

Protein Arginine Methyltransferases in Pancreatic Ductal Adenocarcinoma: New Molecular Targets for Therapy



Abstract
1. Introduction: Pancreatic Ductal Adenocarcinoma and the Need for New Therapeutics
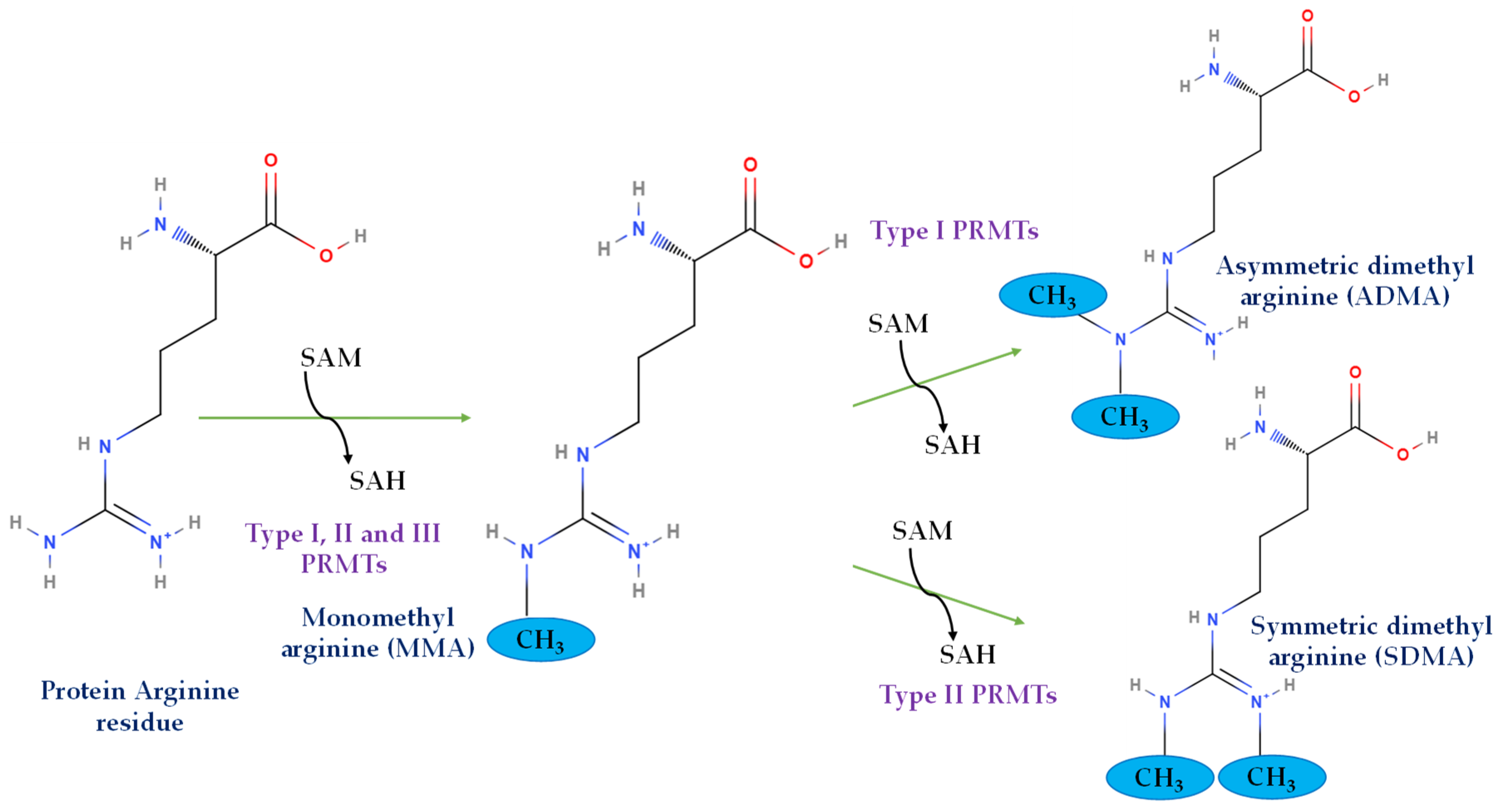
2. Arginine Methylation and PRMTs
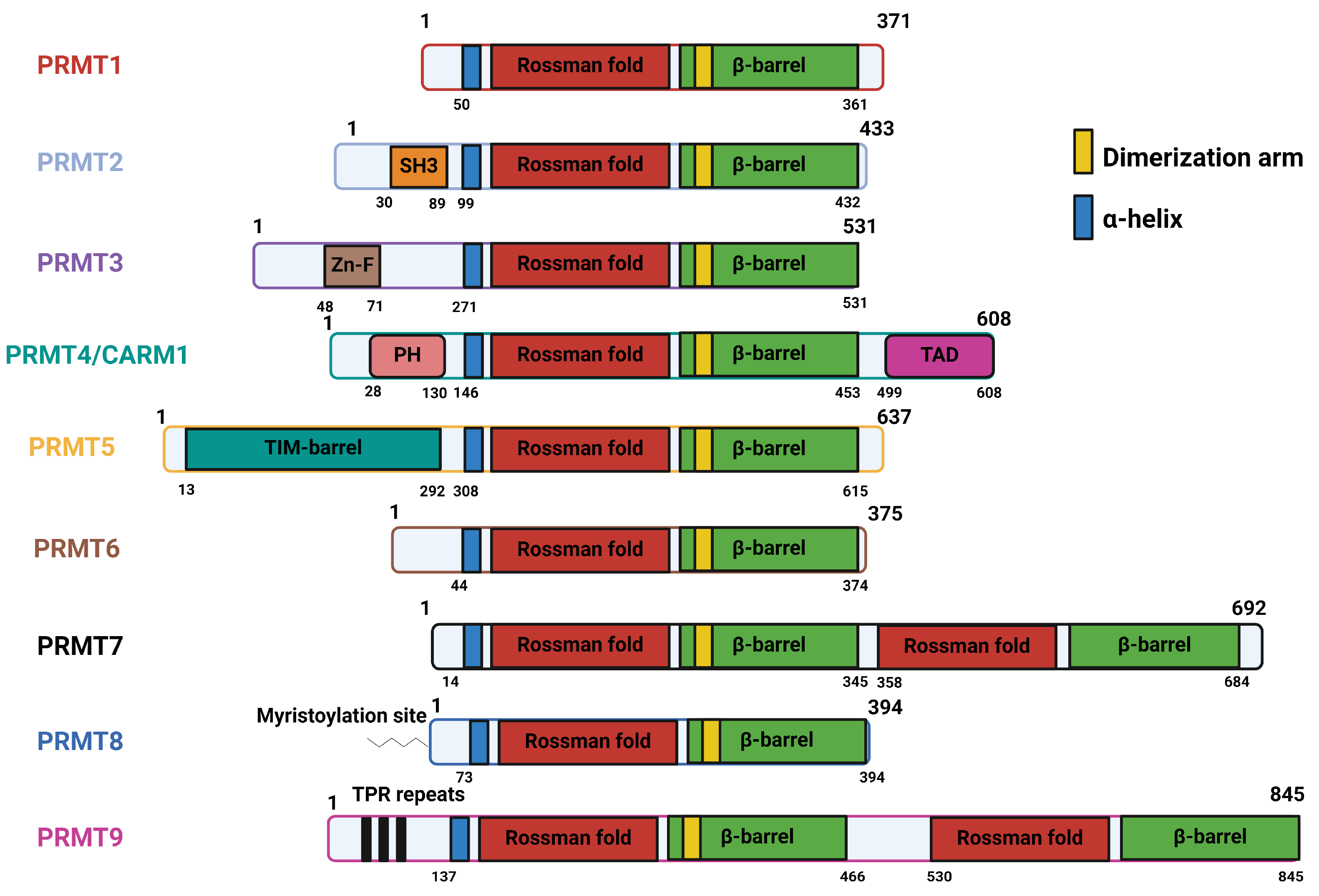
2.1. Structural Basis, Localization, and Motif Preference of PRMTs
2.2. Physiological Role of PRMTs
3. PRMTs Are Involved in the Pathogenesis and Progression of PDAC
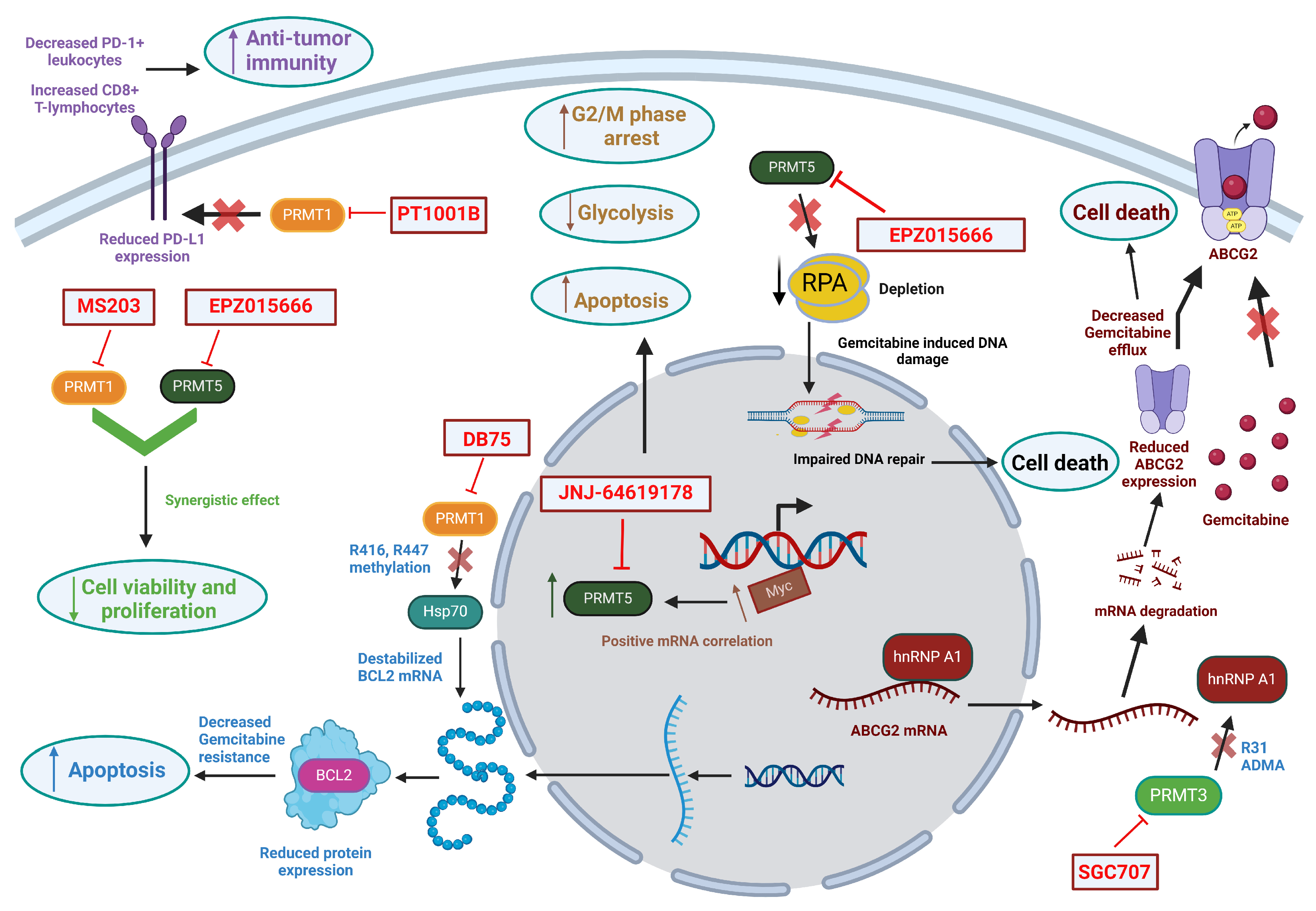
3.1. PRMT1
3.2. PRMT3
3.3. PRMT5
4. Inhibitors of PRMTs
5. Perspectives
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Sarantis, P.; Koustas, E.; Papadimitropoulou, A.; Papavassiliou, A.G.; Karamouzis, M.V. Pancreatic ductal adenocarcinoma: Treatment hurdles, tumor microenvironment and immunotherapy. World J. Gastrointest. Oncol. 2020, 12, 173–181. [Google Scholar] [CrossRef]
- Rahib, L.; Wehner, M.R.; Matrisian, L.M.; Nead, K.T. Estimated Projection of US Cancer Incidence and Death to 2040. JAMA Netw. Open 2021, 4, e214708. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Giaquinto, A.N.; Jemal, A. Cancer statistics, 2024. CA Cancer J. Clin. 2024, 74, 12–49. [Google Scholar] [CrossRef] [PubMed]
- Kleeff, J.; Korc, M.; Apte, M.; La Vecchia, C.; Johnson, C.D.; Biankin, A.V.; Neale, R.E.; Tempero, M.; Tuveson, D.A.; Hruban, R.H.; et al. Pancreatic cancer. Nat. Rev. Dis. Primers 2016, 2, 16022. [Google Scholar] [CrossRef] [PubMed]
- Farr, K.P.; Moses, D.; Haghighi, K.S.; Phillips, P.A.; Hillenbrand, C.M.; Chua, B.H. Imaging Modalities for Early Detection of Pancreatic Cancer: Current State and Future Research Opportunities. Cancers 2022, 14, 2539. [Google Scholar] [CrossRef] [PubMed]
- Morani, A.C.; Hanafy, A.K.; Ramani, N.S.; Katabathina, V.S.; Yedururi, S.; Dasyam, A.K.; Prasad, S.R. Hereditary and Sporadic Pancreatic Ductal Adenocarcinoma: Current Update on Genetics and Imaging. Radiol. Imaging Cancer 2020, 2, e190020. [Google Scholar] [CrossRef] [PubMed]
- Singhi, A.D.; Koay, E.J.; Chari, S.T.; Maitra, A. Early Detection of Pancreatic Cancer: Opportunities and Challenges. Gastroenterology 2019, 156, 2024–2040. [Google Scholar] [CrossRef] [PubMed]
- Halbrook, C.J.; Lyssiotis, C.A.; Pasca di Magliano, M.; Maitra, A. Pancreatic cancer: Advances and challenges. Cell 2023, 186, 1729–1754. [Google Scholar] [CrossRef] [PubMed]
- Conroy, T.; Hammel, P.; Hebbar, M.; Ben Abdelghani, M.; Wei, A.C.; Raoul, J.L.; Chone, L.; Francois, E.; Artru, P.; Biagi, J.J.; et al. FOLFIRINOX or Gemcitabine as Adjuvant Therapy for Pancreatic Cancer. N. Engl. J. Med. 2018, 379, 2395–2406. [Google Scholar] [CrossRef]
- Amrutkar, M.; Gladhaug, I.P. Pancreatic Cancer Chemoresistance to Gemcitabine. Cancers 2017, 9, 157. [Google Scholar] [CrossRef]
- de Jesus, V.H.F.; Camandaroba, M.P.G.; Donadio, M.D.S.; Cabral, A.; Muniz, T.P.; de Moura Leite, L.; Sant’Ana, L.F. Retrospective comparison of the efficacy and the toxicity of standard and modified FOLFIRINOX regimens in patients with metastatic pancreatic adenocarcinoma. J. Gastrointest. Oncol. 2018, 9, 694–707. [Google Scholar] [CrossRef]
- Timmer, F.E.F.; Geboers, B.; Nieuwenhuizen, S.; Dijkstra, M.; Schouten, E.A.C.; Puijk, R.S.; de Vries, J.J.J.; van den Tol, M.P.; Bruynzeel, A.M.E.; Streppel, M.M.; et al. Pancreatic Cancer and Immunotherapy: A Clinical Overview. Cancers 2021, 13, 4138. [Google Scholar] [CrossRef] [PubMed]
- Qian, Y.; Gong, Y.; Fan, Z.; Luo, G.; Huang, Q.; Deng, S.; Cheng, H.; Jin, K.; Ni, Q.; Yu, X.; et al. Molecular alterations and targeted therapy in pancreatic ductal adenocarcinoma. J. Hematol. Oncol. 2020, 13, 130. [Google Scholar] [CrossRef] [PubMed]
- Balachandran, V.P.; Beatty, G.L.; Dougan, S.K. Broadening the Impact of Immunotherapy to Pancreatic Cancer: Challenges and Opportunities. Gastroenterology 2019, 156, 2056–2072. [Google Scholar] [CrossRef]
- Fang, Y.T.; Yang, W.W.; Niu, Y.R.; Sun, Y.K. Recent advances in targeted therapy for pancreatic adenocarcinoma. World J. Gastrointest. Oncol. 2023, 15, 571–595. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network. Integrated Genomic Characterization of Pancreatic Ductal Adenocarcinoma. Cancer Cell 2017, 32, 185–203.e113. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Allen, S.; Blake, J.F.; Bowcut, V.; Briere, D.M.; Calinisan, A.; Dahlke, J.R.; Fell, J.B.; Fischer, J.P.; Gunn, R.J.; et al. Identification of MRTX1133, a Noncovalent, Potent, and Selective KRAS(G12D) Inhibitor. J. Med. Chem. 2022, 65, 3123–3133. [Google Scholar] [CrossRef] [PubMed]
- Kemp, S.B.; Cheng, N.; Markosyan, N.; Sor, R.; Kim, I.K.; Hallin, J.; Shoush, J.; Quinones, L.; Brown, N.V.; Bassett, J.B.; et al. Efficacy of a Small-Molecule Inhibitor of KrasG12D in Immunocompetent Models of Pancreatic Cancer. Cancer Discov. 2023, 13, 298–311. [Google Scholar] [CrossRef]
- Viale, A.; Pettazzoni, P.; Lyssiotis, C.A.; Ying, H.; Sanchez, N.; Marchesini, M.; Carugo, A.; Green, T.; Seth, S.; Giuliani, V.; et al. Oncogene ablation-resistant pancreatic cancer cells depend on mitochondrial function. Nature 2014, 514, 628–632. [Google Scholar] [CrossRef]
- Kapoor, A.; Yao, W.; Ying, H.; Hua, S.; Liewen, A.; Wang, Q.; Zhong, Y.; Wu, C.J.; Sadanandam, A.; Hu, B.; et al. Yap1 activation enables bypass of oncogenic Kras addiction in pancreatic cancer. Cell 2014, 158, 185–197. [Google Scholar] [CrossRef]
