
Multi-Omics Profiling Reveals Phenotypic and Functional Heterogeneity of Neutrophils in COVID-19

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Characterization of the scRNA-seq Data
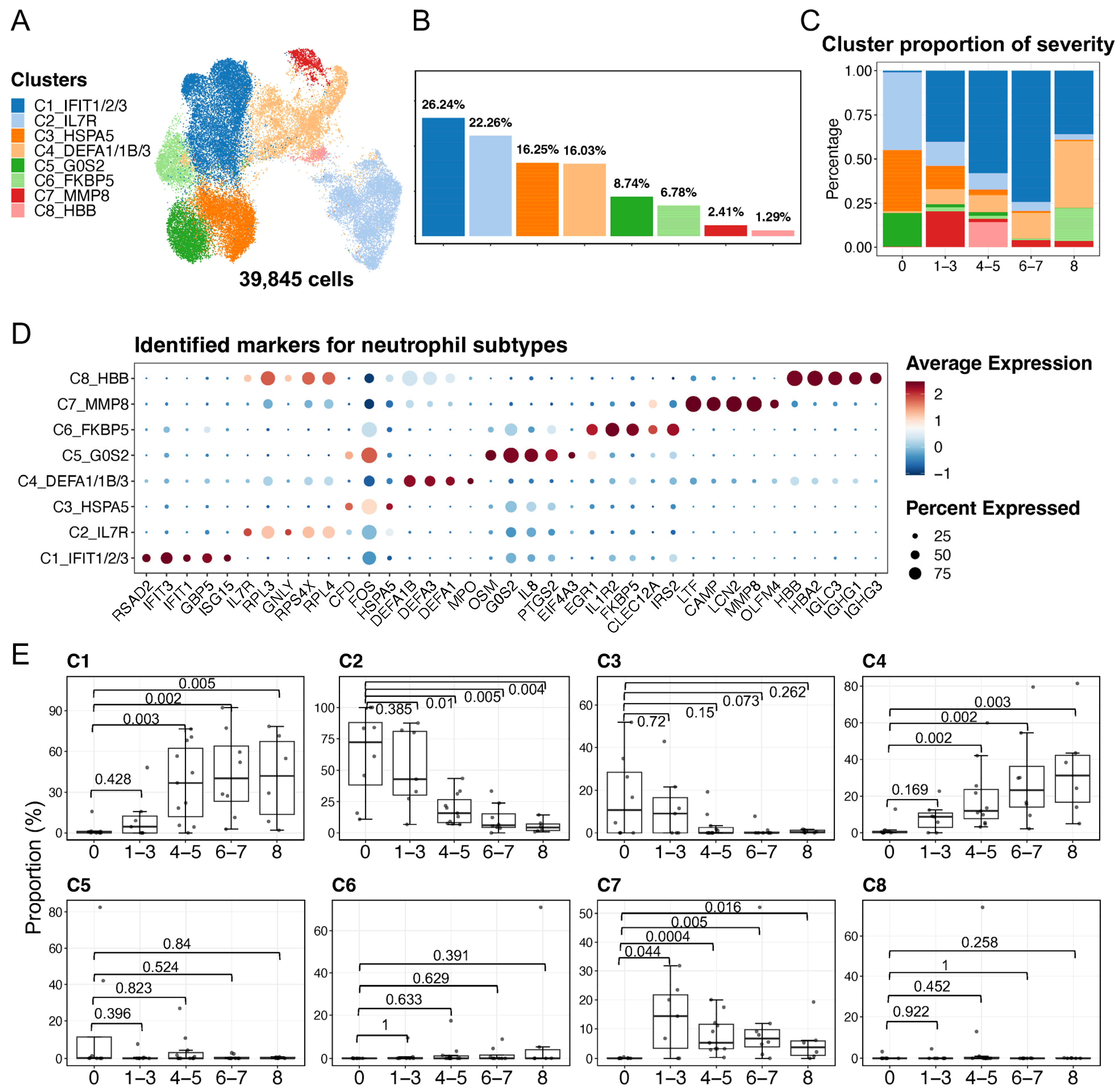
2.2. Identification and Description of Neutrophil Subpopulations
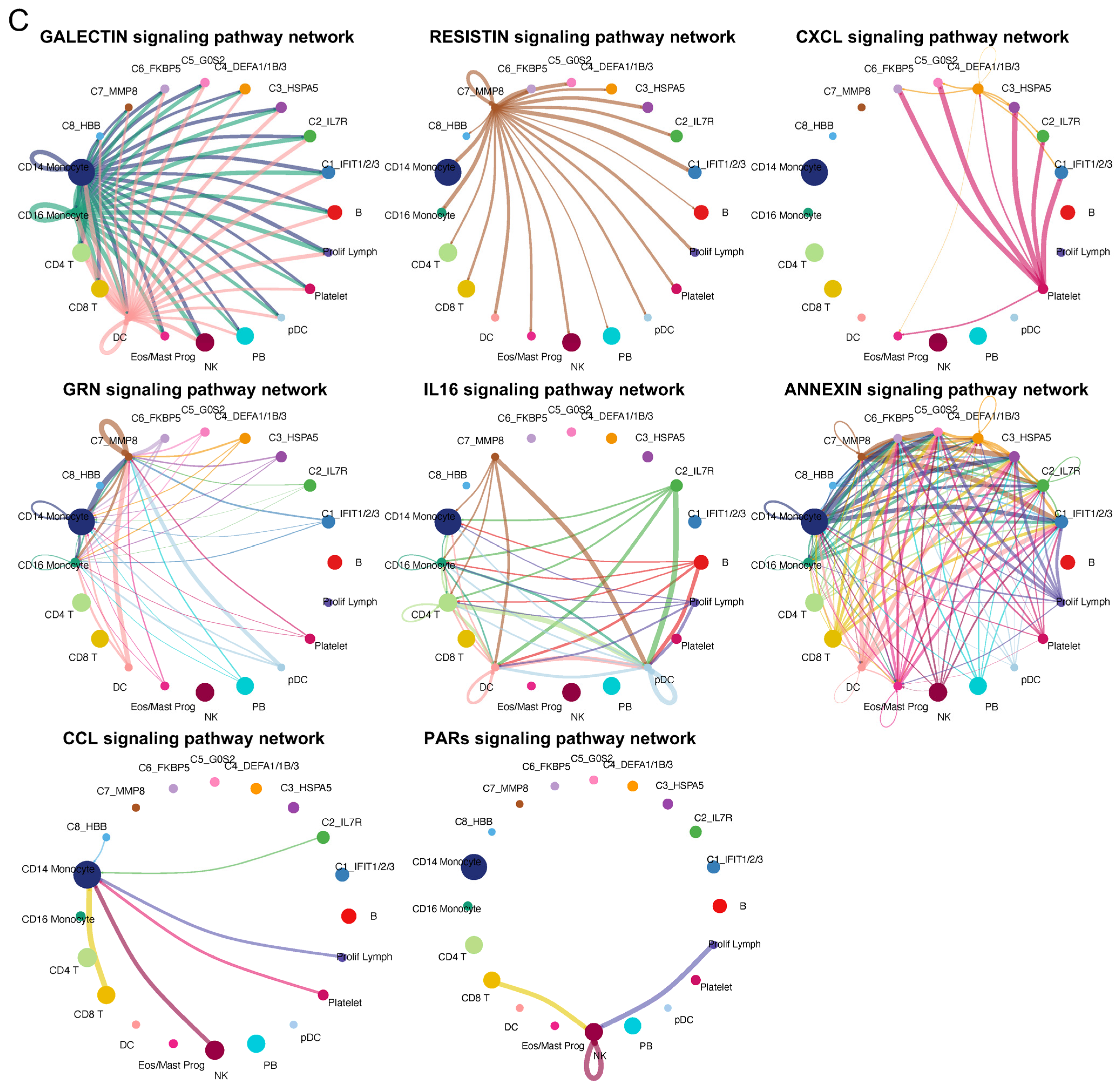
2.3. Diverse Cell–Cell Communications among Neutrophil Subtypes and Other Blood Cell Types
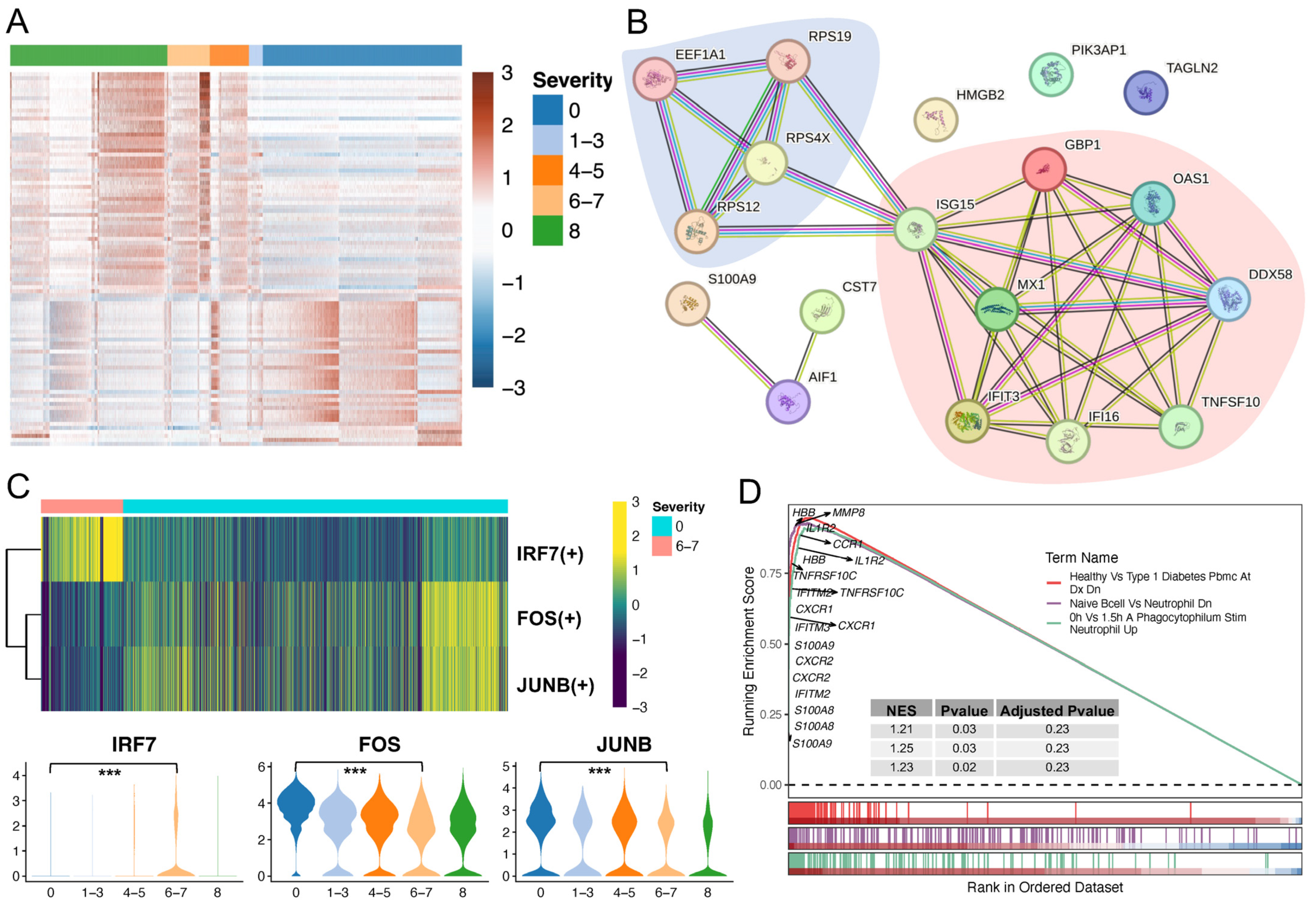
2.4. Neutrophil Profiling Was Altered under Physiological and Pathological Conditions
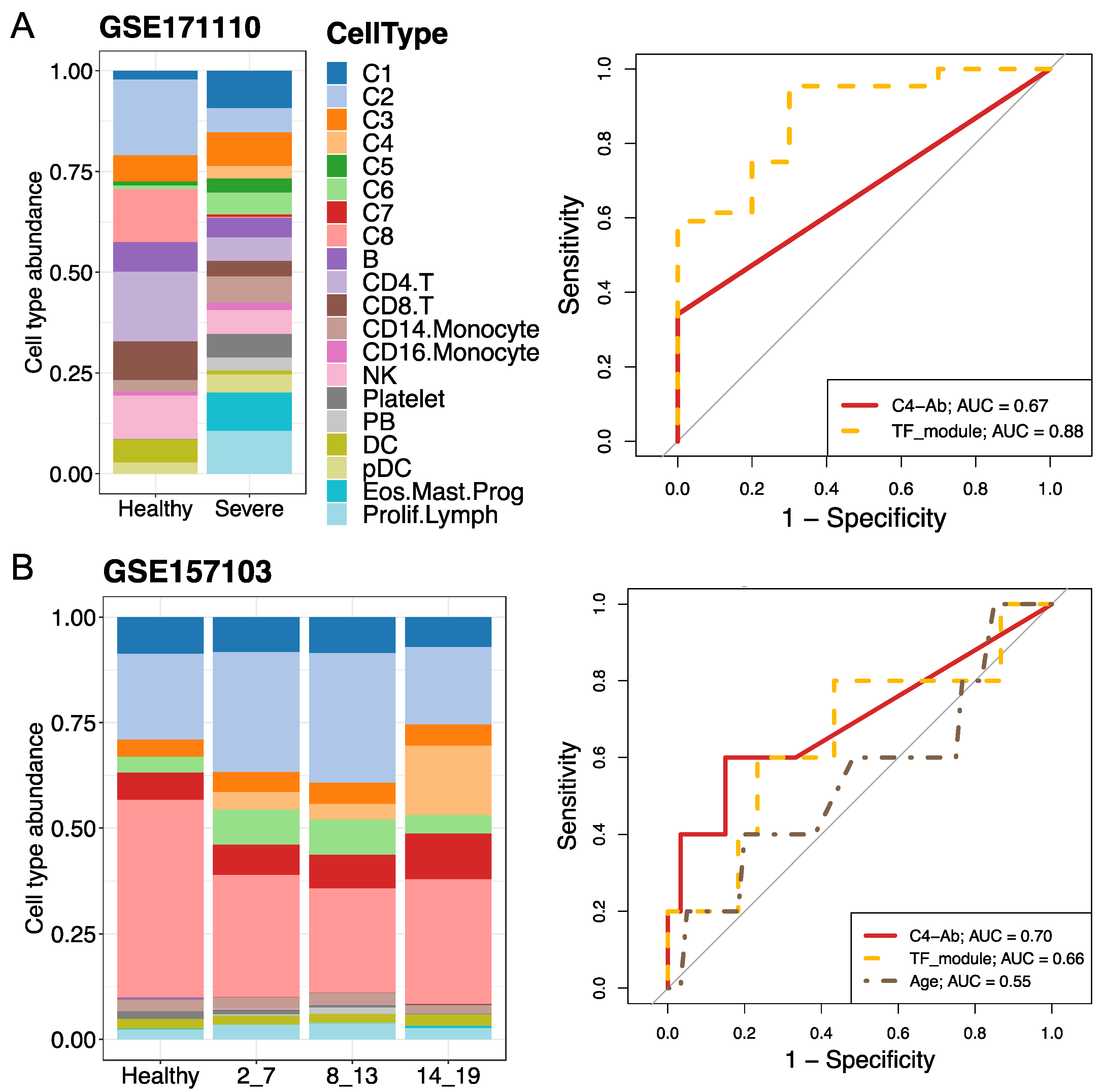
2.5. Validation of C4 Neutrophil Subtype Abundance and Evaluation of Predictors towards Severe COVID-19
3. Discussion
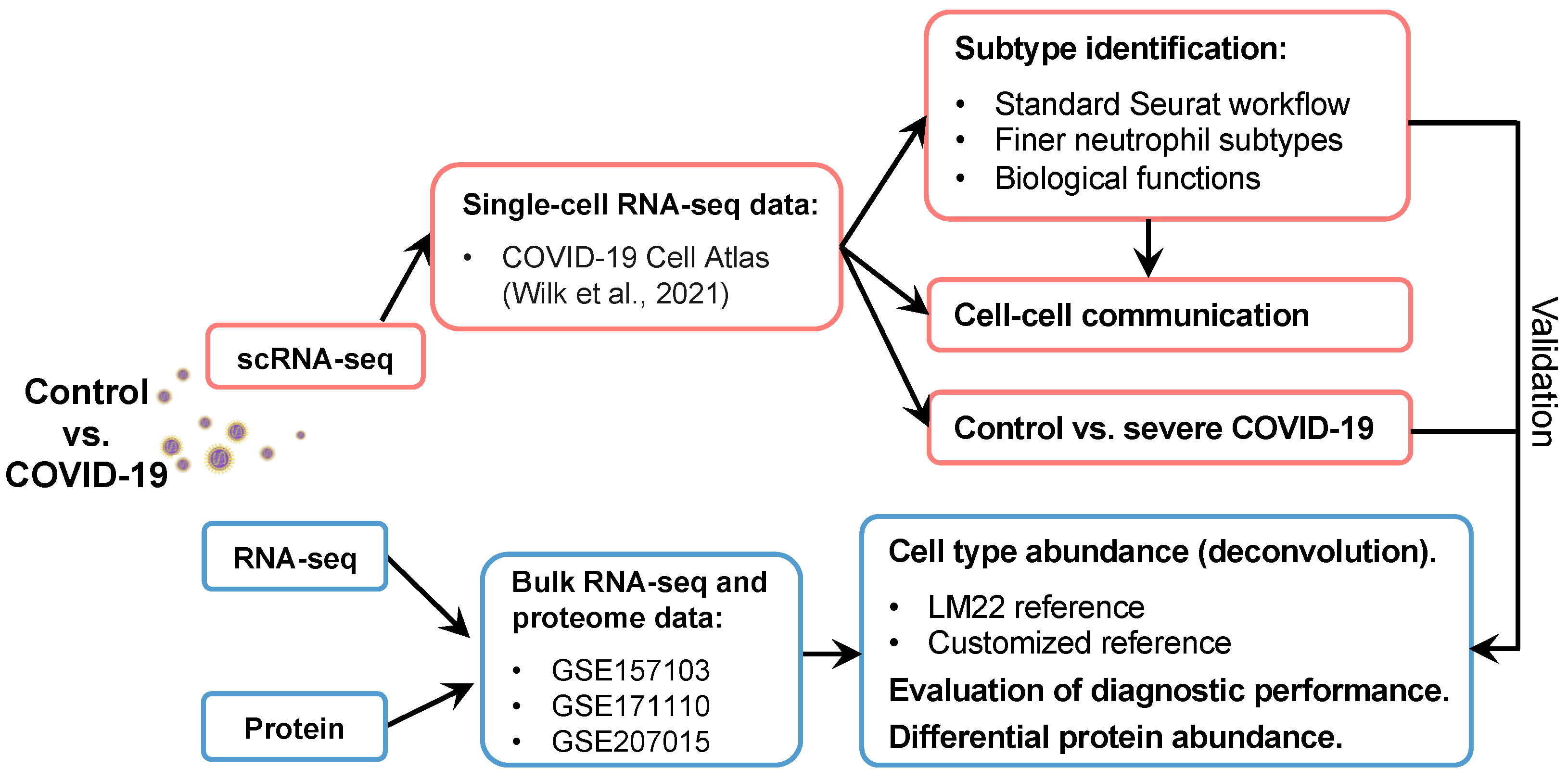
4. Materials and Methods
4.1. Data Collection
4.2. scRNA-seq Data Analysis and Neutrophil Subtype Identification
4.3. Functional Enrichment Analysis
4.4. Cell–Cell Communication Analysis
4.5. Gene Regulation Inferences
4.6. Cellular Deconvolution and Severe Disease Predictions
4.7. Cell Senescence Status Analysis
4.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Veras, F.P.; Pontelli, M.C.; Silva, C.M.; Toller-Kawahisa, J.E.; de Lima, M.; Nascimento, D.C.; Schneider, A.H.; Caetité, D.; Tavares, L.A.; Paiva, I.M. SARS-CoV-2–Triggered Neutrophil Extracellular Traps Mediate COVID-19 Pathology. J. Exp. Med. 2020, 217, e20201129. [Google Scholar] [CrossRef]
- Wilk, A.J.; Rustagi, A.; Zhao, N.Q.; Roque, J.; Martínez-Colón, G.J.; McKechnie, J.L.; Ivison, G.T.; Ranganath, T.; Vergara, R.; Hollis, T.; et al. A Single-Cell Atlas of the Peripheral Immune Response in Patients with Severe COVID-19. Nat. Med. 2020, 26, 1070–1076. [Google Scholar] [CrossRef]
- Aschenbrenner, A.C.; Mouktaroudi, M.; Krämer, B.; Oestreich, M.; Antonakos, N.; Nuesch-Germano, M.; Gkizeli, K.; Bonaguro, L.; Reusch, N.; Baßler, K. Disease Severity-Specific Neutrophil Signatures in Blood Transcriptomes Stratify COVID-19 Patients. Genome Med. 2021, 13, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Meizlish, M.L.; Pine, A.B.; Bishai, J.D.; Goshua, G.; Nadelmann, E.R.; Simonov, M.; Chang, C.-H.; Zhang, H.; Shallow, M.; Bahel, P. A Neutrophil Activation Signature Predicts Critical Illness and Mortality in COVID-19. Blood Adv. 2021, 5, 1164–1177. [Google Scholar] [CrossRef] [PubMed]
