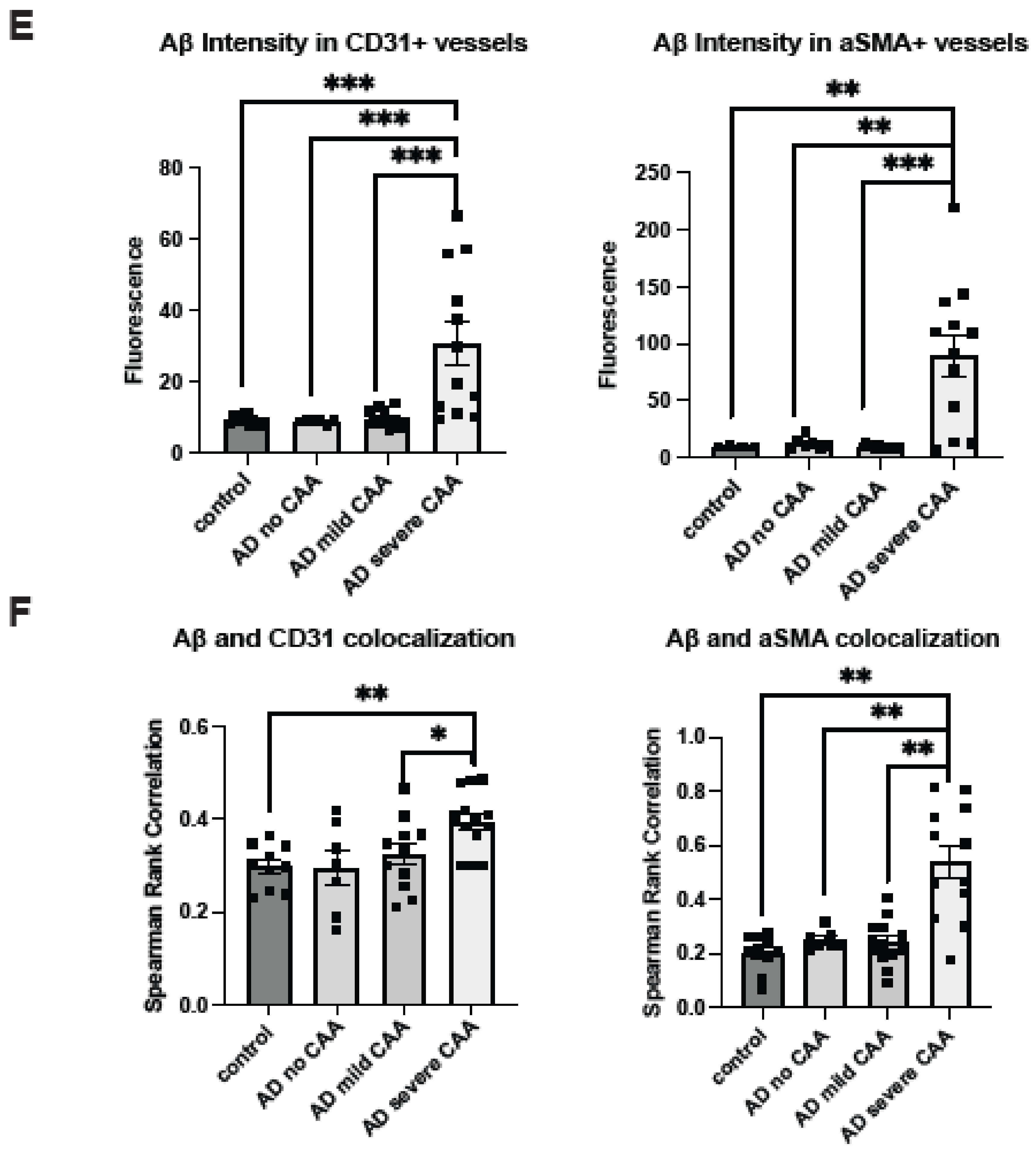
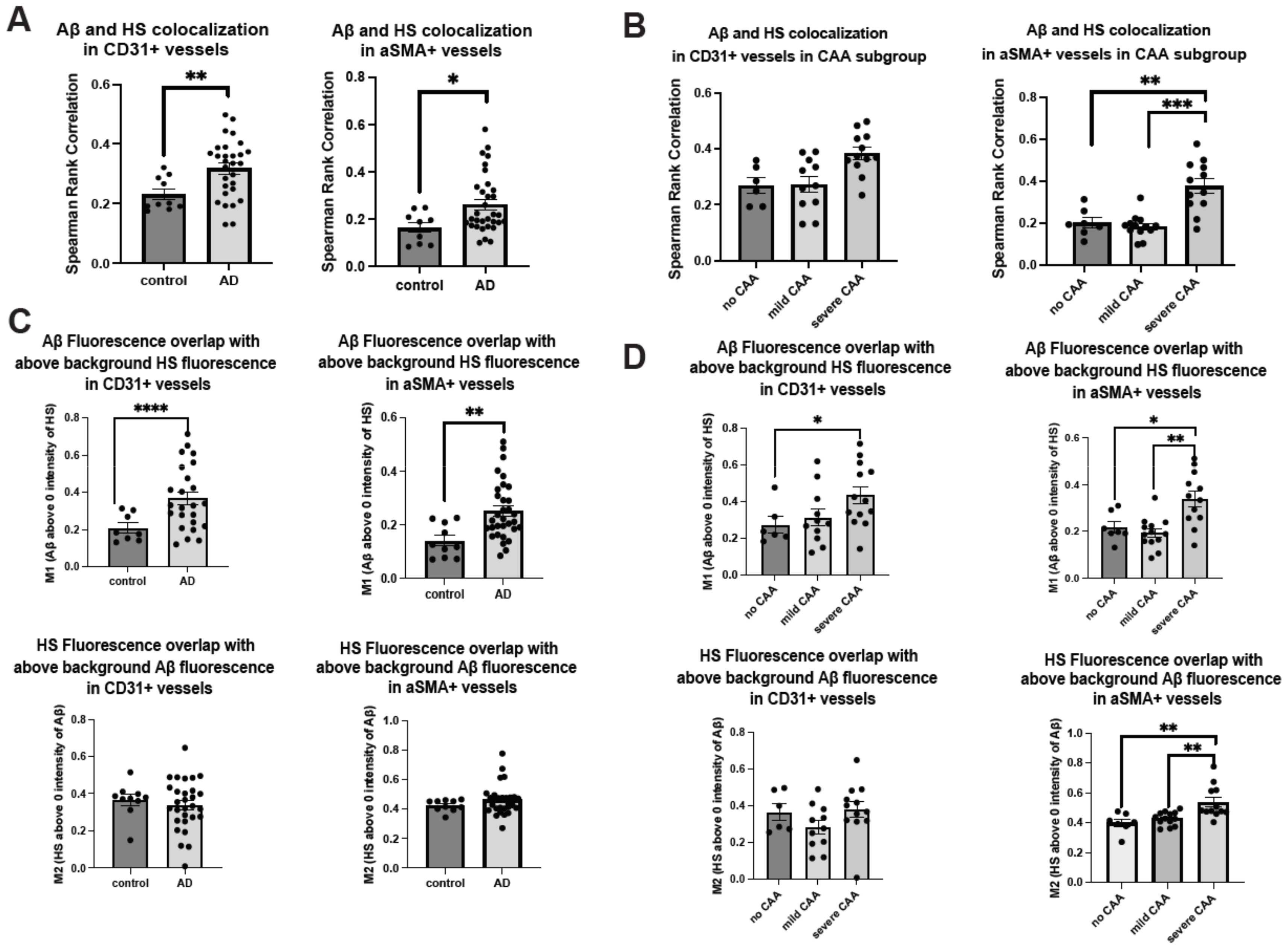
2.1. Cerebrovascular Aβ Accumulation in AD Patients Increases with Severe CAA
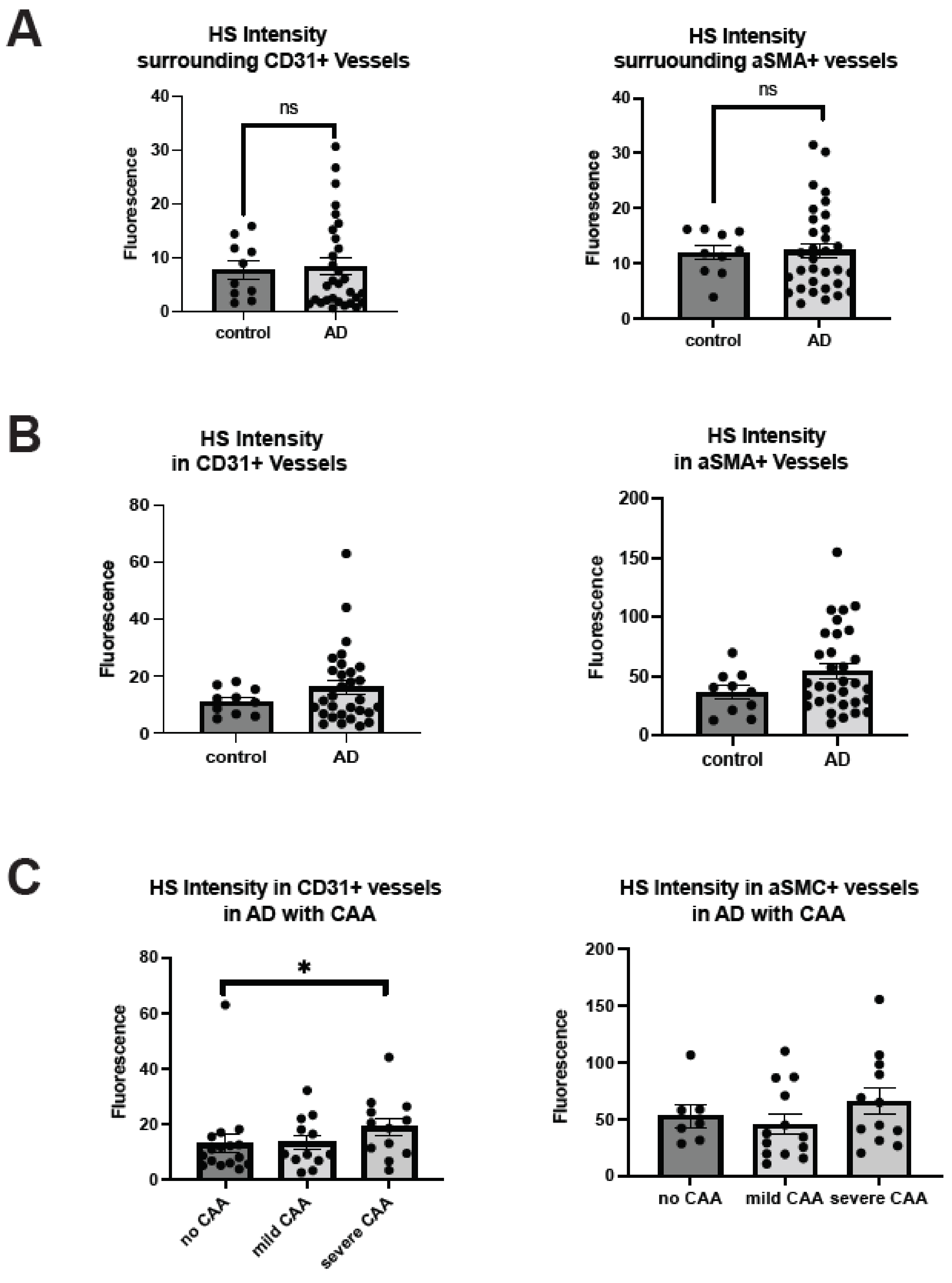
2.2. Cerebrovascular HS Is Elevated in AD Patients with Severe CAA
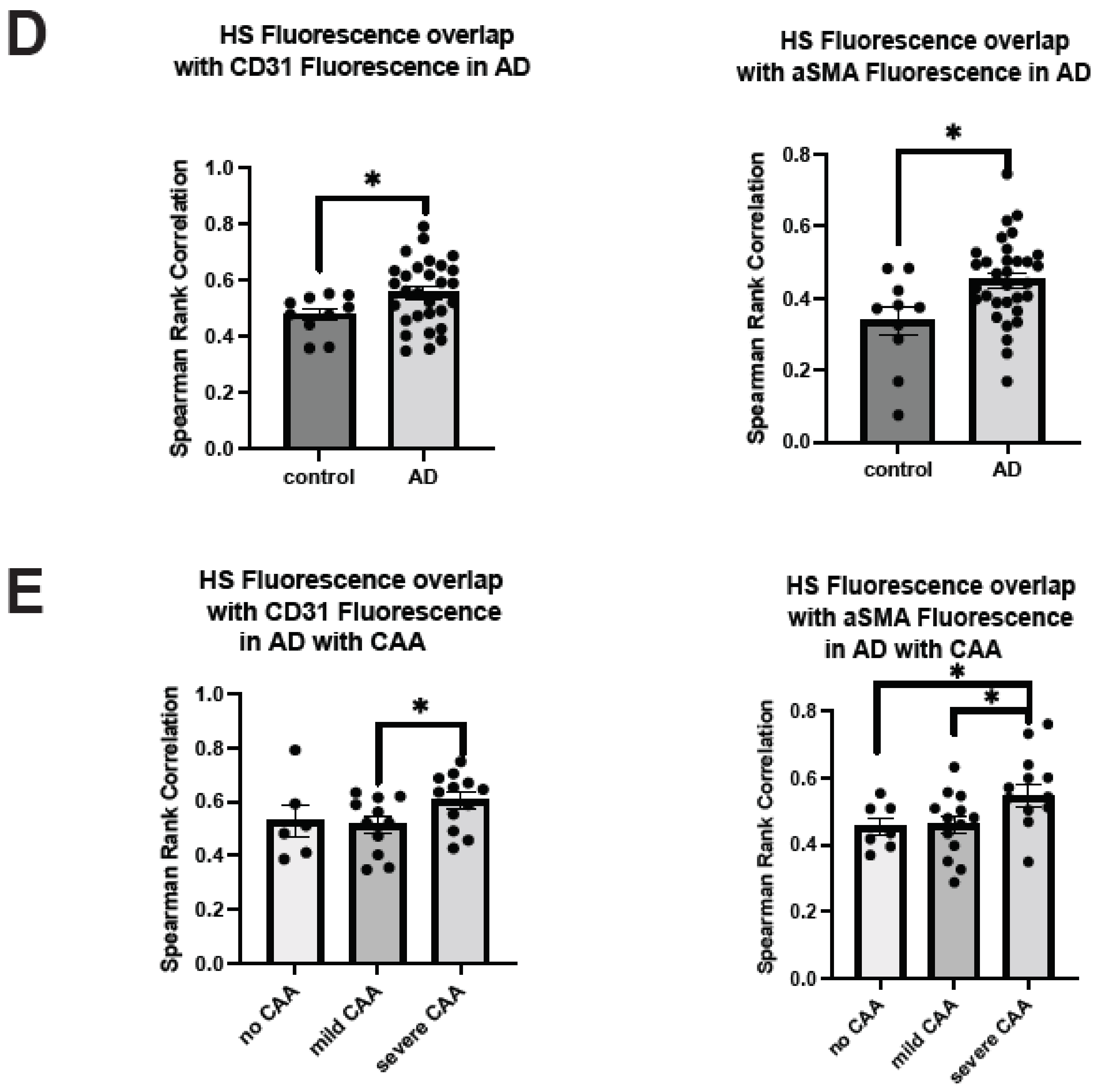
2.3. Co-Deposition of Cerebrovascular HS with Aβ Is Increased in AD
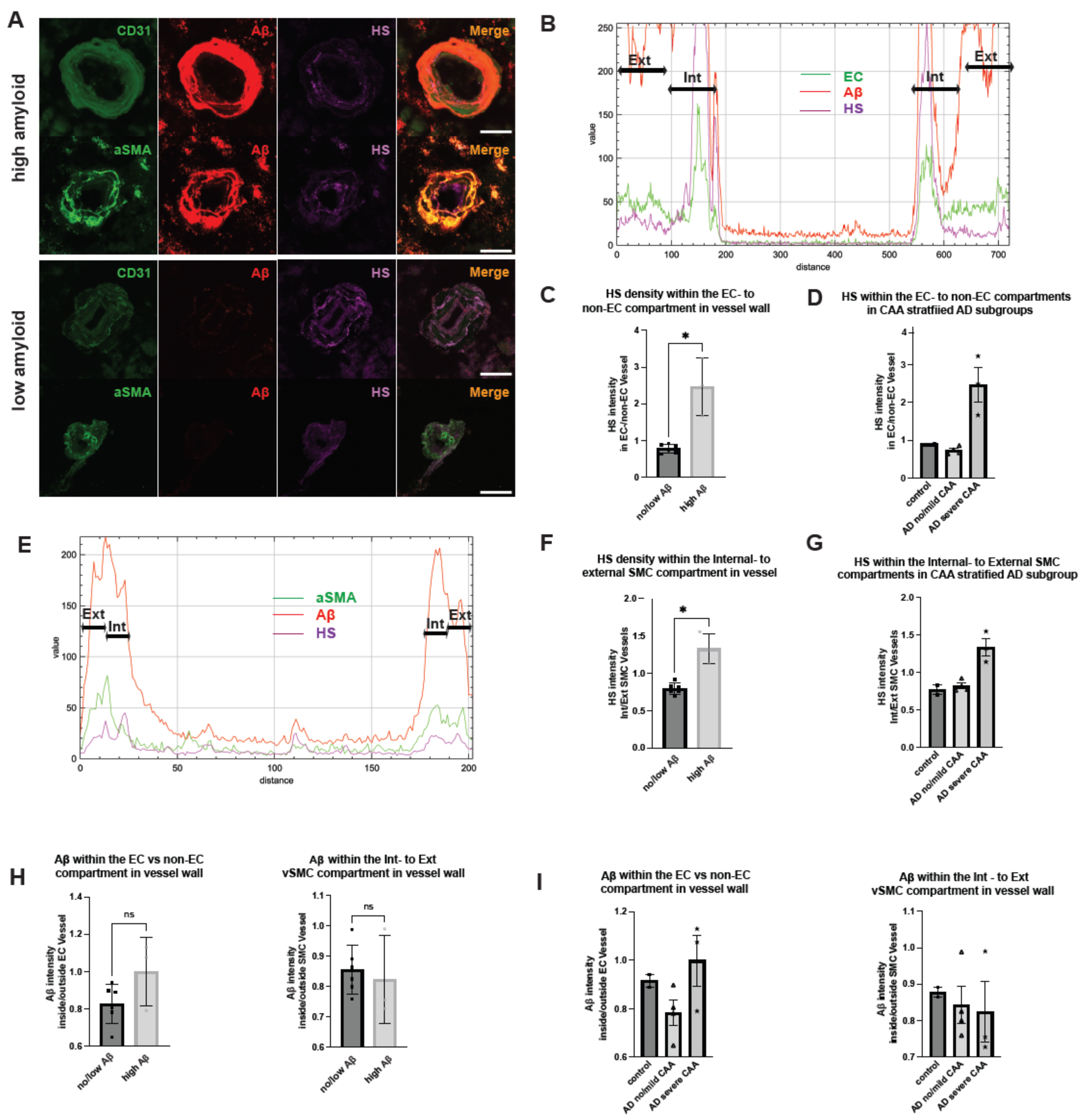
2.4. Polarized HS Expression in Cerebrovasculature Is Reversed in AD with Severe CAA While No Polarization of Vascular Aβ Deposition Is Observed
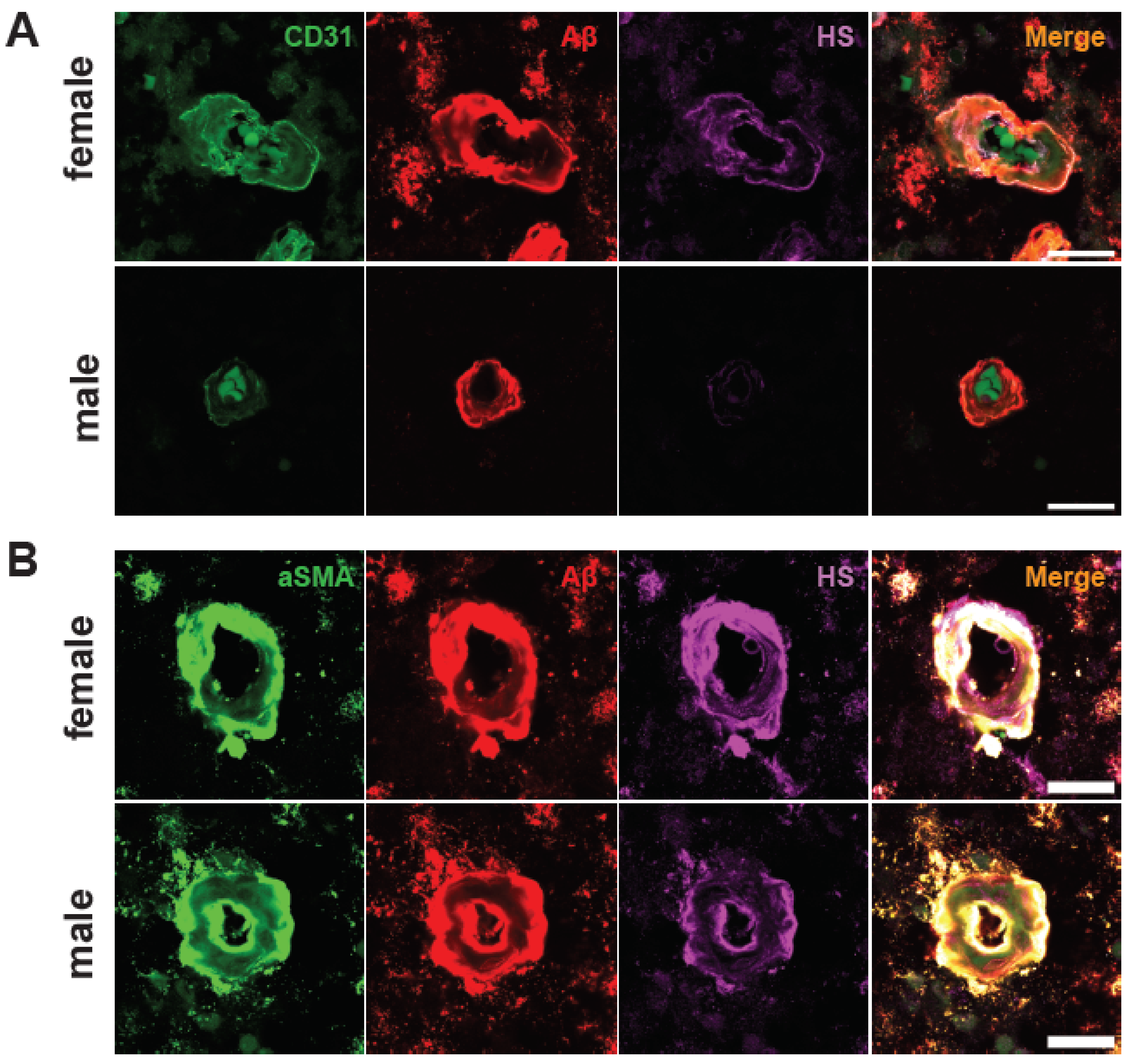
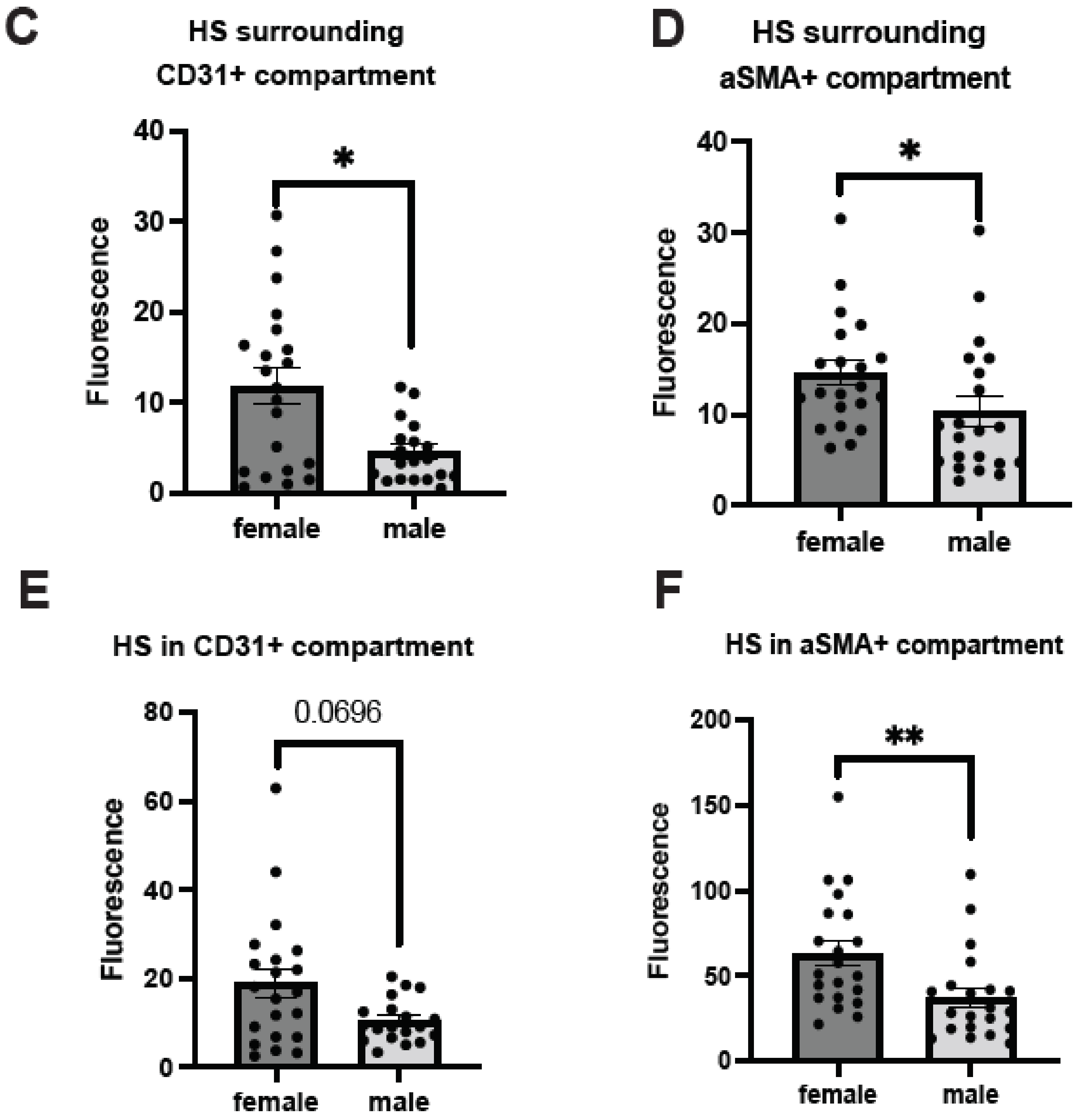
2.5. Male AD Patients Have Lower Parenchymal and Cerebrovascular HS
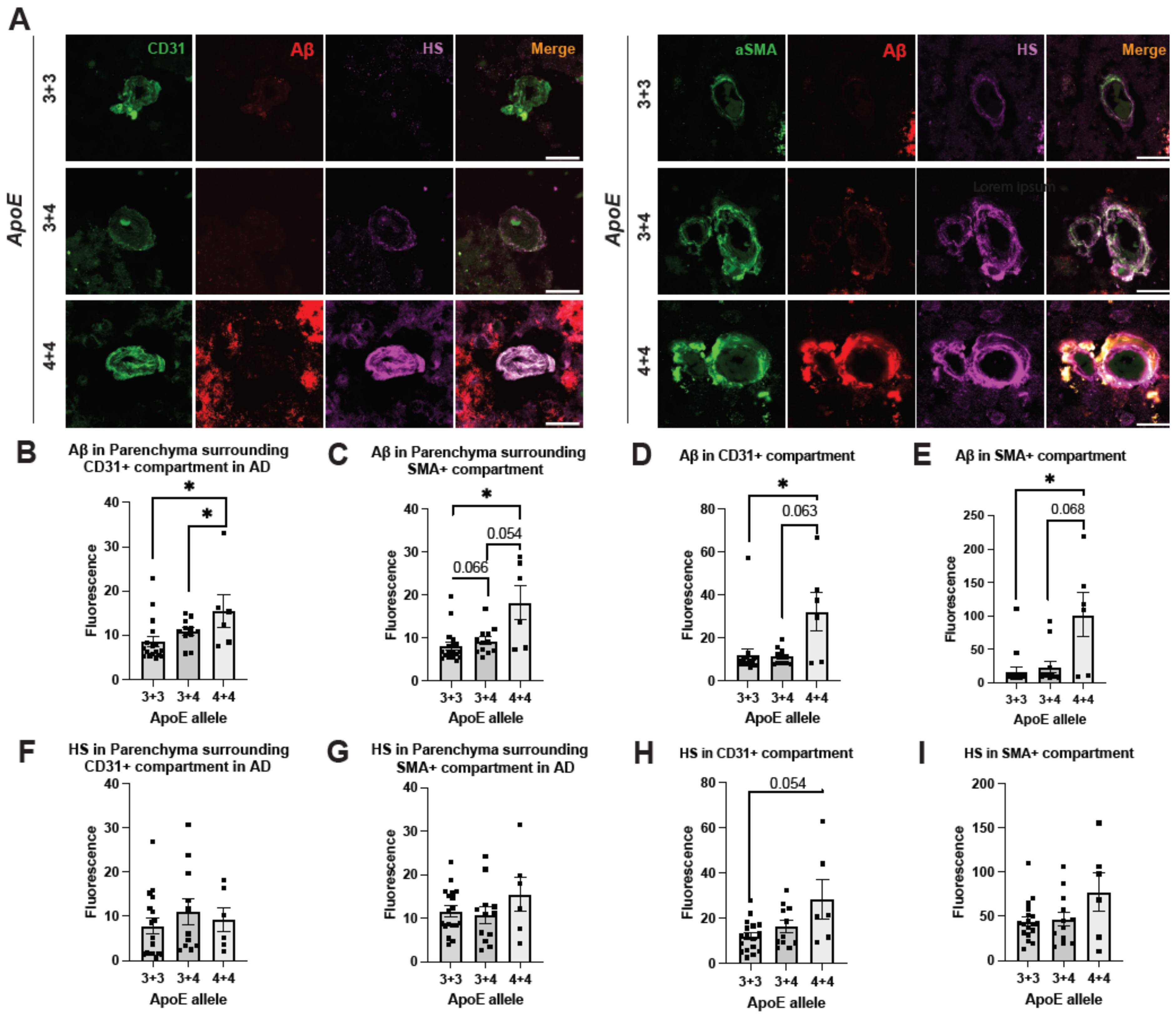
2.6. ApoE4 Correlates with Elevated Cerebrovascular Aβ and a Tendency of Higher HS Expression in AD
3. Discussion
4. Materials and Methods
4.1. Human Brain Tissues
4.2. Immunofluorescence Staining
4.3. Imaging and Image Analyses
4.4. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- DeTure, M.A.; Dickson, D.W. The neuropathological diagnosis of Alzheimer’s disease. Mol. Neurodegener. 2019, 14, 32. [Google Scholar] [CrossRef] [PubMed]
- Keage, H.A.; Carare, R.O.; Friedland, R.P.; Ince, P.G.; Love, S.; Nicoll, J.A.; Wharton, S.B.; Weller, R.O.; Brayne, C. Population studies of sporadic cerebral amyloid angiopathy and dementia: A systematic review. BMC Neurol. 2009, 9, 3. [Google Scholar] [CrossRef] [PubMed]
- Jakel, L.; De Kort, A.M.; Klijn, C.J.M.; Schreuder, F.; Verbeek, M.M. Prevalence of cerebral amyloid angiopathy: A systematic review and meta-analysis. Alzheimer’s Dement. 2022, 18, 10–28. [Google Scholar] [CrossRef] [PubMed]
- Van Dyck, C.H.; Swanson, C.J.; Aisen, P.; Bateman, R.J.; Chen, C.; Gee, M.; Kanekiyo, M.; Li, D.; Reyderman, L.; Cohen, S.; et al. Lecanemab in Early Alzheimer’s Disease. N. Engl. J. Med. 2023, 388, 9–21. [Google Scholar] [CrossRef]
- Greenberg, S.M.; Bacskai, B.J.; Hernandez-Guillamon, M.; Pruzin, J.; Sperling, R.; van Veluw, S.J. Cerebral amyloid angiopathy and Alzheimer disease—One peptide, two pathways. Nat. Rev. Neurol. 2020, 16, 30–42. [Google Scholar] [CrossRef] [PubMed]
- Swanson, C.J.; Zhang, Y.; Dhadda, S.; Wang, J.; Kaplow, J.; Lai, R.Y.K.; Lannfelt, L.; Bradley, H.; Rabe, M.; Koyama, A.; et al. A randomized, double-blind, phase 2b proof-of-concept clinical trial in early Alzheimer’s disease with lecanemab, an anti-Aβ protofibril antibody. Alzheimer’s Res. Ther. 2021, 13, 80. [Google Scholar] [CrossRef] [PubMed]
- Mintun, M.A.; Lo, A.C.; Duggan Evans, C.; Wessels, A.M.; Ardayfio, P.A.; Andersen, S.W.; Shcherbinin, S.; Sparks, J.; Sims, J.R.; Brys, M.; et al. Donanemab in Early Alzheimer’s Disease. N. Engl. J. Med. 2021, 384, 1691–1704. [Google Scholar] [CrossRef] [PubMed]
- Budd Haeberlein, S.; Aisen, P.S.; Barkhof, F.; Chalkias, S.; Chen, T.; Cohen, S.; Dent, G.; Hansson, O.; Harrison, K.; von Hehn, C.; et al. Two Randomized Phase 3 Studies of Aducanumab in Early Alzheimer’s Disease. J. Prev. Alzheimer’s Dis. 2022, 9, 197–210. [Google Scholar] [CrossRef] [PubMed]