- Bryant, K.L.; Stalnecker, C.A.; Zeitouni, D.; Klomp, J.E.; Peng, S.; Tikunov, A.P.; Gunda, V.; Pierobon, M.; Waters, A.M.; George, S.D.; et al. Combination of ERK and autophagy inhibition as a treatment approach for pancreatic cancer. Nat. Med. 2019, 25, 628–640. [Google Scholar] [CrossRef]
- Daemen, A.; Peterson, D.; Sahu, N.; McCord, R.; Du, X.; Liu, B.; Kowanetz, K.; Hong, R.; Moffat, J.; Gao, M.; et al. Metabolite profiling stratifies pancreatic ductal adenocarcinomas into subtypes with distinct sensitivities to metabolic inhibitors. Proc. Natl. Acad. Sci. USA 2015, 112, E4410–E4417. [Google Scholar] [CrossRef] [PubMed]
- Provenzano, P.P.; Cuevas, C.; Chang, A.E.; Goel, V.K.; Von Hoff, D.D.; Hingorani, S.R. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell 2012, 21, 418–429. [Google Scholar] [CrossRef] [PubMed]
- Jacobetz, M.A.; Chan, D.S.; Neesse, A.; Bapiro, T.E.; Cook, N.; Frese, K.K.; Feig, C.; Nakagawa, T.; Caldwell, M.E.; Zecchini, H.I.; et al. Hyaluronan impairs vascular function and drug delivery in a mouse model of pancreatic cancer. Gut 2013, 62, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Bear, A.S.; Vonderheide, R.H.; O’Hara, M.H. Challenges and Opportunities for Pancreatic Cancer Immunotherapy. Cancer Cell 2020, 38, 788–802. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Bedford, M.T. Protein arginine methyltransferases and cancer. Nat. Rev. Cancer 2013, 13, 37–50. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.W.; Cho, Y.; Bae, G.U.; Kim, S.N.; Kim, Y.K. Protein arginine methyltransferases: Promising targets for cancer therapy. Exp. Mol. Med. 2021, 53, 788–808. [Google Scholar] [CrossRef]
- Feustel, K.; Falchook, G.S. Protein Arginine Methyltransferase 5 (PRMT5) Inhibitors in Oncology Clinical Trials: A review. J. Immunother. Precis. Oncol. 2022, 5, 58–67. [Google Scholar] [CrossRef] [PubMed]
- Ramazi, S.; Zahiri, J. Posttranslational modifications in proteins: Resources, tools and prediction methods. Database 2021, 2021, baab012. [Google Scholar] [CrossRef]
- Clarke, S.G. Protein methylation at the surface and buried deep: Thinking outside the histone box. Trends Biochem. Sci. 2013, 38, 243–252. [Google Scholar] [CrossRef]
- Bhat, K.P.; Umit Kaniskan, H.; Jin, J.; Gozani, O. Epigenetics and beyond: Targeting writers of protein lysine methylation to treat disease. Nat. Rev. Drug Discov. 2021, 20, 265–286. [Google Scholar] [CrossRef] [PubMed]
- Lanouette, S.; Mongeon, V.; Figeys, D.; Couture, J.F. The functional diversity of protein lysine methylation. Mol. Syst. Biol. 2014, 10, 724. [Google Scholar] [CrossRef] [PubMed]
- Aziz, N.; Hong, Y.H.; Kim, H.G.; Kim, J.H.; Cho, J.Y. Tumor-suppressive functions of protein lysine methyltransferases. Exp. Mol. Med. 2023, 55, 2475–2497. [Google Scholar] [CrossRef] [PubMed]
- Bedford, M.T.; Clarke, S.G. Protein arginine methylation in mammals: Who, what, and why. Mol. Cell 2009, 33, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Blanc, R.S.; Richard, S. Arginine Methylation: The Coming of Age. Mol. Cell 2017, 65, 8–24. [Google Scholar] [CrossRef] [PubMed]
- Dong, R.; Li, X.; Lai, K.O. Activity and Function of the PRMT8 Protein Arginine Methyltransferase in Neurons. Life 2021, 11, 1132. [Google Scholar] [CrossRef]
- Jarrold, J.; Davies, C.C. PRMTs and Arginine Methylation: Cancer’s Best-Kept Secret? Trends Mol. Med. 2019, 25, 993–1009. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, M.; Toma-Fukai, S.; Kim, J.D.; Fukamizu, A.; Shimizu, T. Protein arginine methyltransferase 7 has a novel homodimer-like structure formed by tandem repeats. FEBS Lett. 2014, 588, 1942–1948. [Google Scholar] [CrossRef] [PubMed]
- Hadjikyriacou, A.; Yang, Y.; Espejo, A.; Bedford, M.T.; Clarke, S.G. Unique Features of Human Protein Arginine Methyltransferase 9 (PRMT9) and Its Substrate RNA Splicing Factor SF3B2. J. Biol. Chem. 2015, 290, 16723–16743. [Google Scholar] [CrossRef]
- Thandapani, P.; O’Connor, T.R.; Bailey, T.L.; Richard, S. Defining the RGG/RG motif. Mol. Cell 2013, 50, 613–623. [Google Scholar] [CrossRef]
- Musiani, D.; Bok, J.; Massignani, E.; Wu, L.; Tabaglio, T.; Ippolito, M.R.; Cuomo, A.; Ozbek, U.; Zorgati, H.; Ghoshdastider, U.; et al. Proteomics profiling of arginine methylation defines PRMT5 substrate specificity. Sci. Signal. 2019, 12, eaat8388. [Google Scholar] [CrossRef]
- Kumar, P.; Joy, J.; Pandey, A.; Gupta, D. PRmePRed: A protein arginine methylation prediction tool. PLoS ONE 2017, 12, e0183318. [Google Scholar] [CrossRef]
- Deng, W.; Wang, Y.; Ma, L.; Zhang, Y.; Ullah, S.; Xue, Y. Computational prediction of methylation types of covalently modified lysine and arginine residues in proteins. Brief. Bioinform. 2017, 18, 647–658. [Google Scholar] [CrossRef]
- Rust, H.L.; Subramanian, V.; West, G.M.; Young, D.D.; Schultz, P.G.; Thompson, P.R. Using unnatural amino acid mutagenesis to probe the regulation of PRMT1. ACS Chem. Biol. 2014, 9, 649–655. [Google Scholar] [CrossRef]
- Wagner, S.; Weber, S.; Kleinschmidt, M.A.; Nagata, K.; Bauer, U.M. SET-mediated promoter hypoacetylation is a prerequisite for coactivation of the estrogen-responsive pS2 gene by PRMT1. J. Biol. Chem. 2006, 281, 27242–27250. [Google Scholar] [CrossRef] [PubMed]
- Fulton, M.D.; Dang, T.; Brown, T.; Zheng, Y.G. Effects of substrate modifications on the arginine dimethylation activities of PRMT1 and PRMT5. Epigenetics 2022, 17, 1–18. [Google Scholar] [CrossRef]
