
The Immunometabolic Gene N-Acetylglucosamine Kinase Is Uniquely Involved in the Heritability of Multiple Sclerosis Severity


Abstract
1. Introduction
2. Results
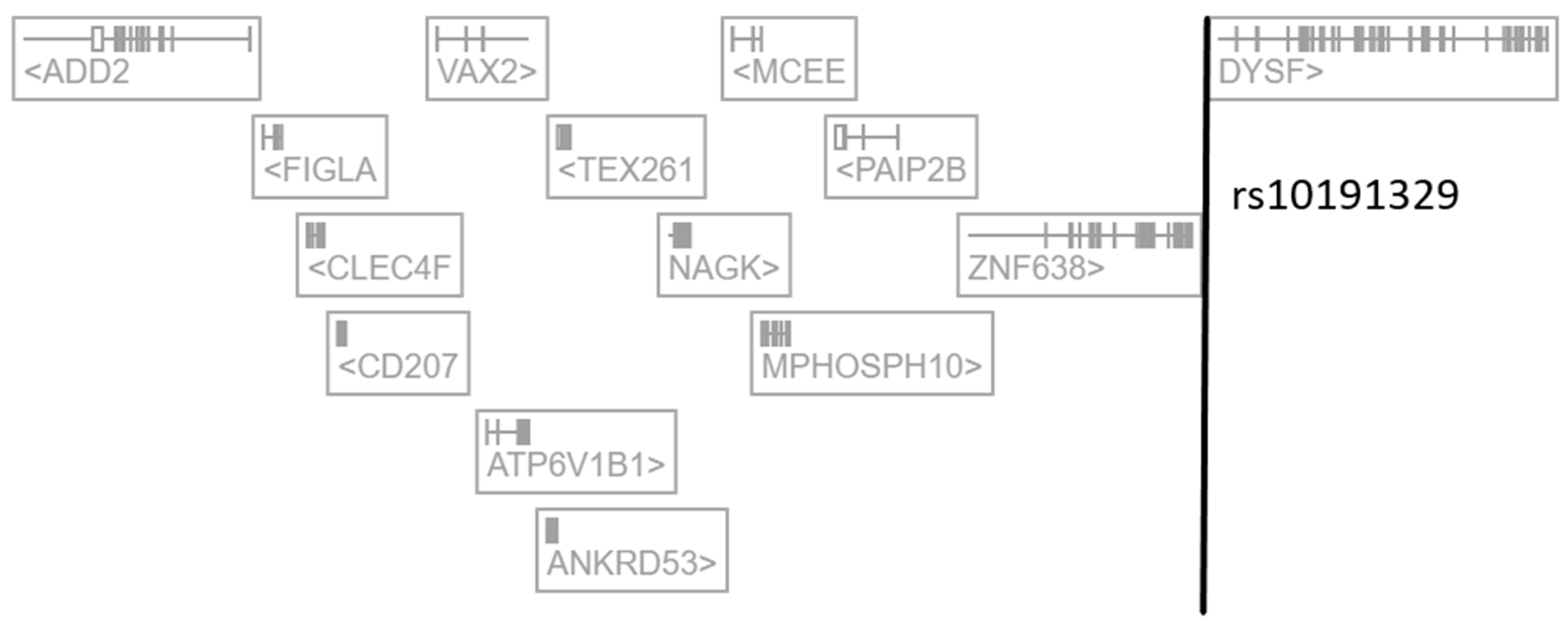
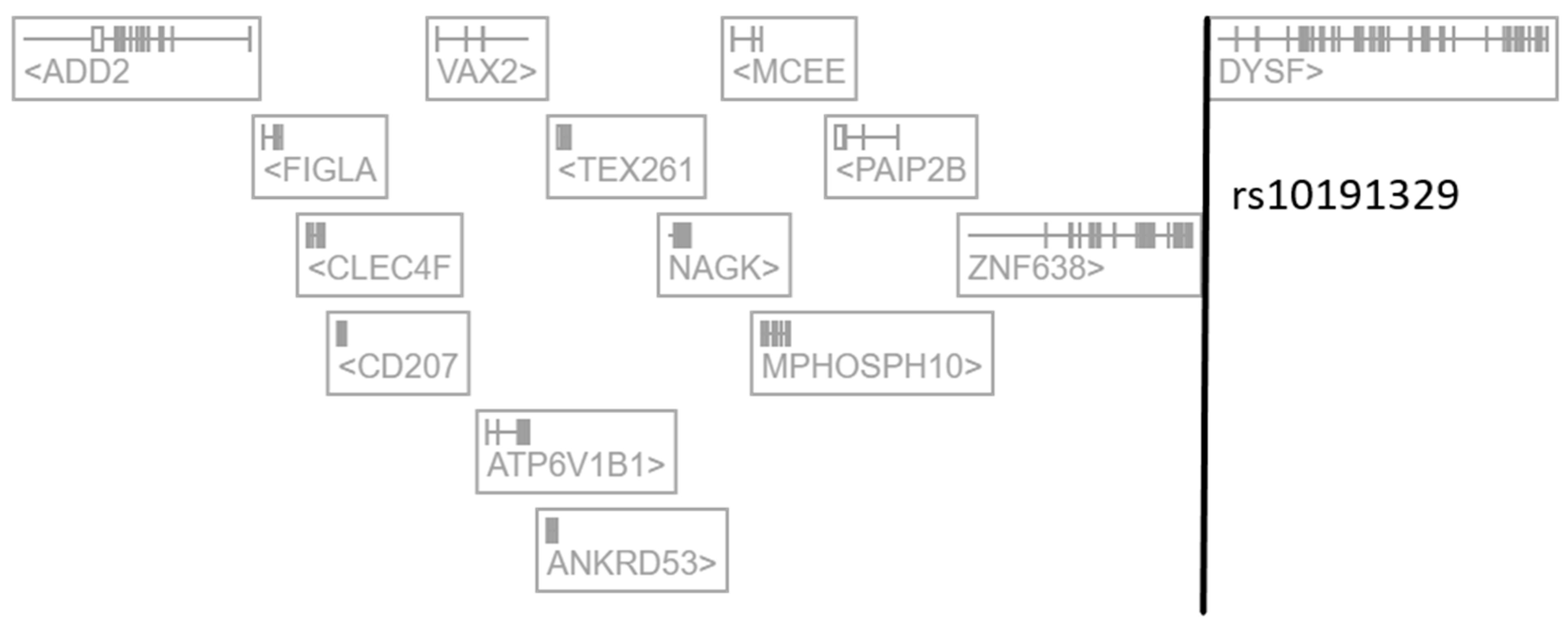
2.1. The Regulatory SNP rs10191329 Targets NAGK in Immune Cells