- Sevigny, J.; Chiao, P.; Bussiere, T.; Weinreb, P.H.; Williams, L.; Maier, M.; Dunstan, R.; Salloway, S.; Chen, T.; Ling, Y.; et al. The antibody aducanumab reduces Aβ plaques in Alzheimer’s disease. Nature 2016, 537, 50–56. [Google Scholar] [CrossRef]
- Salloway, S.; Farlow, M.; McDade, E.; Clifford, D.B.; Wang, G.; Llibre-Guerra, J.J.; Hitchcock, J.M.; Mills, S.L.; Santacruz, A.M.; Aschenbrenner, A.J.; et al. A trial of gantenerumab or solanezumab in dominantly inherited Alzheimer’s disease. Nat. Med. 2021, 27, 1187–1196. [Google Scholar] [CrossRef]
- Piller, C. Report on trial death stokes Alzheimer’s drug fears. Science 2023, 380, 122–123. [Google Scholar] [CrossRef] [PubMed]
- Salloway, S.; Chalkias, S.; Barkhof, F.; Burkett, P.; Barakos, J.; Purcell, D.; Suhy, J.; Forrestal, F.; Tian, Y.; Umans, K.; et al. Amyloid-Related Imaging Abnormalities in 2 Phase 3 Studies Evaluating Aducanumab in Patients with Early Alzheimer Disease. JAMA Neurol. 2022, 79, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Withington, C.G.; Turner, R.S. Amyloid-Related Imaging Abnormalities with Anti-Amyloid Antibodies for the Treatment of Dementia Due to Alzheimer’s Disease. Front. Neurol. 2022, 13, 862369. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Chu, F.; Zhu, F.; Zhu, J. Impact of Anti-amyloid-β Monoclonal Antibodies on the Pathology and Clinical Profile of Alzheimer’s Disease: A Focus on Aducanumab and Lecanemab. Front. Aging Neurosci. 2022, 14, 870517. [Google Scholar] [CrossRef] [PubMed]
- Sarrazin, S.; Lamanna, W.C.; Esko, J.D. Heparan sulfate proteoglycans. Cold Spring Harb. Perspect. Biol. 2011, 3, a004952. [Google Scholar] [CrossRef]
- Qiu, H.; Shi, S.; Yue, J.; Xin, M.; Nairn, A.V.; Lin, L.; Liu, X.; Li, G.; Archer-Hartmann, S.A.; Dela Rosa, M.; et al. A mutant-cell library for systematic analysis of heparan sulfate structure-function relationships. Nat. Methods 2018, 15, 889–899. [Google Scholar] [CrossRef] [PubMed]
- Bishop, J.R.; Schuksz, M.; Esko, J.D. Heparan sulphate proteoglycans fine-tune mammalian physiology. Nature 2007, 446, 1030–1037. [Google Scholar] [CrossRef] [PubMed]
- Li, J.P.; Kusche-Gullberg, M. Heparan Sulfate: Biosynthesis, Structure, and Function. Int. Rev. Cell Mol. Biol. 2016, 325, 215–273. [Google Scholar] [CrossRef] [PubMed]
- Kraushaar, D.C.; Rai, S.; Condac, E.; Nairn, A.; Zhang, S.; Yamaguchi, Y.; Moremen, K.; Dalton, S.; Wang, L. Heparan sulfate facilitates FGF and BMP signaling to drive mesoderm differentiation of mouse embryonic stem cells. J. Biol. Chem. 2012, 287, 22691–22700. [Google Scholar] [CrossRef]
- Zhang, B.; Xiao, W.; Qiu, H.; Zhang, F.; Moniz, H.A.; Jaworski, A.; Condac, E.; Gutierrez-Sanchez, G.; Heiss, C.; Clugston, R.D.; et al. Heparan sulfate deficiency disrupts developmental angiogenesis and causes congenital diaphragmatic hernia. J. Clin. Investig. 2014, 124, 209–221. [Google Scholar] [CrossRef]
- Rai, S.; Alsaidan, O.A.; Yang, H.; Cai, H.; Wang, L. Heparan sulfate inhibits transforming growth factor β signaling and functions in cis and in trans to regulate prostate stem/progenitor cell activities. Glycobiology 2020, 30, 381–395. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Fuster, M.; Sriramarao, P.; Esko, J.D. Endothelial heparan sulfate deficiency impairs L-selectin- and chemokine-mediated neutrophil trafficking during inflammatory responses. Nat. Immunol. 2005, 6, 902–910. [Google Scholar] [CrossRef] [PubMed]