- Hartley, A.V.; Lu, T. Modulating the modulators: Regulation of protein arginine methyltransferases by post-translational modifications. Drug Discov. Today 2020, 25, 1735–1743. [Google Scholar] [CrossRef] [PubMed]
- Feng, Q.; He, B.; Jung, S.Y.; Song, Y.; Qin, J.; Tsai, S.Y.; Tsai, M.J.; O’Malley, B.W. Biochemical control of CARM1 enzymatic activity by phosphorylation. J. Biol. Chem. 2009, 284, 36167–36174. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Zhao, X.; Perna, F.; Wang, L.; Koppikar, P.; Abdel-Wahab, O.; Harr, M.W.; Levine, R.L.; Xu, H.; Tefferi, A.; et al. JAK2V617F-mediated phosphorylation of PRMT5 downregulates its methyltransferase activity and promotes myeloproliferation. Cancer Cell 2011, 19, 283–294. [Google Scholar] [CrossRef]
- Halabelian, L.; Barsyte-Lovejoy, D. Structure and Function of Protein Arginine Methyltransferase PRMT7. Life 2021, 11, 768. [Google Scholar] [CrossRef]
- Driehuis, E.; van Hoeck, A.; Moore, K.; Kolders, S.; Francies, H.E.; Gulersonmez, M.C.; Stigter, E.C.A.; Burgering, B.; Geurts, V.; Gracanin, A.; et al. Pancreatic cancer organoids recapitulate disease and allow personalized drug screening. Proc. Natl. Acad. Sci. USA 2019, 116, 26580–26590. [Google Scholar] [CrossRef] [PubMed]
- Singhroy, D.N.; Mesplede, T.; Sabbah, A.; Quashie, P.K.; Falgueyret, J.P.; Wainberg, M.A. Automethylation of protein arginine methyltransferase 6 (PRMT6) regulates its stability and its anti-HIV-1 activity. Retrovirology 2013, 10, 73. [Google Scholar] [CrossRef] [PubMed]
- Thiebaut, C.; Eve, L.; Poulard, C.; Le Romancer, M. Structure, Activity, and Function of PRMT1. Life 2021, 11, 1147. [Google Scholar] [CrossRef]
- Cura, V.; Cavarelli, J. Structure, Activity and Function of the PRMT2 Protein Arginine Methyltransferase. Life 2021, 11, 1263. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Gary, J.D.; Clarke, S.; Herschman, H.R. PRMT 3, a type I protein arginine N-methyltransferase that differs from PRMT1 in its oligomerization, subcellular localization, substrate specificity, and regulation. J. Biol. Chem. 1998, 273, 16935–16945. [Google Scholar] [CrossRef] [PubMed]
- Frankel, A.; Clarke, S. PRMT3 is a distinct member of the protein arginine N-methyltransferase family. Conferral of substrate specificity by a zinc-finger domain. J. Biol. Chem. 2000, 275, 32974–32982. [Google Scholar] [CrossRef]
- Suresh, S.; Huard, S.; Dubois, T. CARM1/PRMT4: Making Its Mark beyond Its Function as a Transcriptional Coactivator. Trends Cell Biol. 2021, 31, 402–417. [Google Scholar] [CrossRef] [PubMed]
- Motolani, A.; Martin, M.; Sun, M.; Lu, T. The Structure and Functions of PRMT5 in Human Diseases. Life 2021, 11, 1074. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Kadumuri, R.V.; Singh, A.K.; Chavali, S.; Dhayalan, A. Structure, Activity and Function of the Protein Arginine Methyltransferase 6. Life 2021, 11, 951. [Google Scholar] [CrossRef]
- Kolbel, K.; Ihling, C.; Bellmann-Sickert, K.; Neundorf, I.; Beck-Sickinger, A.G.; Sinz, A.; Kuhn, U.; Wahle, E. Type I Arginine Methyltransferases PRMT1 and PRMT-3 Act Distributively. J. Biol. Chem. 2009, 284, 8274–8282. [Google Scholar] [CrossRef]
- Wang, M.; Fuhrmann, J.; Thompson, P.R. Protein arginine methyltransferase 5 catalyzes substrate dimethylation in a distributive fashion. Biochemistry 2014, 53, 7884–7892. [Google Scholar] [CrossRef] [PubMed]
- Lakowski, T.M.; Frankel, A. A kinetic study of human protein arginine N-methyltransferase 6 reveals a distributive mechanism. J. Biol. Chem. 2008, 283, 10015–10025. [Google Scholar] [CrossRef] [PubMed]
- Pahlich, S.; Zakaryan, R.P.; Gehring, H. Protein arginine methylation: Cellular functions and methods of analysis. Biochim. Biophys. Acta 2006, 1764, 1890–1903. [Google Scholar] [CrossRef] [PubMed]
- Chung, J.; Karkhanis, V.; Baiocchi, R.A.; Sif, S. Protein arginine methyltransferase 5 (PRMT5) promotes survival of lymphoma cells via activation of WNT/beta-catenin and AKT/GSK3beta proliferative signaling. J. Biol. Chem. 2019, 294, 7692–7710. [Google Scholar] [CrossRef] [PubMed]
- Dowhan, D.H.; Harrison, M.J.; Eriksson, N.A.; Bailey, P.; Pearen, M.A.; Fuller, P.J.; Funder, J.W.; Simpson, E.R.; Leedman, P.J.; Tilley, W.D.; et al. Protein arginine methyltransferase 6-dependent gene expression and splicing: Association with breast cancer outcomes. Endocr. Relat. Cancer 2012, 19, 509–526. [Google Scholar] [CrossRef] [PubMed]
- Bezzi, M.; Teo, S.X.; Muller, J.; Mok, W.C.; Sahu, S.K.; Vardy, L.A.; Bonday, Z.Q.; Guccione, E. Regulation of constitutive and alternative splicing by PRMT5 reveals a role for Mdm4 pre-mRNA in sensing defects in the spliceosomal machinery. Genes Dev. 2013, 27, 1903–1916. [Google Scholar] [CrossRef] [PubMed]
- Brobbey, C.; Liu, L.; Yin, S.; Gan, W. The Role of Protein Arginine Methyltransferases in DNA Damage Response. Int. J. Mol. Sci. 2022, 23, 9780. [Google Scholar] [CrossRef]
- Wei, X.; Yang, J.; Adair, S.J.; Ozturk, H.; Kuscu, C.; Lee, K.Y.; Kane, W.J.; O’Hara, P.E.; Liu, D.; Demirlenk, Y.M.; et al. Targeted CRISPR screening identifies PRMT5 as synthetic lethality combinatorial target with gemcitabine in pancreatic cancer cells. Proc. Natl. Acad. Sci. USA 2020, 117, 28068–28079. [Google Scholar] [CrossRef]
- Le Romancer, M.; Treilleux, I.; Leconte, N.; Robin-Lespinasse, Y.; Sentis, S.; Bouchekioua-Bouzaghou, K.; Goddard, S.; Gobert-Gosse, S.; Corbo, L. Regulation of estrogen rapid signaling through arginine methylation by PRMT1. Mol. Cell 2008, 31, 212–221. [Google Scholar] [CrossRef]