- Schulte-Schrepping, J.; Reusch, N.; Paclik, D.; Baßler, K.; Schlickeiser, S.; Zhang, B.; Krämer, B.; Krammer, T.; Brumhard, S.; Bonaguro, L. Severe COVID-19 Is Marked by a Dysregulated Myeloid Cell Compartment. Cell 2020, 182, 1419–1440. [Google Scholar] [CrossRef] [PubMed]
- Diao, B.; Wang, C.; Tan, Y.; Chen, X.; Liu, Y.; Ning, L.; Chen, L.; Li, M.; Liu, Y.; Wang, G. Reduction and Functional Exhaustion of T Cells in Patients with Coronavirus Disease 2019 (COVID-19). Front. Immunol. 2020, 11, 827. [Google Scholar] [CrossRef] [PubMed]
- Overmyer, K.A.; Shishkova, E.; Miller, I.J.; Balnis, J.; Bernstein, M.N.; Peters-Clarke, T.M.; Meyer, J.G.; Quan, Q.; Muehlbauer, L.K.; Trujillo, E.A. Large-Scale Multi-Omic Analysis of COVID-19 Severity. Cell Syst. 2021, 12, 23–40. [Google Scholar] [CrossRef] [PubMed]
- Shen, B.; Yi, X.; Sun, Y.; Bi, X.; Du, J.; Zhang, C.; Quan, S.; Zhang, F.; Sun, R.; Qian, L. Proteomic and Metabolomic Characterization of COVID-19 Patient Sera. Cell 2020, 182, 59–72. [Google Scholar] [CrossRef] [PubMed]
- Ren, X.; Wen, W.; Fan, X.; Hou, W.; Su, B.; Cai, P.; Li, J.; Liu, Y.; Tang, F.; Zhang, F.; et al. COVID-19 Immune Features Revealed by a Large-Scale Single-Cell Transcriptome Atlas. Cell 2021, 184, 1895–1913.e19. [Google Scholar] [CrossRef]
- Ericson, J.A.; Duffau, P.; Yasuda, K.; Ortiz-Lopez, A.; Rothamel, K.; Rifkin, I.R.; Monach, P.A.; Consortium, I. Gene Expression during the Generation and Activation of Mouse Neutrophils: Implication of Novel Functional and Regulatory Pathways. PLoS ONE 2014, 9, e108553. [Google Scholar] [CrossRef]
- Wilk, A.J.; Lee, M.J.; Wei, B.; Parks, B.; Pi, R.; Martínez-Colón, G.J.; Ranganath, T.; Zhao, N.Q.; Taylor, S.; Becker, W.; et al. Multi-Omic Profiling Reveals Widespread Dysregulation of Innate Immunity and Hematopoiesis in COVID-19. J. Exp. Med. 2021, 218, e20210582. [Google Scholar] [CrossRef]
- Hughes, M.J.; McGettrick, H.M.; Sapey, E. Shared Mechanisms of Multimorbidity in COPD, Atherosclerosis and Type-2 Diabetes: The Neutrophil as a Potential Inflammatory Target. Eur. Respir. Rev. 2020, 29, 190102. [Google Scholar] [CrossRef]
- Ng, L.G.; Ostuni, R.; Hidalgo, A. Heterogeneity of Neutrophils. Nat. Rev. Immunol. 2019, 19, 255–265. [Google Scholar] [CrossRef]
- Eash, K.J.; Greenbaum, A.M.; Gopalan, P.K.; Link, D.C. CXCR2 and CXCR4 Antagonistically Regulate Neutrophil Trafficking from Murine Bone Marrow. J. Clin. Investig. 2010, 120, 2423–2431. [Google Scholar] [CrossRef]