2.2. Monocytes, Macrophages and Myeloid Dendritic Cells Are the Immune Cell Types Exhibiting the Highest NAGK mRNA Levels
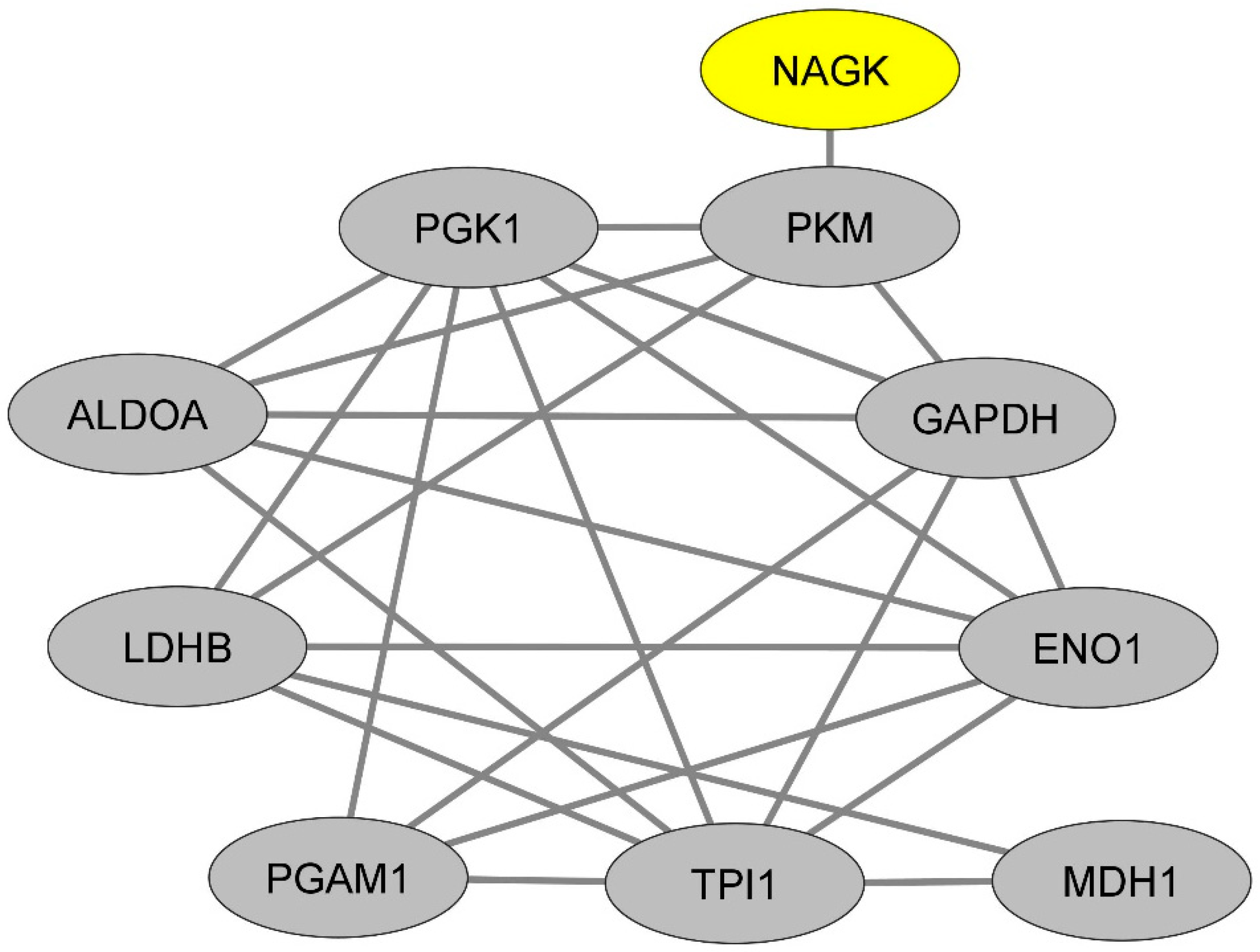
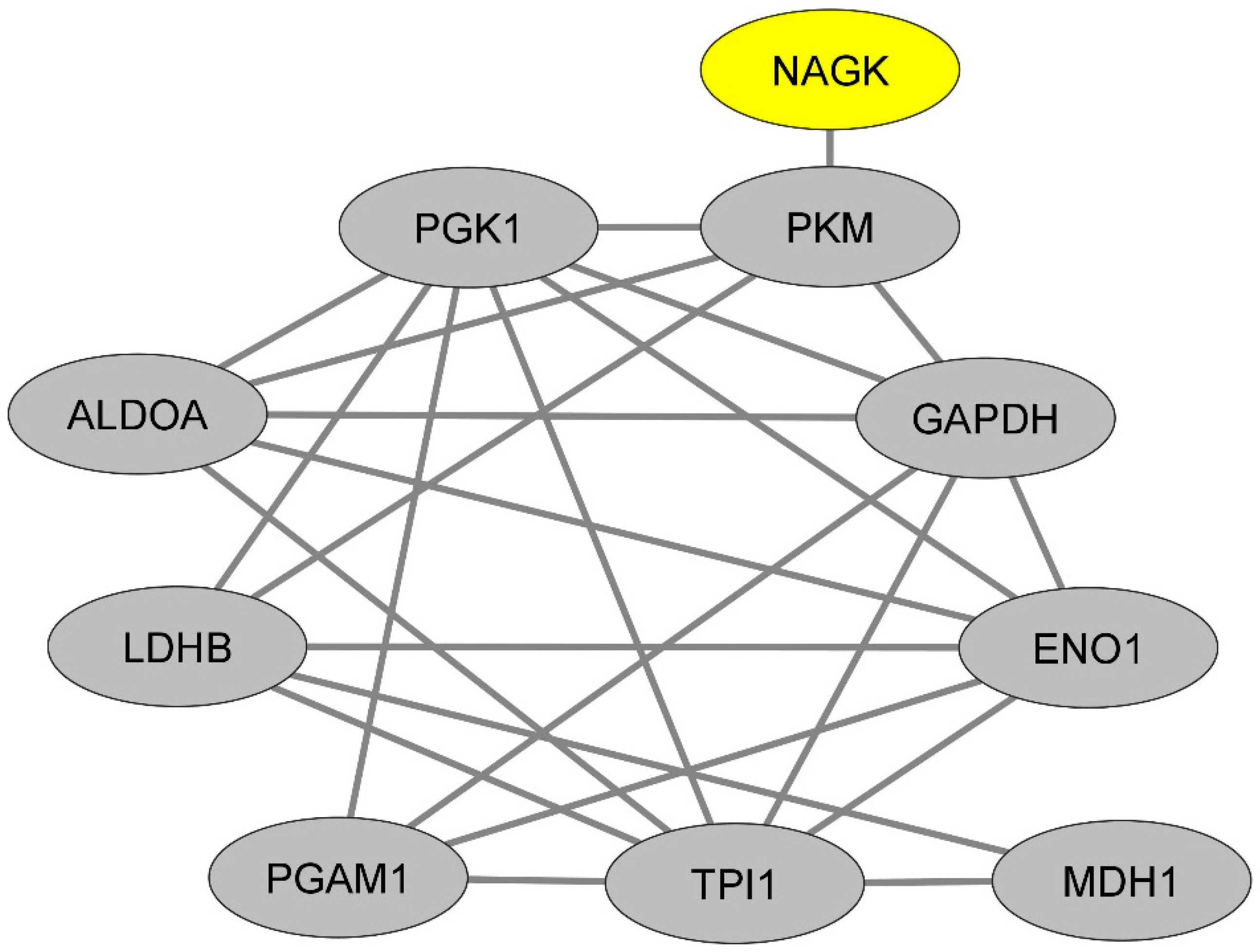
2.3. In Monocytes, NAGK Physically Interacts with a Dense Network of Proteins Involved in the Glycogenesis and Neoglucogenesis Pathway
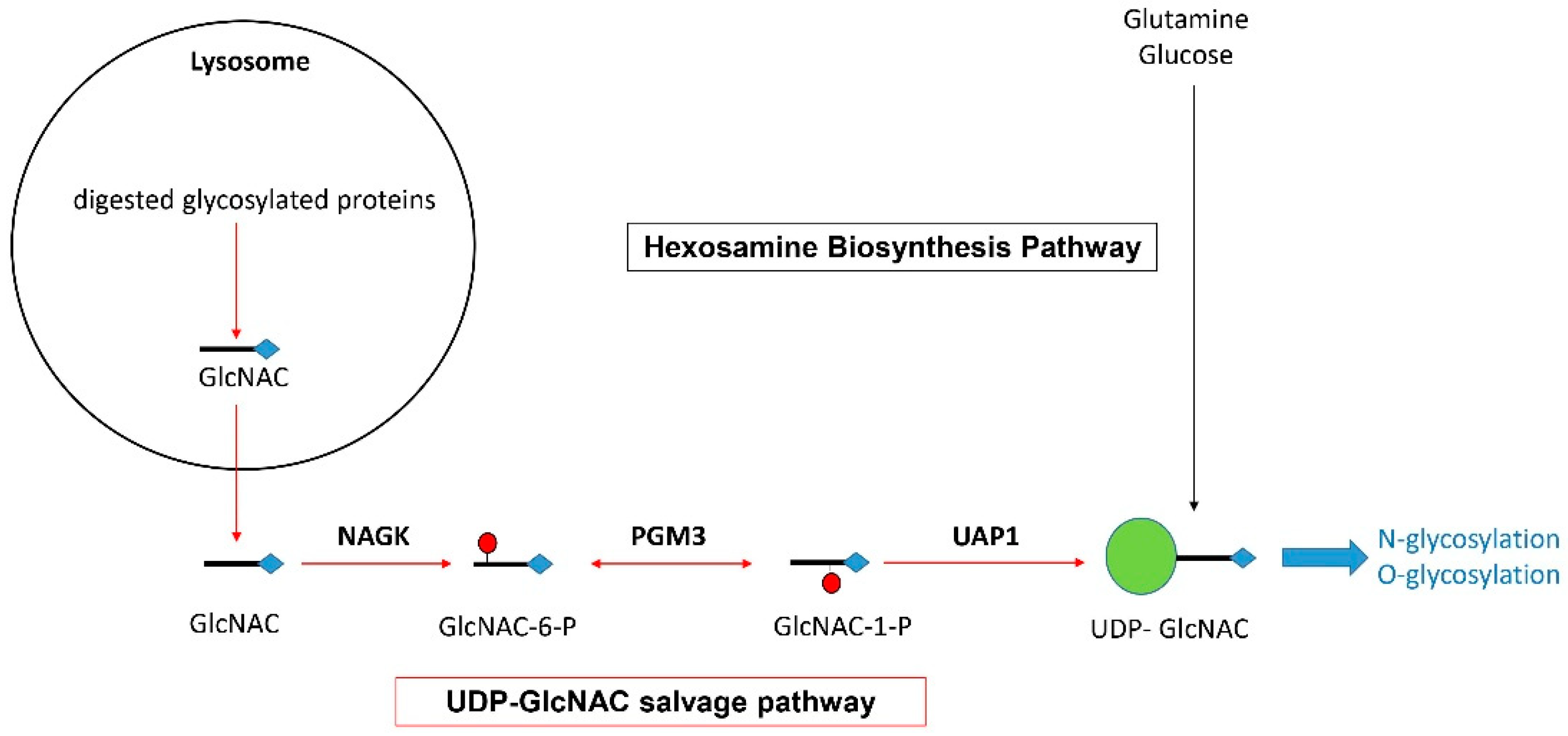