- Rezai-Zadeh, N.; Zhang, X.; Namour, F.; Fejer, G.; Wen, Y.D.; Yao, Y.L.; Gyory, I.; Wright, K.; Seto, E. Targeted recruitment of a histone H4-specific methyltransferase by the transcription factor YY1. Genes Dev. 2003, 17, 1019–1029. [Google Scholar] [CrossRef]
- Yadav, N.; Cheng, D.; Richard, S.; Morel, M.; Iyer, V.R.; Aldaz, C.M.; Bedford, M.T. CARM1 promotes adipocyte differentiation by coactivating PPARgamma. EMBO Rep. 2008, 9, 193–198. [Google Scholar] [CrossRef]
- Reintjes, A.; Fuchs, J.E.; Kremser, L.; Lindner, H.H.; Liedl, K.R.; Huber, L.A.; Valovka, T. Asymmetric arginine dimethylation of RelA provides a repressive mark to modulate TNFalpha/NF-kappaB response. Proc. Natl. Acad. Sci. USA 2016, 113, 4326–4331. [Google Scholar] [CrossRef]
- Yao, B.; Gui, T.; Zeng, X.; Deng, Y.; Wang, Z.; Wang, Y.; Yang, D.; Li, Q.; Xu, P.; Hu, R.; et al. PRMT1-mediated H4R3me2a recruits SMARCA4 to promote colorectal cancer progression by enhancing EGFR signaling. Genome Med. 2021, 13, 58. [Google Scholar] [CrossRef] [PubMed]
- Hou, Z.; Peng, H.; Ayyanathan, K.; Yan, K.P.; Langer, E.M.; Longmore, G.D.; Rauscher, F.J., 3rd. The LIM protein AJUBA recruits protein arginine methyltransferase 5 to mediate SNAIL-dependent transcriptional repression. Mol. Cell. Biol. 2008, 28, 3198–3207. [Google Scholar] [CrossRef]
- Pal, S.; Yun, R.; Datta, A.; Lacomis, L.; Erdjument-Bromage, H.; Kumar, J.; Tempst, P.; Sif, S. mSin3A/histone deacetylase 2- and PRMT5-containing Brg1 complex is involved in transcriptional repression of the Myc target gene cad. Mol. Cell. Biol. 2003, 23, 7475–7487. [Google Scholar] [CrossRef]
- Chen, H.; Lorton, B.; Gupta, V.; Shechter, D. A TGFbeta-PRMT5-MEP50 axis regulates cancer cell invasion through histone H3 and H4 arginine methylation coupled transcriptional activation and repression. Oncogene 2017, 36, 373–386. [Google Scholar] [CrossRef] [PubMed]
- Migliori, V.; Muller, J.; Phalke, S.; Low, D.; Bezzi, M.; Mok, W.C.; Sahu, S.K.; Gunaratne, J.; Capasso, P.; Bassi, C.; et al. Symmetric dimethylation of H3R2 is a newly identified histone mark that supports euchromatin maintenance. Nat. Struct. Mol. Biol. 2012, 19, 136–144. [Google Scholar] [CrossRef]
- Guccione, E.; Richard, S. The regulation, functions and clinical relevance of arginine methylation. Nat. Rev. Mol. Cell. Biol. 2019, 20, 642–657. [Google Scholar] [CrossRef] [PubMed]
- Meister, G.; Fischer, U. Assisted RNP assembly: SMN and PRMT5complexes cooperate in the formation ofspliceosomal UsnRNPs. EMBO J. 2002, 21, 5853–5863. [Google Scholar] [CrossRef]
- Larsen, S.C.; Sylvestersen, K.B.; Mund, A.; Lyon, D.; Mullari, M.; Madsen, M.V.; Daniel, J.A.; Jensen, L.J.; Nielsen, M.L. Proteome-wide analysis of arginine monomethylation reveals widespread occurrence in human cells. Sci. Signal. 2016, 9, rs9. [Google Scholar] [CrossRef]
- Cheng, D.; Cote, J.; Shaaban, S.; Bedford, M.T. The arginine methyltransferase CARM1 regulates the coupling of transcription and mRNA processing. Mol. Cell 2007, 25, 71–83. [Google Scholar] [CrossRef] [PubMed]
- Tradewell, M.L.; Yu, Z.; Tibshirani, M.; Boulanger, M.C.; Durham, H.D.; Richard, S. Arginine methylation by PRMT1 regulates nuclear-cytoplasmic localization and toxicity of FUS/TLS harbouring ALS-linked mutations. Hum. Mol. Genet. 2012, 21, 136–149. [Google Scholar] [CrossRef] [PubMed]
- Murata, K.; Lu, W.; Hashimoto, M.; Ono, N.; Muratani, M.; Nishikata, K.; Kim, J.D.; Ebihara, S.; Ishida, J.; Fukamizu, A. PRMT1 Deficiency in Mouse Juvenile Heart Induces Dilated Cardiomyopathy and Reveals Cryptic Alternative Splicing Products. iScience 2018, 8, 200–213. [Google Scholar] [CrossRef]
- Yu, Z.; Chen, T.; Hebert, J.; Li, E.; Richard, S. A mouse PRMT1 null allele defines an essential role for arginine methylation in genome maintenance and cell proliferation. Mol. Cell. Biol. 2009, 29, 2982–2996. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Vogel, G.; Coulombe, Y.; Dubeau, D.; Spehalski, E.; Hebert, J.; Ferguson, D.O.; Masson, J.Y.; Richard, S. The MRE11 GAR motif regulates DNA double-strand break processing and ATR activation. Cell Res. 2012, 22, 305–320. [Google Scholar] [CrossRef] [PubMed]
- Clarke, T.L.; Sanchez-Bailon, M.P.; Chiang, K.; Reynolds, J.J.; Herrero-Ruiz, J.; Bandeiras, T.M.; Matias, P.M.; Maslen, S.L.; Skehel, J.M.; Stewart, G.S.; et al. PRMT5-Dependent Methylation of the TIP60 Coactivator RUVBL1 Is a Key Regulator of Homologous Recombination. Mol. Cell 2017, 65, 900–916 e907. [Google Scholar] [CrossRef]
- Hamard, P.J.; Santiago, G.E.; Liu, F.; Karl, D.L.; Martinez, C.; Man, N.; Mookhtiar, A.K.; Duffort, S.; Greenblatt, S.; Verdun, R.E.; et al. PRMT5 Regulates DNA Repair by Controlling the Alternative Splicing of Histone-Modifying Enzymes. Cell Rep. 2018, 24, 2643–2657. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.W.; Kim, S.N.; Myung, N.; Song, D.; Han, G.; Bae, G.U.; Bedford, M.T.; Kim, Y.K. PRMT5 promotes DNA repair through methylation of 53BP1 and is regulated by Src-mediated phosphorylation. Commun. Biol. 2020, 3, 428. [Google Scholar] [CrossRef]
- He, W.; Ma, X.; Yang, X.; Zhao, Y.; Qiu, J.; Hang, H. A role for the arginine methylation of Rad9 in checkpoint control and cellular sensitivity to DNA damage. Nucleic Acids Res. 2011, 39, 4719–4727. [Google Scholar] [CrossRef]