- Köhler, A.; De Filippo, K.; Hasenberg, M.; Van Den Brandt, C.; Nye, E.; Hosking, M.P.; Lane, T.E.; Männ, L.; Ransohoff, R.M.; Hauser, A.E. G-CSF–Mediated Thrombopoietin Release Triggers Neutrophil Motility and Mobilization from Bone Marrow via Induction of Cxcr2 Ligands. Blood J. Am. Soc. Hematol. 2011, 117, 4349–4357. [Google Scholar] [CrossRef]
- Ley, K.; Hoffman, H.M.; Kubes, P.; Cassatella, M.A.; Zychlinsky, A.; Hedrick, C.C.; Catz, S.D. Neutrophils: New Insights and Open Questions. Sci. Immunol. 2018, 3, eaat4579. [Google Scholar] [CrossRef]
- Lang, S.; Bruderek, K.; Kaspar, C.; Höing, B.; Kanaan, O.; Dominas, N.; Hussain, T.; Droege, F.; Eyth, C.; Hadaschik, B. Clinical Relevance and Suppressive Capacity of Human Myeloid-Derived Suppressor Cell SubsetsCD16+/CD11b+ PMN-MDSC in Head and Neck Cancer. Clin. Cancer Res. 2018, 24, 4834–4844. [Google Scholar] [CrossRef]
- Sagiv, J.Y.; Michaeli, J.; Assi, S.; Mishalian, I.; Kisos, H.; Levy, L.; Damti, P.; Lumbroso, D.; Polyansky, L.; Sionov, R.V. Phenotypic Diversity and Plasticity in Circulating Neutrophil Subpopulations in Cancer. Cell Rep. 2015, 10, 562–573. [Google Scholar] [CrossRef]
- Brandau, S.; Trellakis, S.; Bruderek, K.; Schmaltz, D.; Steller, G.; Elian, M.; Suttmann, H.; Schenck, M.; Welling, J.; Zabel, P. Myeloid-derived Suppressor Cells in the Peripheral Blood of Cancer Patients Contain a Subset of Immature Neutrophils with Impaired Migratory Properties. J. Leukoc. Biol. 2011, 89, 311–317. [Google Scholar]
- Xue, R.; Zhang, Q.; Cao, Q.; Kong, R.; Xiang, X.; Liu, H.; Feng, M.; Wang, F.; Cheng, J.; Li, Z.; et al. Liver Tumour Immune Microenvironment Subtypes and Neutrophil Heterogeneity. Nature 2022, 612, 141–147. [Google Scholar] [CrossRef]
- Jaillon, S.; Ponzetta, A.; Di Mitri, D.; Santoni, A.; Bonecchi, R.; Mantovani, A. Neutrophil Diversity and Plasticity in Tumour Progression and Therapy. Nat. Rev. Cancer 2020, 20, 485–503. [Google Scholar] [CrossRef]
- Toumi, H.; F’guyer, S.; Best, T.M. The Role of Neutrophils in Injury and Repair Following Muscle Stretch. J. Anat. 2006, 208, 459–470. [Google Scholar] [CrossRef]
- Pizza, F.X.; Peterson, J.M.; Baas, J.H.; Koh, T.J. Neutrophils Contribute to Muscle Injury and Impair Its Resolution after Lengthening Contractions in Mice. J. Physiol. 2005, 562, 899–913. [Google Scholar] [CrossRef]