- Talsma, D.T.; Katta, K.; Ettema, M.A.B.; Kel, B.; Kusche-Gullberg, M.; Daha, M.R.; Stegeman, C.A.; van den Born, J.; Wang, L. Endothelial heparan sulfate deficiency reduces inflammation and fibrosis in murine diabetic nephropathy. Lab. Investig. 2018, 98, 427–438. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, H.; Ghazizadeh, M.; Sato, S.; Oguro, T.; Kawanami, O. Interaction between β-amyloid protein and heparan sulfate proteoglycans from the cerebral capillary basement membrane in Alzheimer’s disease. J. Clin. Neurosci. 2009, 16, 277–282. [Google Scholar] [CrossRef] [PubMed]
- Huynh, M.B.; Ouidja, M.O.; Chantepie, S.; Carpentier, G.; Maiza, A.; Zhang, G.; Vilares, J.; Raisman-Vozari, R.; Papy-Garcia, D. Glycosaminoglycans from Alzheimer’s disease hippocampus have altered capacities to bind and regulate growth factors activities and to bind tau. PLoS ONE 2019, 14, e0209573. [Google Scholar] [CrossRef] [PubMed]
- Hosono-Fukao, T.; Ohtake-Niimi, S.; Nishitsuji, K.; Hossain, M.M.; van Kuppevelt, T.H.; Michikawa, M.; Uchimura, K. RB4CD12 epitope expression and heparan sulfate disaccharide composition in brain vasculature. J. Neurosci. Res. 2011, 89, 1840–1848. [Google Scholar] [CrossRef] [PubMed]
- Snow, A.D.; Lara, S.; Nochlin, D.; Wight, T.N. Cationic dyes reveal proteoglycans structurally integrated within the characteristic lesions of Alzheimer’s disease. Acta Neuropathol. 1989, 78, 113–123. [Google Scholar] [CrossRef]
- Snow, A.D.; Mar, H.; Nochlin, D.; Kimata, K.; Kato, M.; Suzuki, S.; Hassell, J.; Wight, T.N. The presence of heparan sulfate proteoglycans in the neuritic plaques and congophilic angiopathy in Alzheimer’s disease. Am. J. Pathol. 1988, 133, 456–463. [Google Scholar] [PubMed]
- Snow, A.D.; Mar, H.; Nochlin, D.; Sekiguchi, R.T.; Kimata, K.; Koike, Y.; Wight, T.N. Early accumulation of heparan sulfate in neurons and in the β-amyloid protein-containing lesions of Alzheimer’s disease and Down’s syndrome. Am. J. Pathol. 1990, 137, 1253–1270. [Google Scholar] [PubMed]
- Snow, A.D.; Sekiguchi, R.T.; Nochlin, D.; Kalaria, R.N.; Kimata, K. Heparan sulfate proteoglycan in diffuse plaques of hippocampus but not of cerebellum in Alzheimer’s disease brain. Am. J. Pathol. 1994, 144, 337–347. [Google Scholar] [PubMed]
- Snow, A.D.; Wight, T.N. Proteoglycans in the pathogenesis of Alzheimer’s disease and other amyloidoses. Neurobiol. Aging 1989, 10, 481–497. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.H.; Kim, Y.H.; Hebisch, M.; Sliwinski, C.; Lee, S.; D’Avanzo, C.; Chen, H.; Hooli, B.; Asselin, C.; Muffat, J.; et al. A three-dimensional human neural cell culture model of Alzheimer’s disease. Nature 2014, 515, 274–278. [Google Scholar] [CrossRef]
- Ozsan McMillan, I.; Li, J.P.; Wang, L. Heparan sulfate proteoglycan in Alzheimer’s disease: Aberrant expression and functions in molecular pathways related to amyloid-β metabolism. Am. J. Physiol. Cell Physiol. 2023, 324, C893–C909. [Google Scholar] [CrossRef]