- Rehman, I.; Basu, S.M.; Das, S.K.; Bhattacharjee, S.; Ghosh, A.; Pommier, Y.; Das, B.B. PRMT5-mediated arginine methylation of TDP1 for the repair of topoisomerase I covalent complexes. Nucleic Acids Res. 2018, 46, 5601–5617. [Google Scholar] [CrossRef]
- El-Andaloussi, N.; Valovka, T.; Toueille, M.; Steinacher, R.; Focke, F.; Gehrig, P.; Covic, M.; Hassa, P.O.; Schar, P.; Hubscher, U.; et al. Arginine methylation regulates DNA polymerase beta. Mol. Cell 2006, 22, 51–62. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.H.; Bedford, M.T.; Stallcup, M.R. Regulated recruitment of tumor suppressor BRCA1 to the p21 gene by coactivator methylation. Genes Dev. 2011, 25, 176–188. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, S.; Kennemer, A.; Patrick, K.; Tsichlis, P.; Guerau-de-Arellano, M. Protein Arginine Methyltransferase 5 in T Lymphocyte Biology. Trends Immunol. 2020, 41, 918–931. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Kim, H.; Feng, Y.; Li, Y.; Tamiya, H.; Tocci, S.; Ronai, Z.A. PRMT5 control of cGAS/STING and NLRC5 pathways defines melanoma response to antitumor immunity. Sci. Transl. Med. 2020, 12, eaaz5683. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Frankel, A.; Cook, R.J.; Kim, S.; Paik, W.K.; Williams, K.R.; Clarke, S.; Herschman, H.R. PRMT1 is the predominant type I protein arginine methyltransferase in mammalian cells. J. Biol. Chem. 2000, 275, 7723–7730. [Google Scholar] [CrossRef] [PubMed]
- Choucair, A.; Pham, T.H.; Omarjee, S.; Jacquemetton, J.; Kassem, L.; Tredan, O.; Rambaud, J.; Marangoni, E.; Corbo, L.; Treilleux, I.; et al. The arginine methyltransferase PRMT1 regulates IGF-1 signaling in breast cancer. Oncogene 2019, 38, 4015–4027. [Google Scholar] [CrossRef] [PubMed]
- Malbeteau, L.; Jacquemetton, J.; Languilaire, C.; Corbo, L.; Le Romancer, M.; Poulard, C. PRMT1, a Key Modulator of Unliganded Progesterone Receptor Signaling in Breast Cancer. Int. J. Mol. Sci. 2022, 23, 9509. [Google Scholar] [CrossRef] [PubMed]
- Avasarala, S.; Van Scoyk, M.; Karuppusamy Rathinam, M.K.; Zerayesus, S.; Zhao, X.; Zhang, W.; Pergande, M.R.; Borgia, J.A.; DeGregori, J.; Port, J.D.; et al. PRMT1 Is a Novel Regulator of Epithelial-Mesenchymal-Transition in Non-small Cell Lung Cancer. J. Biol. Chem. 2015, 290, 13479–13489. [Google Scholar] [CrossRef]
- Liao, H.W.; Hsu, J.M.; Xia, W.; Wang, H.L.; Wang, Y.N.; Chang, W.C.; Arold, S.T.; Chou, C.K.; Tsou, P.H.; Yamaguchi, H.; et al. PRMT1-mediated methylation of the EGF receptor regulates signaling and cetuximab response. J. Clin. Investig. 2015, 125, 4529–4543. [Google Scholar] [CrossRef]
- Song, C.; Chen, T.; He, L.; Ma, N.; Li, J.A.; Rong, Y.F.; Fang, Y.; Liu, M.; Xie, D.; Lou, W. PRMT1 promotes pancreatic cancer growth and predicts poor prognosis. Cell. Oncol. 2020, 43, 51–62. [Google Scholar] [CrossRef]
- Wang, Y.; Hsu, J.M.; Kang, Y.; Wei, Y.; Lee, P.C.; Chang, S.J.; Hsu, Y.H.; Hsu, J.L.; Wang, H.L.; Chang, W.C.; et al. Oncogenic Functions of Gli1 in Pancreatic Adenocarcinoma Are Supported by Its PRMT1-Mediated Methylation. Cancer Res. 2016, 76, 7049–7058. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Jia, Z.; Xie, D.; Zhao, T.; Tan, Z.; Zhang, S.; Kong, F.; Wei, D.; Xie, K. Methylation of HSP70 Orchestrates Its Binding to and Stabilization of BCL2 mRNA and Renders Pancreatic Cancer Cells Resistant to Therapeutics. Cancer Res. 2020, 80, 4500–4513. [Google Scholar] [CrossRef] [PubMed]
- Giuliani, V.; Miller, M.A.; Liu, C.Y.; Hartono, S.R.; Class, C.A.; Bristow, C.A.; Suzuki, E.; Sanz, L.A.; Gao, G.; Gay, J.P.; et al. PRMT1-dependent regulation of RNA metabolism and DNA damage response sustains pancreatic ductal adenocarcinoma. Nat. Commun. 2021, 12, 4626. [Google Scholar] [CrossRef] [PubMed]
- Zheng, N.N.; Zhou, M.; Sun, F.; Huai, M.X.; Zhang, Y.; Qu, C.Y.; Shen, F.; Xu, L.M. Combining protein arginine methyltransferase inhibitor and anti-programmed death-ligand-1 inhibits pancreatic cancer progression. World J. Gastroenterol. 2020, 26, 3737–3749. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Chen, Y.; Lin, Z.; Chen, C.; Dong, Y. Overexpressing PRMT1 Inhibits Proliferation and Invasion in Pancreatic Cancer by Inverse Correlation of ZEB1. IUBMB Life 2018, 70, 1032–1039. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Liu, H.; Ye, M.; Jiang, M.; Chen, X.; Song, G.; Ji, H.; Wang, Z.W.; Zhu, X. Methylation of BRD4 by PRMT1 regulates BRD4 phosphorylation and promotes ovarian cancer invasion. Cell Death Dis. 2023, 14, 624. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Wang, D.; Lu, J.; Huang, B.; Wang, Y.; Dong, M.; Fan, D.; Li, H.; Gao, Y.; Hou, P.; et al. Methylation of EZH2 by PRMT1 regulates its stability and promotes breast cancer metastasis. Cell Death Differ. 2020, 27, 3226–3242. [Google Scholar] [CrossRef] [PubMed]
- Zheng, D.; Chen, D.; Lin, F.; Wang, X.; Lu, L.; Luo, S.; Chen, J.; Xu, X. LncRNA NNT-AS1 promote glioma cell proliferation and metastases through miR-494-3p/PRMT1 axis. Cell Cycle 2020, 19, 1621–1631. [Google Scholar] [CrossRef]
- Hsu, M.C.; Tsai, Y.L.; Lin, C.H.; Pan, M.R.; Shan, Y.S.; Cheng, T.Y.; Cheng, S.H.; Chen, L.T.; Hung, W.C. Protein arginine methyltransferase 3-induced metabolic reprogramming is a vulnerable target of pancreatic cancer. J. Hematol. Oncol. 2019, 12, 79. [Google Scholar] [CrossRef]
- Jiang, W.; Newsham, I.F. The tumor suppressor DAL-1/4.1B and protein methylation cooperate in inducing apoptosis in MCF-7 breast cancer cells. Mol. Cancer 2006, 5, 4. [Google Scholar] [CrossRef]