- Hedrick, C.C.; Malanchi, I. Neutrophils in Cancer: Heterogeneous and Multifaceted. Nat. Rev. Immunol. 2022, 22, 173–187. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, Q.; Mackay, C.R.; Ng, L.G.; Kwok, I. Neutrophil Subsets and Their Differential Roles in Viral Respiratory Diseases. J. Leukoc. Biol. 2022, 111, 1159–1173. [Google Scholar] [CrossRef]
- Grassi, L.; Pourfarzad, F.; Ullrich, S.; Merkel, A.; Were, F.; Carrillo-de-Santa-Pau, E.; Yi, G.; Hiemstra, I.H.; Tool, A.T.J.; Mul, E. Dynamics of Transcription Regulation in Human Bone Marrow Myeloid Differentiation to Mature Blood Neutrophils. Cell Rep. 2018, 24, 2784–2794. [Google Scholar] [CrossRef]
- Evrard, M.; Kwok, I.W.H.; Chong, S.Z.; Teng, K.W.W.; Becht, E.; Chen, J.; Sieow, J.L.; Penny, H.L.; Ching, G.C.; Devi, S. Developmental Analysis of Bone Marrow Neutrophils Reveals Populations Specialized in Expansion, Trafficking, and Effector Functions. Immunity 2018, 48, 364–379. [Google Scholar] [CrossRef]
- Chen, L.; Ge, B.; Casale, F.P.; Vasquez, L.; Kwan, T.; Garrido-Martín, D.; Watt, S.; Yan, Y.; Kundu, K.; Ecker, S. Genetic Drivers of Epigenetic and Transcriptional Variation in Human Immune Cells. Cell 2016, 167, 1398–1414. [Google Scholar] [CrossRef]
- Ecker, S.; Chen, L.; Pancaldi, V.; Bagger, F.O.; Fernández, J.M.; Carrillo de Santa Pau, E.; Juan, D.; Mann, A.L.; Watt, S.; Casale, F.P. Genome-Wide Analysis of Differential Transcriptional and Epigenetic Variability across Human Immune Cell Types. Genome Biol. 2017, 18, 1–17. [Google Scholar] [CrossRef]
- Egan, C.E.; Sukhumavasi, W.; Bierly, A.L.; Denkers, E.Y. Understanding the Multiple Functions of Gr-1+ Cell Subpopulations during Microbial Infection. Immunol. Res. 2008, 40, 35–48. [Google Scholar] [CrossRef]
- Tate, M.D.; Deng, Y.-M.; Jones, J.E.; Anderson, G.P.; Brooks, A.G.; Reading, P.C. Neutrophils Ameliorate Lung Injury and the Development of Severe Disease during Influenza Infection. J. Immunol. 2009, 183, 7441–7450. [Google Scholar] [CrossRef]
- Hufford, M.M.; Richardson, G.; Zhou, H.; Manicassamy, B.; García-Sastre, A.; Enelow, R.I.; Braciale, T.J. Influenza-Infected Neutrophils within the Infected Lungs Act as Antigen Presenting Cells for Anti-Viral CD8+ T Cells. PLoS ONE 2012, 7, e46581. [Google Scholar] [CrossRef]
- Tripathi, S.; Verma, A.; Kim, E.-J.; White, M.R.; Hartshorn, K.L. LL-37 Modulates Human Neutrophil Responses to Influenza A Virus. J. Leukoc. Biol. 2014, 96, 931–938. [Google Scholar] [CrossRef]