- Zhu, Y.; Gandy, L.; Zhang, F.; Liu, J.; Wang, C.; Blair, L.J.; Linhardt, R.J.; Wang, L. Heparan Sulfate Proteoglycans in Tauopathy. Biomolecules 2022, 12, 1792. [Google Scholar] [CrossRef]
- Lindahl, B.; Westling, C.; Gimenez-Gallego, G.; Lindahl, U.; Salmivirta, M. Common binding sites for β-amyloid fibrils and fibroblast growth factor-2 in heparan sulfate from human cerebral cortex. J. Biol. Chem. 1999, 274, 30631–30635. [Google Scholar] [CrossRef] [PubMed]
- Cui, H.; Hung, A.C.; Freeman, C.; Narkowicz, C.; Jacobson, G.A.; Small, D.H. Size and sulfation are critical for the effect of heparin on APP processing and Aβ production. J. Neurochem. 2012, 123, 447–457. [Google Scholar] [CrossRef] [PubMed]
- Watson, D.J.; Lander, A.D.; Selkoe, D.J. Heparin-binding properties of the amyloidogenic peptides Aβ and amylin. Dependence on aggregation state and inhibition by Congo red. J. Biol. Chem. 1997, 272, 31617–31624. [Google Scholar] [CrossRef] [PubMed]
- Castillo, G.M.; Lukito, W.; Wight, T.N.; Snow, A.D. The sulfate moieties of glycosaminoglycans are critical for the enhancement of β-amyloid protein fibril formation. J. Neurochem. 1999, 72, 1681–1687. [Google Scholar] [CrossRef]
- Perry, G.; Siedlak, S.L.; Richey, P.; Kawai, M.; Cras, P.; Kalaria, R.N.; Galloway, P.G.; Scardina, J.M.; Cordell, B.; Greenberg, B.D.; et al. Association of heparan sulfate proteoglycan with the neurofibrillary tangles of Alzheimer’s disease. J. Neurosci. 1991, 11, 3679–3683. [Google Scholar] [CrossRef]
- Perlmutter, L.S.; Chui, H.C.; Saperia, D.; Athanikar, J. Microangiopathy and the colocalization of heparan sulfate proteoglycan with amyloid in senile plaques of Alzheimer’s disease. Brain Res. 1990, 508, 13–19. [Google Scholar] [CrossRef]
- Sandwall, E.; O’Callaghan, P.; Zhang, X.; Lindahl, U.; Lannfelt, L.; Li, J.P. Heparan sulfate mediates amyloid-β internalization and cytotoxicity. Glycobiology 2010, 20, 533–541. [Google Scholar] [CrossRef]
- Kuwabara, K.; Nishitsuji, K.; Uchimura, K.; Hung, S.C.; Mizuguchi, M.; Nakajima, H.; Mikawa, S.; Kobayashi, N.; Saito, H.; Sakashita, N. Cellular interaction and cytotoxicity of the iowa mutation of apolipoprotein A-I (ApoA-IIowa) amyloid mediated by sulfate moieties of heparan sulfate. J. Biol. Chem. 2015, 290, 24210–24221. [Google Scholar] [CrossRef] [PubMed]
- Stopschinski, B.E.; Holmes, B.B.; Miller, G.M.; Manon, V.A.; Vaquer-Alicea, J.; Prueitt, W.L.; Hsieh-Wilson, L.C.; Diamond, M.I. Specific glycosaminoglycan chain length and sulfation patterns are required for cell uptake of tau versus alpha-synuclein and β-amyloid aggregates. J. Biol. Chem. 2018, 293, 10826–10840. [Google Scholar] [CrossRef]
- Chen, L.; Sanderson, R.D. Heparanase regulates levels of syndecan-1 in the nucleus. PLoS ONE 2009, 4, e4947. [Google Scholar] [CrossRef] [PubMed]
- Clausen, T.M.; Sandoval, D.R.; Spliid, C.B.; Pihl, J.; Perrett, H.R.; Painter, C.D.; Narayanan, A.; Majowicz, S.A.; Kwong, E.M.; McVicar, R.N.; et al. SARS-CoV-2 Infection Depends on Cellular Heparan Sulfate and ACE2. Cell 2020, 183, 1043–1057.e15. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wang, B.; O’Callaghan, P.; Hjertstrom, E.; Jia, J.; Gong, F.; Zcharia, E.; Nilsson, L.N.; Lannfelt, L.; Vlodavsky, I.; et al. Heparanase overexpression impairs inflammatory response and macrophage-mediated clearance of amyloid-β in murine brain. Acta Neuropathol. 2012, 124, 465–478. [Google Scholar] [CrossRef] [PubMed]