- Zhi, R.; Wu, K.; Zhang, J.; Liu, H.; Niu, C.; Li, S.; Fu, L. PRMT3 regulates the progression of invasive micropapillary carcinoma of the breast. Cancer Sci. 2023, 114, 1912–1928. [Google Scholar] [CrossRef] [PubMed]
- Hsu, M.C.; Pan, M.R.; Chu, P.Y.; Tsai, Y.L.; Tsai, C.H.; Shan, Y.S.; Chen, L.T.; Hung, W.C. Protein Arginine Methyltransferase 3 Enhances Chemoresistance in Pancreatic Cancer by Methylating hnRNPA1 to Increase ABCG2 Expression. Cancers 2018, 11, 8. [Google Scholar] [CrossRef] [PubMed]
- Stopa, N.; Krebs, J.E.; Shechter, D. The PRMT5 arginine methyltransferase: Many roles in development, cancer and beyond. Cell. Mol. Life Sci. 2015, 72, 2041–2059. [Google Scholar] [CrossRef] [PubMed]
- Berglund, L.; Bjorling, E.; Oksvold, P.; Fagerberg, L.; Asplund, A.; Szigyarto, C.A.; Persson, A.; Ottosson, J.; Wernerus, H.; Nilsson, P.; et al. A genecentric Human Protein Atlas for expression profiles based on antibodies. Mol. Cell Proteom. 2008, 7, 2019–2027. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.K.C.; Grimmond, S.M.; McArthur, G.A.; Sheppard, K.E. PRMT5: An Emerging Target for Pancreatic Adenocarcinoma. Cancers 2021, 13, 5136. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.; Hu, Q.; Xu, J.; Ji, S.; Dai, W.; Liu, W.; Xu, W.; Sun, Q.; Zhang, Z.; Ni, Q.; et al. PRMT5 enhances tumorigenicity and glycolysis in pancreatic cancer via the FBW7/cMyc axis. Cell Commun. Signal 2019, 17, 30. [Google Scholar] [CrossRef] [PubMed]
- Welcker, M.; Orian, A.; Jin, J.; Grim, J.E.; Harper, J.W.; Eisenman, R.N.; Clurman, B.E. The Fbw7 tumor suppressor regulates glycogen synthase kinase 3 phosphorylation-dependent c-Myc protein degradation. Proc. Natl. Acad. Sci. USA 2004, 101, 9085–9090. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Bellon, M.; Ju, M.; Zhao, L.; Wei, M.; Fu, L.; Nicot, C. Clinical significance of FBXW7 loss of function in human cancers. Mol. Cancer 2022, 21, 87. [Google Scholar] [CrossRef] [PubMed]
- Orben, F.; Lankes, K.; Schneeweis, C.; Hassan, Z.; Jakubowsky, H.; Krauss, L.; Boniolo, F.; Schneider, C.; Schafer, A.; Murr, J.; et al. Epigenetic drug screening defines a PRMT5 inhibitor-sensitive pancreatic cancer subtype. JCI Insight 2022, 7, e151353. [Google Scholar] [CrossRef]
- Ge, L.; Wang, H.; Xu, X.; Zhou, Z.; He, J.; Peng, W.; Du, F.; Zhang, Y.; Gong, A.; Xu, M. PRMT5 promotes epithelial-mesenchymal transition via EGFR-beta-catenin axis in pancreatic cancer cells. J. Cell. Mol. Med. 2020, 24, 1969–1979. [Google Scholar] [CrossRef]
- Hsu, J.M.; Chen, C.T.; Chou, C.K.; Kuo, H.P.; Li, L.Y.; Lin, C.Y.; Lee, H.J.; Wang, Y.N.; Liu, M.; Liao, H.W.; et al. Crosstalk between Arg 1175 methylation and Tyr 1173 phosphorylation negatively modulates EGFR-mediated ERK activation. Nat Cell Biol 2011, 13, 174–181. [Google Scholar] [CrossRef] [PubMed]
- Gao, G.; Zhang, L.; Villarreal, O.D.; He, W.; Su, D.; Bedford, E.; Moh, P.; Shen, J.; Shi, X.; Bedford, M.T.; et al. PRMT1 loss sensitizes cells to PRMT5 inhibition. Nucleic Acids Res. 2019, 47, 5038–5048. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Chen, Z.H.; Savarese, T.M. Codeletion of the genes for p16INK4, methylthioadenosine phosphorylase, interferon-alpha1, interferon-beta1, and other 9p21 markers in human malignant cell lines. Cancer Genet. Cytogenet. 1996, 86, 22–28. [Google Scholar] [CrossRef]
- Zappia, V.; Della Ragione, F.; Pontoni, G.; Gragnaniello, V.; Carteni-Farina, M. Human 5′-deoxy-5′-methylthioadenosine phosphorylase: Kinetic studies and catalytic mechanism. Adv. Exp. Med. Biol. 1988, 250, 165–177. [Google Scholar] [CrossRef] [PubMed]
- Kryukov, G.V.; Wilson, F.H.; Ruth, J.R.; Paulk, J.; Tsherniak, A.; Marlow, S.E.; Vazquez, F.; Weir, B.A.; Fitzgerald, M.E.; Tanaka, M.; et al. MTAP deletion confers enhanced dependency on the PRMT5 arginine methyltransferase in cancer cells. Science 2016, 351, 1214–1218. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Richard, S. Cellular pathways influenced by protein arginine methylation: Implications for cancer. Mol. Cell 2021, 81, 4357–4368. [Google Scholar] [CrossRef] [PubMed]
- Guccione, E.; Schwarz, M.; Di Tullio, F.; Mzoughi, S. Cancer synthetic vulnerabilities to protein arginine methyltransferase inhibitors. Curr. Opin. Pharmacol. 2021, 59, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Qian, K.; Ho, M.C.; Zheng, Y.G. Small Molecule Inhibitors of Protein Arginine Methyltransferases. Expert. Opin. Investig. Drugs 2016, 25, 335–358. [Google Scholar] [CrossRef] [PubMed]
- Cheng, D.; Yadav, N.; King, R.W.; Swanson, M.S.; Weinstein, E.J.; Bedford, M.T. Small molecule regulators of protein arginine methyltransferases. J. Biol. Chem. 2004, 279, 23892–23899. [Google Scholar] [CrossRef]
- Spannhoff, A.; Heinke, R.; Bauer, I.; Trojer, P.; Metzger, E.; Gust, R.; Schule, R.; Brosch, G.; Sippl, W.; Jung, M. Target-based approach to inhibitors of histone arginine methyltransferases. J. Med. Chem. 2007, 50, 2319–2325. [Google Scholar] [CrossRef]
- Sun, Y.; Wang, Z.; Yang, H.; Zhu, X.; Wu, H.; Ma, L.; Xu, F.; Hong, W.; Wang, H. The Development of Tetrazole Derivatives as Protein Arginine Methyltransferase I (PRMT I) Inhibitors. Int. J. Mol. Sci. 2019, 20, 3840. [Google Scholar] [CrossRef]
- Eram, M.S.; Shen, Y.; Szewczyk, M.; Wu, H.; Senisterra, G.; Li, F.; Butler, K.V.; Kaniskan, H.U.; Speed, B.A.; Dela Sena, C.; et al. A Potent, Selective, and Cell-Active Inhibitor of Human Type I Protein Arginine Methyltransferases. ACS Chem. Biol. 2016, 11, 772–781. [Google Scholar] [CrossRef] [PubMed]