- Noyola, E.; Noor, A.; Sweeney, N.; Chan, J.; Ramesh, R.; Calixte, R.; Krilov, L.R. Prevalence of Bandemia in Respiratory Viral Infections: A Pediatric Emergency Room Experience. Front. Pediatr. 2020, 8, 576676. [Google Scholar] [CrossRef]
- Cabrera, L.E.; Pekkarinen, P.T.; Alander, M.; Nowlan, K.H.A.; Nguyen, N.A.; Jokiranta, S.; Kuivanen, S.; Patjas, A.; Mero, S.; Pakkanen, S.H. Characterization of Low-Density Granulocytes in COVID-19. PLoS Pathog. 2021, 17, e1009721. [Google Scholar] [CrossRef]
- Silvestre-Roig, C.; Hidalgo, A.; Soehnlein, O. Neutrophil Heterogeneity: Implications for Homeostasis and Pathogenesis. Blood J. Am. Soc. Hematol. 2016, 127, 2173–2181. [Google Scholar] [CrossRef]
- Hao, Y.; Hao, S.; Andersen-Nissen, E.; Mauck III, W.M.; Zheng, S.; Butler, A.; Lee, M.J.; Wilk, A.J.; Darby, C.; Zager, M.; et al. Integrated Analysis of Multimodal Single-Cell Data. Cell 2021, 184, 3573–3587.e29. [Google Scholar] [CrossRef]
- Zappia, L.; Oshlack, A. Clustering Trees: A Visualization for Evaluating Clusterings at Multiple Resolutions. Gigascience 2018, 7, giy083. [Google Scholar] [CrossRef]
- Liu, B.; Li, C.; Li, Z.; Wang, D.; Ren, X.; Zhang, Z. An Entropy-Based Metric for Assessing the Purity of Single Cell Populations. Nat. Commun. 2020, 11, 3155. [Google Scholar] [CrossRef]
- Xie, X.; Shi, Q.; Wu, P.; Zhang, X.; Kambara, H.; Su, J.; Yu, H.; Park, S.-Y.; Guo, R.; Ren, Q.; et al. Single-Cell Transcriptome Profiling Reveals Neutrophil Heterogeneity in Homeostasis and Infection. Nat. Immunol. 2020, 21, 1119–1133. [Google Scholar] [CrossRef]
- Cowland, J.B.; Johnsen, A.H.; Borregaard, N. HCAP-18, a Cathelin/pro-Bactenecin-like Protein of Human Neutrophil Specific Granules. FEBS Lett. 1995, 368, 173–176. [Google Scholar] [CrossRef]
- Cieutat, A.-M.; Lobel, P.; August, J.T.; Kjeldsen, L.; Sengeløv, H.; Borregaard, N.; Bainton, D.F. Azurophilic Granules of Human Neutrophilic Leukocytes Are Deficient in Lysosome-Associated Membrane Proteins but Retain the Mannose 6-Phosphate Recognition Marker. Blood 1998, 91, 1044–1058. [Google Scholar] [CrossRef]
- Lood, C.; Blanco, L.P.; Purmalek, M.M.; Carmona-Rivera, C.; De Ravin, S.S.; Smith, C.K.; Malech, H.L.; Ledbetter, J.A.; Elkon, K.B.; Kaplan, M.J. Neutrophil Extracellular Traps Enriched in Oxidized Mitochondrial DNA Are Interferogenic and Contribute to Lupus-like Disease. Nat. Med. 2016, 22, 146–153. [Google Scholar] [CrossRef]