- Jendresen, C.B.; Cui, H.; Zhang, X.; Vlodavsky, I.; Nilsson, L.N.; Li, J.P. Overexpression of heparanase lowers the amyloid burden in amyloid-β precursor protein transgenic mice. J. Biol. Chem. 2015, 290, 5053–5064. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.C.; Zhao, N.; Yamaguchi, Y.; Cirrito, J.R.; Kanekiyo, T.; Holtzman, D.M.; Bu, G. Neuronal heparan sulfates promote amyloid pathology by modulating brain amyloid-β clearance and aggregation in Alzheimer’s disease. Sci. Transl. Med. 2016, 8, 332ra344. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Arnold, K.; Dhurandahare, V.M.; Xu, Y.; Pagadala, V.; Labra, E.; Jeske, W.; Fareed, J.; Gearing, M.; Liu, J. Analysis of 3-O-Sulfated Heparan Sulfate Using Isotopically Labeled Oligosaccharide Calibrants. Anal. Chem. 2022, 94, 2950–2957. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Patel, V.N.; Song, X.; Xu, Y.; Kaminski, A.M.; Doan, V.U.; Su, G.; Liao, Y.; Mah, D.; Zhang, F.; et al. Increased 3-O-sulfated heparan sulfate in Alzheimer’s disease brain is associated with genetic risk gene HS3ST1. Sci. Adv. 2023, 9, eadf6232. [Google Scholar] [CrossRef]
- Stoler-Barak, L.; Moussion, C.; Shezen, E.; Hatzav, M.; Sixt, M.; Alon, R. Blood vessels pattern heparan sulfate gradients between their apical and basolateral aspects. PLoS ONE 2014, 9, e85699. [Google Scholar] [CrossRef]
- Reijmer, Y.D.; Fotiadis, P.; Riley, G.A.; Xiong, L.; Charidimou, A.; Boulouis, G.; Ayres, A.M.; Schwab, K.; Rosand, J.; Gurol, M.E.; et al. Progression of Brain Network Alterations in Cerebral Amyloid Angiopathy. Stroke 2016, 47, 2470–2475. [Google Scholar] [CrossRef]
- Liu, C.C.; Liu, C.C.; Kanekiyo, T.; Xu, H.; Bu, G. Apolipoprotein E and Alzheimer disease: Risk, mechanisms and therapy. Nat. Rev. Neurol. 2013, 9, 106–118. [Google Scholar] [CrossRef] [PubMed]
- Buckley, R.F.; Mormino, E.C.; Rabin, J.S.; Hohman, T.J.; Landau, S.; Hanseeuw, B.J.; Jacobs, H.I.L.; Papp, K.V.; Amariglio, R.E.; Properzi, M.J.; et al. Sex Differences in the Association of Global Amyloid and Regional Tau Deposition Measured by Positron Emission Tomography in Clinically Normal Older Adults. JAMA Neurol. 2019, 76, 542–551. [Google Scholar] [CrossRef] [PubMed]
- Nelson, P.T.; Pious, N.M.; Jicha, G.A.; Wilcock, D.M.; Fardo, D.W.; Estus, S.; Rebeck, G.W. APOE-epsilon2 and APOE-epsilon4 correlate with increased amyloid accumulation in cerebral vasculature. J. Neuropathol. Exp. Neurol. 2013, 72, 708–715. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, Y.; Deguchi, N.; Takagi, C.; Tanaka, M.; Dhanasekaran, P.; Nakano, M.; Handa, T.; Phillips, M.C.; Lund-Katz, S.; Saito, H. Role of the N- and C-terminal domains in binding of apolipoprotein E isoforms to heparan sulfate and dermatan sulfate: A surface plasmon resonance study. Biochemistry 2008, 47, 6702–6710. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Zhao, J.; Atagi, Y.; Nielsen, H.M.; Liu, C.C.; Zheng, H.; Shinohara, M.; Kanekiyo, T.; Bu, G. Apolipoprotein E lipoprotein particles inhibit amyloid-β uptake through cell surface heparan sulphate proteoglycan. Mol. Neurodegener. 2016, 11, 37. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, S.M.; Rebeck, G.W.; Vonsattel, J.P.; Gomez-Isla, T.; Hyman, B.T. Apolipoprotein E epsilon 4 and cerebral hemorrhage associated with amyloid angiopathy. Ann. Neurol. 1995, 38, 254–259. [Google Scholar] [CrossRef] [PubMed]