- Fedoriw, A.; Rajapurkar, S.R.; O’Brien, S.; Gerhart, S.V.; Mitchell, L.H.; Adams, N.D.; Rioux, N.; Lingaraj, T.; Ribich, S.A.; Pappalardi, M.B.; et al. Anti-tumor Activity of the Type I PRMT Inhibitor, GSK3368715, Synergizes with PRMT5 Inhibition through MTAP Loss. Cancer Cell 2019, 36, 100–114.e125. [Google Scholar] [CrossRef]
- El-Khoueiry, A.B.; Clarke, J.; Neff, T.; Crossman, T.; Ratia, N.; Rathi, C.; Noto, P.; Tarkar, A.; Garrido-Laguna, I.; Calvo, E.; et al. Phase 1 study of GSK3368715, a type I PRMT inhibitor, in patients with advanced solid tumors. Br. J. Cancer 2023, 129, 309–317. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Szewczyk, M.M.; Eram, M.S.; Smil, D.; Kaniskan, H.U.; de Freitas, R.F.; Senisterra, G.; Li, F.; Schapira, M.; Brown, P.J.; et al. Discovery of a Potent, Selective, and Cell-Active Dual Inhibitor of Protein Arginine Methyltransferase 4 and Protein Arginine Methyltransferase 6. J. Med. Chem. 2016, 59, 9124–9139. [Google Scholar] [CrossRef]
- Shen, Y.; Li, F.; Szewczyk, M.M.; Halabelian, L.; Park, K.S.; Chau, I.; Dong, A.; Zeng, H.; Chen, H.; Meng, F.; et al. Discovery of a First-in-Class Protein Arginine Methyltransferase 6 (PRMT6) Covalent Inhibitor. J. Med. Chem. 2020, 63, 5477–5487. [Google Scholar] [CrossRef]
- Alinari, L.; Mahasenan, K.V.; Yan, F.; Karkhanis, V.; Chung, J.H.; Smith, E.M.; Quinion, C.; Smith, P.L.; Kim, L.; Patton, J.T.; et al. Selective inhibition of protein arginine methyltransferase 5 blocks initiation and maintenance of B-cell transformation. Blood 2015, 125, 2530–2543. [Google Scholar] [CrossRef] [PubMed]
- Chan-Penebre, E.; Kuplast, K.G.; Majer, C.R.; Boriack-Sjodin, P.A.; Wigle, T.J.; Johnston, L.D.; Rioux, N.; Munchhof, M.J.; Jin, L.; Jacques, S.L.; et al. A selective inhibitor of PRMT5 with in vivo and in vitro potency in MCL models. Nat. Chem. Biol. 2015, 11, 432–437. [Google Scholar] [CrossRef]
- Mitchell, L.H.; Drew, A.E.; Ribich, S.A.; Rioux, N.; Swinger, K.K.; Jacques, S.L.; Lingaraj, T.; Boriack-Sjodin, P.A.; Waters, N.J.; Wigle, T.J.; et al. Aryl Pyrazoles as Potent Inhibitors of Arginine Methyltransferases: Identification of the First PRMT6 Tool Compound. ACS Med. Chem. Lett. 2015, 6, 655–659. [Google Scholar] [CrossRef]
- Iyamu, I.D.; Al-Hamashi, A.A.; Huang, R. A Pan-Inhibitor for Protein Arginine Methyltransferase Family Enzymes. Biomolecules 2021, 11, 854. [Google Scholar] [CrossRef]
- Szewczyk, M.M.; Ishikawa, Y.; Organ, S.; Sakai, N.; Li, F.; Halabelian, L.; Ackloo, S.; Couzens, A.L.; Eram, M.; Dilworth, D.; et al. Pharmacological inhibition of PRMT7 links arginine monomethylation to the cellular stress response. Nat. Commun. 2020, 11, 2396. [Google Scholar] [CrossRef]
- Dong, J.; Duan, J.; Hui, Z.; Garrido, C.; Deng, Z.; Xie, T.; Ye, X.Y. An updated patent review of protein arginine N-methyltransferase inhibitors (2019–2022). Expert. Opin. Ther. Pat. 2022, 32, 1185–1205. [Google Scholar] [CrossRef] [PubMed]
- Yin, S.; Liu, L.; Brobbey, C.; Palanisamy, V.; Ball, L.E.; Olsen, S.K.; Ostrowski, M.C.; Gan, W. PRMT5-mediated arginine methylation activates AKT kinase to govern tumorigenesis. Nat. Commun. 2021, 12, 3444. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Zhang, X.O.; Rozen, E.J.; Sun, X.; Sallis, B.; Verdejo-Torres, O.; Wigglesworth, K.; Moon, D.; Huang, T.; Cavaretta, J.P.; et al. PRMT5 activates AKT via methylation to promote tumor metastasis. Nat. Commun. 2022, 13, 3955. [Google Scholar] [CrossRef]
- Kugeratski, F.G.; Hodge, K.; Lilla, S.; McAndrews, K.M.; Zhou, X.; Hwang, R.F.; Zanivan, S.; Kalluri, R. Quantitative proteomics identifies the core proteome of exosomes with syntenin-1 as the highest abundant protein and a putative universal biomarker. Nat. Cell Biol. 2021, 23, 631–641. [Google Scholar] [CrossRef] [PubMed]
- Hinzman, C.P.; Singh, B.; Bansal, S.; Li, Y.; Iliuk, A.; Girgis, M.; Herremans, K.M.; Trevino, J.G.; Singh, V.K.; Banerjee, P.P.; et al. A multi-omics approach identifies pancreatic cancer cell extracellular vesicles as mediators of the unfolded protein response in normal pancreatic epithelial cells. J. Extracell. Vesicles 2022, 11, e12232. [Google Scholar] [CrossRef] [PubMed]
- Hannafon, B.N.; Ding, W.Q. Intercellular Communication by Exosome-Derived microRNAs in Cancer. Int. J. Mol. Sci. 2013, 14, 14240–14269. [Google Scholar] [CrossRef]
- Hoshino, A.; Costa-Silva, B.; Shen, T.L.; Rodrigues, G.; Hashimoto, A.; Tesic Mark, M.; Molina, H.; Kohsaka, S.; Di Giannatale, A.; Ceder, S.; et al. Tumour exosome integrins determine organotropic metastasis. Nature 2015, 527, 329–335. [Google Scholar] [CrossRef] [PubMed]
- Radzisheuskaya, A.; Shliaha, P.V.; Grinev, V.; Lorenzini, E.; Kovalchuk, S.; Shlyueva, D.; Gorshkov, V.; Hendrickson, R.C.; Jensen, O.N.; Helin, K. PRMT5 methylome profiling uncovers a direct link to splicing regulation in acute myeloid leukemia. Nat. Struct. Mol. Biol. 2019, 26, 999–1012. [Google Scholar] [CrossRef]
- Wang, Q.; Li, Z.; Zhang, S.; Li, Y.; Wang, Y.; Fang, Z.; Ma, Y.; Liu, Z.; Zhang, W.; Li, D.; et al. Global profiling of arginine dimethylation in regulating protein phase separation by a steric effect-based chemical-enrichment method. Proc. Natl. Acad. Sci. USA 2022, 119, e2205255119. [Google Scholar] [CrossRef]