- Garcia-Romo, G.S.; Caielli, S.; Vega, B.; Connolly, J.; Allantaz, F.; Xu, Z.; Punaro, M.; Baisch, J.; Guiducci, C.; Coffman, R.L. Netting Neutrophils Are Major Inducers of Type I IFN Production in Pediatric Systemic Lupus Erythematosus. Sci. Transl. Med. 2011, 3, 73ra20. [Google Scholar] [CrossRef]
- Lecot, P.; Ardin, M.; Dussurgey, S.; Alcazer, V.; Moudombi, L.; Pereira Abrantes, M.; Hubert, M.; Swalduz, A.; Hernandez-Vargas, H.; Viari, A.; et al. Gene Signature of Circulating Platelet-Bound Neutrophils Is Associated with Poor Prognosis in Cancer Patients. Int. J. Cancer 2022, 151, 138–152. [Google Scholar] [CrossRef]
- Prame Kumar, K.; Nicholls, A.J.; Wong, C.H.Y. Partners in Crime: Neutrophils and Monocytes/Macrophages in Inflammation and Disease. Cell Tissue Res. 2018, 371, 551–565. [Google Scholar] [CrossRef]
- Hasan, M.Z.; Islam, S.; Matsumoto, K.; Kawai, T. SARS-CoV-2 Infection Initiates Interleukin-17-Enriched Transcriptional Response in Different Cells from Multiple Organs. Sci. Rep. 2021, 11, 16814. [Google Scholar] [CrossRef]
- Khanmohammadi, S.; Rezaei, N. Role of Toll-like Receptors in the Pathogenesis of COVID-19. J. Med. Virol. 2021, 93, 2735–2739. [Google Scholar] [CrossRef]
- Yamada, T.; Sato, S.; Sotoyama, Y.; Orba, Y.; Sawa, H.; Yamauchi, H.; Sasaki, M.; Takaoka, A. RIG-I Triggers a Signaling-Abortive Anti-SARS-CoV-2 Defense in Human Lung Cells. Nat. Immunol. 2021, 22, 820–828. [Google Scholar] [CrossRef]
- Kouwaki, T.; Nishimura, T.; Wang, G.; Oshiumi, H. RIG-I-like Receptor-Mediated Recognition of Viral Genomic RNA of Severe Acute Respiratory Syndrome Coronavirus-2 and Viral Escape from the Host Innate Immune Responses. Front. Immunol. 2021, 12, 700926. [Google Scholar] [CrossRef]
- Rivera, E.G.; Patnaik, A.; Salvemini, J.; Jain, S.; Lee, K.; Lozeau, D.; Yao, Q. SARS-CoV-2/COVID-19 and Its Relationship with NOD2 and Ubiquitination. Clin. Immunol. 2022, 238, 109027. [Google Scholar] [CrossRef] [PubMed]
- Almeida-da-Silva, C.L.C.; Savio, L.E.B.; Coutinho-Silva, R.; Ojcius, D.M. The Role of NOD-like Receptors in Innate Immunity. Front. Immunol. 2023, 14, 1122586. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S. Toll-like Receptor 4 in COVID-19: Friend or Foe? Future Virol. 2022, 17, 415–417. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Gable, A.L.; Nastou, K.C.; Lyon, D.; Kirsch, R.; Pyysalo, S.; Doncheva, N.T.; Legeay, M.; Fang, T.; Bork, P. The STRING Database in 2021: Customizable Protein–Protein Networks, and Functional Characterization of User-Uploaded Gene/Measurement Sets. Nucleic Acids Res. 2021, 49, D605–D612. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Yu, Y.; Trimpert, J.; Benthani, F.; Mairhofer, M.; Richter-Pechanska, P.; Wyler, E.; Belenki, D.; Kaltenbrunner, S.; Pammer, M. Virus-Induced Senescence Is a Driver and Therapeutic Target in COVID-19. Nature 2021, 599, 283–289. [Google Scholar] [CrossRef] [PubMed]
- Camell, C.D.; Yousefzadeh, M.J.; Zhu, Y.; Prata, L.G.P.L.; Huggins, M.A.; Pierson, M.; Zhang, L.; O’Kelly, R.D.; Pirtskhalava, T.; Xun, P. Senolytics Reduce Coronavirus-Related Mortality in Old Mice. Science 2021, 373, eabe4832. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.; Jeon, H.-M.; Lee, J. Tissue Factor Links Inflammation, Thrombosis, and Senescence in COVID-19. Sci. Rep. 2022, 12, 19842. [Google Scholar] [CrossRef] [PubMed]
- Mansanguan, C.; Maneerat, Y. PPBP Gene as a Biomarker for Coronary Heart Disease Risk in Postmenopausal Thai Women. PeerJ 2022, 10, e13615. [Google Scholar] [CrossRef] [PubMed]
- Kuo, T.; McQueen, A.; Chen, T.-C.; Wang, J.-C. Regulation of Glucose Homeostasis by Glucocorticoids. In Glucocorticoid Signal: From Molecules to Mice to Man; Springer: New York, NY, USA, 2015; pp. 99–126. [Google Scholar] [CrossRef]
- El-Gedaily, A.; Schoedon, G.; Schneemann, M.; Schaffner, A. Constitutive and Regulated Expression of Platelet Basic Protein in Human Monocytes. J. Leucoc. Biol. 2004, 75, 495–503. [Google Scholar] [CrossRef] [PubMed]
- Spick, M.; Campbell, A.; Baricevic-Jones, I.; von Gerichten, J.; Lewis, H.-M.; Frampas, C.F.; Longman, K.; Stewart, A.; Dunn-Walters, D.; Skene, D.J. Multi-Omics Reveals Mechanisms of Partial Modulation of COVID-19 Dysregulation by Glucocorticoid Treatment. Int. J. Mol. Sci. 2022, 23, 12079. [Google Scholar] [CrossRef]
- Gomez-Lopez, N.; Romero, R.; Escobar, M.F.; Carvajal, J.A.; Echavarria, M.P.; Albornoz, L.L.; Nasner, D.; Miller, D.; Gallo, D.M.; Galaz, J.; et al. Pregnancy-Specific Responses to COVID-19 Revealed by High-Throughput Proteomics of Human Plasma. Commun. Med. 2023, 3, 48. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.I. ClusterProfiler 4.0: A Universal Enrichment Tool for Interpreting Omics Data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.; Guerrero-Juarez, C.F.; Zhang, L.; Chang, I.; Ramos, R.; Kuan, C.-H.; Myung, P.; Plikus, M.V.; Nie, Q. Inference and Analysis of Cell-Cell Communication Using CellChat. Nat. Commun. 2021, 12, 1088. [Google Scholar] [CrossRef] [PubMed]