- Sperling, R.A.; Jack, C.R., Jr.; Black, S.E.; Frosch, M.P.; Greenberg, S.M.; Hyman, B.T.; Scheltens, P.; Carrillo, M.C.; Thies, W.; Bednar, M.M.; et al. Amyloid-related imaging abnormalities in amyloid-modifying therapeutic trials: Recommendations from the Alzheimer’s Association Research Roundtable Workgroup. Alzheimer’s Dement. 2011, 7, 367–385. [Google Scholar] [CrossRef] [PubMed]
- Kakuda, N.; Miyasaka, T.; Iwasaki, N.; Nirasawa, T.; Wada-Kakuda, S.; Takahashi-Fujigasaki, J.; Murayama, S.; Ihara, Y.; Ikegawa, M. Distinct deposition of amyloid-β species in brains with Alzheimer’s disease pathology visualized with MALDI imaging mass spectrometry. Acta Neuropathol. Commun. 2017, 5, 73. [Google Scholar] [CrossRef]
- Gravina, S.A.; Ho, L.; Eckman, C.B.; Long, K.E.; Otvos, L., Jr.; Younkin, L.H.; Suzuki, N.; Younkin, S.G. Amyloid β protein (Aβ) in Alzheimer’s disease brain. Biochemical and immunocytochemical analysis with antibodies specific for forms ending at Aβ40 or Aβ42(43). J. Biol. Chem. 1995, 270, 7013–7016. [Google Scholar] [CrossRef]
- Roher, A.E.; Lowenson, J.D.; Clarke, S.; Woods, A.S.; Cotter, R.J.; Gowing, E.; Ball, M.J. β-Amyloid-(1-42) is a major component of cerebrovascular amyloid deposits: Implications for the pathology of Alzheimer disease. Proc. Natl. Acad. Sci. USA 1993, 90, 10836–10840. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.L.; Papayannopoulos, I.A.; Styles, J.; Bobin, S.A.; Lin, Y.Y.; Biemann, K.; Iqbal, K. Peptide compositions of the cerebrovascular and senile plaque core amyloid deposits of Alzheimer’s disease. Arch. Biochem. Biophys. 1993, 301, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Madine, J.; Pandya, M.J.; Hicks, M.R.; Rodger, A.; Yates, E.A.; Radford, S.E.; Middleton, D.A. Site-specific identification of an Aβ fibril-heparin interaction site by using solid-state NMR spectroscopy. Angew. Chem. Int. Ed. Engl. 2012, 51, 13140–13143. [Google Scholar] [CrossRef] [PubMed]
- Madine, J.; Clayton, J.C.; Yates, E.A.; Middleton, D.A. Exploiting a 13C-labelled heparin analogue for in situ solid-state NMR investigations of peptide-glycan interactions within amyloid fibrils. Org. Biomol. Chem. 2009, 7, 2414–2420. [Google Scholar] [CrossRef] [PubMed]
- Snow, A.D.; Cummings, J.A.; Lake, T. The Unifying Hypothesis of Alzheimer’s Disease: Heparan Sulfate Proteoglycans/Glycosaminoglycans Are Key as First Hypothesized over 30 Years Ago. Front. Aging Neurosci. 2021, 13, 710683. [Google Scholar] [CrossRef] [PubMed]
- Tarasoff-Conway, J.M.; Carare, R.O.; Osorio, R.S.; Glodzik, L.; Butler, T.; Fieremans, E.; Axel, L.; Rusinek, H.; Nicholson, C.; Zlokovic, B.V.; et al. Clearance systems in the brain-implications for Alzheimer disease. Nat. Rev. Neurol. 2015, 11, 457–470. [Google Scholar] [CrossRef] [PubMed]