- Tsai, W.C.; Gayatri, S.; Reineke, L.C.; Sbardella, G.; Bedford, M.T.; Lloyd, R.E. Arginine Demethylation of G3BP1 Promotes Stress Granule Assembly. J. Biol. Chem. 2016, 291, 22671–22685. [Google Scholar] [CrossRef] [PubMed]
- Hofweber, M.; Hutten, S.; Bourgeois, B.; Spreitzer, E.; Niedner-Boblenz, A.; Schifferer, M.; Ruepp, M.D.; Simons, M.; Niessing, D.; Madl, T.; et al. Phase Separation of FUS Is Suppressed by Its Nuclear Import Receptor and Arginine Methylation. Cell 2018, 173, 706–719.e713. [Google Scholar] [CrossRef]
- Liu, X.M.; Ma, L.; Schekman, R. Selective sorting of microRNAs into exosomes by phase-separated YBX1 condensates. eLife 2021, 10, e71982. [Google Scholar] [CrossRef] [PubMed]
- Suetsugu, A.; Honma, K.; Saji, S.; Moriwaki, H.; Ochiya, T.; Hoffman, R.M. Imaging exosome transfer from breast cancer cells to stroma at metastatic sites in orthotopic nude-mouse models. Adv. Drug Deliv. Rev. 2013, 65, 383–390. [Google Scholar] [CrossRef] [PubMed]
- Taylor, D.D.; Gercel-Taylor, C. MicroRNA signatures of tumor-derived exosomes as diagnostic biomarkers of ovarian cancer. Gynecol. Oncol. 2008, 110, 13–21. [Google Scholar] [CrossRef]
- Bhandari, K.; Kong, J.S.; Morris, K.; Xu, C.; Ding, W.Q. Protein Arginine Methylation Patterns in Plasma Small Extracellular Vesicles Are Altered in Patients with Early-Stage Pancreatic Ductal Adenocarcinoma. Cancers 2024, 16, 654. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PRMTs | Endocrine Region | Exocrine Region |
|---|---|---|
| PRMT1 | Medium | Low |
| PRMT2 | Low | Medium |
| PRMT3 | Medium | High to medium |
| PRMT4 | Low | Medium to low |
| PRMT5 | Low | Low to medium |
| PRMT6 | Medium | High |
| PRMT7 | Medium | Medium |
| PRMT9 | Negative | Low |
| Enzyme | Structural Features |
|---|---|
| PRMT1 | Contain three canonical domains: N-terminal methyltransferase domain (Rossman fold), containing AdoMet binding pocket C-terminal ß-barrel domain, forming cylindrical structure corresponding to the arginine-substrate binding sites α-helical dimerization arm, an N-terminal part of the ß-barrel domain [53] |
| PRMT2 | Three canonical domains, along with a unique Src homology 3 domain (SH3 domain) towards N-terminal extremity [54] |
| PRMT3 | Three canonical domains, along with a unique C2H2Zn finger domain at the N-terminus for substrate binding [55,56] |
| PRMT4/CARM1 | Three canonical domains, along with a C-terminal TAD domain and PH-like homology at the N-terminus for substrate recognition [57] |
| PRMT5 | Three canonical domains, with N-terminal TIM barrel, which is essential for formation of complex with MEP50 [58] |
| PRMT6 | Three canonical domains; no unique feature [59] |
| PRMT7 | Three canonical domains, with two tandem methyltransferase domains due to gene duplication [38] |
| PRMT8 | Three canonical domains, with a myristoylation site at the N-terminus that mediates its anchorage to the plasma membrane [36] |
| PRMT9 | Three canonical domains, with two tandem methyltransferase domains and N-terminal TPR repeats [39] |
| PRMT | Cellular Localization * | Enzyme Type | Methylation Product | Motif Preference |
|---|---|---|---|---|
| PRMT1 | Cytoplasm, Nucleus | I | MMA/ADMA | RGG or RxR |
| PRMT2 | Nucleus, Cytoplasm | I | MMA/ADMA | RGG/RG |
| PRMT3 | Cytoplasm, Nucleus | I | MMA/ADMA | RGG/RG |
| PRMT4/CARM1 | Nucleus, Cytoplasm | I | MMA/ADMA | PGM |
| PRMT5 | Cytoplasm, Nucleus | II | MMA/SDMA | GRG, PGM |
| PRMT6 | Nucleus, Cytoplasm | I | MMA/ADMA | RxR |
| PRMT7 | Cytoplasm, Nucleus | III | MMA | RxR |
| PRMT8 | Plasma membrane | I | MMA/ADMA | RGG or RxR |
| PRMT9 | Cytoplasm, Nucleus | II | MMA/SDMA | GAR |
| Inhibitor | Target | Clinical Trial ID | Phase | Tumor |
|---|---|---|---|---|
| GSK3368715 | Type I PRMT | NCT03666988 | Phase I | Solid Tumors and Diffuse Large B-cell Lymphoma |
| AMG 193 | PRMT5 | NCT05094336 | Phase I/II | MTAP-null solid tumors |
| JNJ-64619178 | PRMT5 | NCT03573310 | Phase I | Advanced solid tumors, B cell non-Hodgkin lymphoma (NHL) |
| PF-06939999 | PRMT5 | NCT03854227 | Phase I | Advanced solid tumors |
| PRT543 | PRMT5 | NCT03886831 | Phase I | Relapsed or refractory solid tumors, lymphoma, and leukemia |
| PRT811 | PRMT5 | NCT04089449 | Phase I | High grade gliomas, anaplastic astrocytoma, and advanced solid tumors |
| GSK3326595/EPZ015666 | PRMT5 | NCT04676516 NCT02783300 NCT03614728 | Phase I/II Phase I Phase I/II | Early-stage breast cancer Advanced solid tumors, NHL Chronic myelomonocytic leukemia, Adult acute myeloid leukemia |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bhandari, K.; Ding, W.-Q. Protein Arginine Methyltransferases in Pancreatic Ductal Adenocarcinoma: New Molecular Targets for Therapy. Int. J. Mol. Sci. 2024, 25, 3958. https://doi.org/10.3390/ijms25073958
Bhandari K, Ding W-Q. Protein Arginine Methyltransferases in Pancreatic Ductal Adenocarcinoma: New Molecular Targets for Therapy. International Journal of Molecular Sciences. 2024; 25(7):3958. https://doi.org/10.3390/ijms25073958
Chicago/Turabian StyleBhandari, Kritisha, and Wei-Qun Ding. 2024. "Protein Arginine Methyltransferases in Pancreatic Ductal Adenocarcinoma: New Molecular Targets for Therapy" International Journal of Molecular Sciences 25, no. 7: 3958. https://doi.org/10.3390/ijms25073958
APA StyleBhandari, K., & Ding, W.-Q. (2024). Protein Arginine Methyltransferases in Pancreatic Ductal Adenocarcinoma: New Molecular Targets for Therapy. International Journal of Molecular Sciences, 25(7), 3958. https://doi.org/10.3390/ijms25073958