- Van de Sande, B.; Flerin, C.; Davie, K.; De Waegeneer, M.; Hulselmans, G.; Aibar, S.; Seurinck, R.; Saelens, W.; Cannoodt, R.; Rouchon, Q.; et al. A Scalable SCENIC Workflow for Single-Cell Gene Regulatory Network Analysis. Nat. Protoc. 2020, 15, 2247–2276. [Google Scholar] [CrossRef] [PubMed]
- Newman, A.M.; Steen, C.B.; Liu, C.L.; Gentles, A.J.; Chaudhuri, A.A.; Scherer, F.; Khodadoust, M.S.; Esfahani, M.S.; Luca, B.A.; Steiner, D.; et al. Determining Cell Type Abundance and Expression from Bulk Tissues with Digital Cytometry. Nat. Biotechnol. 2019, 37, 773–782. [Google Scholar] [CrossRef] [PubMed]
- Gorgoulis, V.; Adams, P.D.; Alimonti, A.; Bennett, D.C.; Bischof, O.; Bishop, C.; Campisi, J.; Collado, M.; Evangelou, K.; Ferbeyre, G. Cellular Senescence: Defining a Path Forward. Cell 2019, 179, 813–827. [Google Scholar] [CrossRef] [PubMed]
- Fridman, A.L.; Tainsky, M.A. Critical Pathways in Cellular Senescence and Immortalization Revealed by Gene Expression Profiling. Oncogene 2008, 27, 5975–5987. [Google Scholar] [CrossRef]
- Li, W.-X.; An, S.-Q.; Dai, S.-X.; Zhou, Z.-M.; Zeng, X.; Deng, G.-H.; Huang, Y.-Y.; Shen, L.-Y.; Xu, A.-Q.; Lin, Y.; et al. Multi-Omics Integrated Analysis Reveals a Specific Phenotype of CD8+ T Cell May Contribute to Immunothromosis via Th17 Response in Severe and Critical COVID-19. bioRxiv 2022. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, L.; Nishi, H.; Kinoshita, K. Multi-Omics Profiling Reveals Phenotypic and Functional Heterogeneity of Neutrophils in COVID-19. Int. J. Mol. Sci. 2024, 25, 3841. https://doi.org/10.3390/ijms25073841
Zhang L, Nishi H, Kinoshita K. Multi-Omics Profiling Reveals Phenotypic and Functional Heterogeneity of Neutrophils in COVID-19. International Journal of Molecular Sciences. 2024; 25(7):3841. https://doi.org/10.3390/ijms25073841
Chicago/Turabian StyleZhang, Lin, Hafumi Nishi, and Kengo Kinoshita. 2024. "Multi-Omics Profiling Reveals Phenotypic and Functional Heterogeneity of Neutrophils in COVID-19" International Journal of Molecular Sciences 25, no. 7: 3841. https://doi.org/10.3390/ijms25073841
APA StyleZhang, L., Nishi, H., & Kinoshita, K. (2024). Multi-Omics Profiling Reveals Phenotypic and Functional Heterogeneity of Neutrophils in COVID-19. International Journal of Molecular Sciences, 25(7), 3841. https://doi.org/10.3390/ijms25073841