- Carare, R.O.; Bernardes-Silva, M.; Newman, T.A.; Page, A.M.; Nicoll, J.A.; Perry, V.H.; Weller, R.O. Solutes, but not cells, drain from the brain parenchyma along basement membranes of capillaries and arteries: Significance for cerebral amyloid angiopathy and neuroimmunology. Neuropathol. Appl. Neurobiol. 2008, 34, 131–144. [Google Scholar] [CrossRef] [PubMed]
- Albargothy, N.J.; Johnston, D.A.; MacGregor-Sharp, M.; Weller, R.O.; Verma, A.; Hawkes, C.A.; Carare, R.O. Convective influx/glymphatic system: Tracers injected into the CSF enter and leave the brain along separate periarterial basement membrane pathways. Acta Neuropathol. 2018, 136, 139–152. [Google Scholar] [CrossRef]
- Keable, A.; Fenna, K.; Yuen, H.M.; Johnston, D.A.; Smyth, N.R.; Smith, C.; Al-Shahi Salman, R.; Samarasekera, N.; Nicoll, J.A.; Attems, J.; et al. Deposition of amyloid β in the walls of human leptomeningeal arteries in relation to perivascular drainage pathways in cerebral amyloid angiopathy. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2016, 1862, 1037–1046. [Google Scholar] [CrossRef]
- Murali, S.; Leong, D.F.; Lee, J.J.; Cool, S.M.; Nurcombe, V. Comparative assessment of the effects of gender-specific heparan sulfates on mesenchymal stem cells. J. Biol. Chem. 2011, 286, 17755–17765. [Google Scholar] [CrossRef]
- Casasnovas, J.; Damron, C.L.; Jarrell, J.; Orr, K.S.; Bone, R.N.; Archer-Hartmann, S.; Azadi, P.; Kua, K.L. Offspring of Obese Dams Exhibit Sex-Differences in Pancreatic Heparan Sulfate Glycosaminoglycans and Islet Insulin Secretion. Front. Endocrinol. 2021, 12, 658439. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, I.; Noguchi, N.; Nata, K.; Yamada, S.; Kaneiwa, T.; Mizumoto, S.; Ikeda, T.; Sugihara, K.; Asano, M.; Yoshikawa, T.; et al. Important role of heparan sulfate in postnatal islet growth and insulin secretion. Biochem. Biophys. Res. Commun. 2009, 383, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Akhter, F.; Persaud, A.; Zaokari, Y.; Zhao, Z.; Zhu, D. Vascular Dementia and Underlying Sex Differences. Front. Aging Neurosci. 2021, 13, 720715. [Google Scholar] [CrossRef] [PubMed]
- Saunders, A.M.; Strittmatter, W.J.; Schmechel, D.; George-Hyslop, P.H.; Pericak-Vance, M.A.; Joo, S.H.; Rosi, B.L.; Gusella, J.F.; Crapper-MacLachlan, D.R.; Alberts, M.J.; et al. Association of apolipoprotein E allele epsilon 4 with late-onset familial and sporadic Alzheimer’s disease. Neurology 1993, 43, 1467–1472. [Google Scholar] [CrossRef] [PubMed]
- Corder, E.H.; Saunders, A.M.; Strittmatter, W.J.; Schmechel, D.E.; Gaskell, P.C.; Small, G.W.; Roses, A.D.; Haines, J.L.; Pericak-Vance, M.A. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 1993, 261, 921–923. [Google Scholar] [CrossRef] [PubMed]
- Farrer, L.A.; Cupples, L.A.; Haines, J.L.; Hyman, B.; Kukull, W.A.; Mayeux, R.; Myers, R.H.; Pericak-Vance, M.A.; Risch, N.; van Duijn, C.M. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. APOE and Alzheimer Disease Meta Analysis Consortium. JAMA 1997, 278, 1349–1356. [Google Scholar] [CrossRef] [PubMed]
- Kok, E.; Haikonen, S.; Luoto, T.; Huhtala, H.; Goebeler, S.; Haapasalo, H.; Karhunen, P.J. Apolipoprotein E-dependent accumulation of Alzheimer disease-related lesions begins in middle age. Ann. Neurol. 2009, 65, 650–657. [Google Scholar] [CrossRef] [PubMed]
- Morris, J.C.; Roe, C.M.; Xiong, C.; Fagan, A.M.; Goate, A.M.; Holtzman, D.M.; Mintun, M.A. APOE predicts amyloid-β but not tau Alzheimer pathology in cognitively normal aging. Ann. Neurol. 2010, 67, 122–131. [Google Scholar] [CrossRef] [PubMed]
- Hawkes, C.A.; Sullivan, P.M.; Hands, S.; Weller, R.O.; Nicoll, J.A.; Carare, R.O. Disruption of arterial perivascular drainage of amyloid-β from the brains of mice expressing the human APOE epsilon4 allele. PLoS ONE 2012, 7, e41636. [Google Scholar] [CrossRef]
- Robert, J.; Button, E.B.; Yuen, B.; Gilmour, M.; Kang, K.; Bahrabadi, A.; Stukas, S.; Zhao, W.; Kulic, I.; Wellington, C.L. Clearance of β-amyloid is facilitated by apolipoprotein E and circulating high-density lipoproteins in bioengineered human vessels. eLife 2017, 6, e29595. [Google Scholar] [CrossRef]
- DeMattos, R.B.; Cirrito, J.R.; Parsadanian, M.; May, P.C.; O’Dell, M.A.; Taylor, J.W.; Harmony, J.A.; Aronow, B.J.; Bales, K.R.; Paul, S.M.; et al. ApoE and clusterin cooperatively suppress Aβ levels and deposition: Evidence that ApoE regulates extracellular Aβ metabolism in vivo. Neuron 2004, 41, 193–202. [Google Scholar] [CrossRef] [PubMed]
- Kanekiyo, T.; Bu, G. The low-density lipoprotein receptor-related protein 1 and amyloid-β clearance in Alzheimer’s disease. Front. Aging Neurosci. 2014, 6, 93. [Google Scholar] [CrossRef] [PubMed]
- Mawuenyega, K.G.; Sigurdson, W.; Ovod, V.; Munsell, L.; Kasten, T.; Morris, J.C.; Yarasheski, K.E.; Bateman, R.J. Decreased clearance of CNS β-amyloid in Alzheimer’s disease. Science 2010, 330, 1774. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Rusinek, H.; Butler, T.; Glodzik, L.; Pirraglia, E.; Babich, J.; Mozley, P.D.; Nehmeh, S.; Pahlajani, S.; Wang, X.; et al. Decreased CSF clearance and increased brain amyloid in Alzheimer’s disease. Fluids Barriers CNS 2022, 19, 21. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient Diagnosis | Total Number | Gender (M/F) | Age at Death (Ave ± SEM) |
|---|---|---|---|
| None AD | 10 | 4/6 | 71.00 ± 5.62 |
| AD, no CAA | 7 | 3/4 | 76.43 ± 3.24 |
| AD, mild CAA | 13 | 8/5 | 76.46 ± 3.43 |
| AD, severe CAA | 12 | 7/5 | 77.75 ± 2.17 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
McMillan, I.O.; Gearing, M.; Wang, L. Vascular Heparan Sulfate and Amyloid-β in Alzheimer’s Disease Patients. Int. J. Mol. Sci. 2024, 25, 3964. https://doi.org/10.3390/ijms25073964
McMillan IO, Gearing M, Wang L. Vascular Heparan Sulfate and Amyloid-β in Alzheimer’s Disease Patients. International Journal of Molecular Sciences. 2024; 25(7):3964. https://doi.org/10.3390/ijms25073964
Chicago/Turabian StyleMcMillan, Ilayda Ozsan, Marla Gearing, and Lianchun Wang. 2024. "Vascular Heparan Sulfate and Amyloid-β in Alzheimer’s Disease Patients" International Journal of Molecular Sciences 25, no. 7: 3964. https://doi.org/10.3390/ijms25073964
APA StyleMcMillan, I. O., Gearing, M., & Wang, L. (2024). Vascular Heparan Sulfate and Amyloid-β in Alzheimer’s Disease Patients. International Journal of Molecular Sciences, 25(7), 3964. https://doi.org/10.3390/ijms25073964