2.4. NAGK Is Potentially Involved in the Glycosylation of Key MS-Related Innate Immune Molecules
3. Discussion
4. Materials and Methods
4.1. Exploration of eQTL Datasets
4.2. Mining of Transcriptomic Datasets
4.3. Mining of Proteomic Datasets
4.4. Enrichment Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Patsopoulos, N.A.; Baranzini, S.E.; Santaniello, A.; Shoostari, P.; Cotsapas, C.; Wong, G.; Beecham, A.H.; James, T.; Replogle, J.; Vlachos, I.S.; et al. Multiple Sclerosis Genomic Map Implicates Peripheral Immune Cells and Microglia in Susceptibility. Science 2019, 365, eaav7188. [Google Scholar] [CrossRef]
- Sayao, A.L.; Devonshire, V.; Tremlett, H. Longitudinal Follow-up of “Benign” Multiple Sclerosis at 20 Years. Neurology 2007, 68, 496–500. [Google Scholar] [CrossRef] [PubMed]
- Ellenberger, D.; Flachenecker, P.; Haas, J.; Hellwig, K.; Paul, F.; Stahmann, A.; Warnke, C.; Zettl, U.K.; Rommer, P.S. Is Benign MS Really Benign? What a Meaningful Classification beyond the EDSS Must Take into Consideration. Mult. Scler. Relat. Disord. 2020, 46, 102485. [Google Scholar] [CrossRef] [PubMed]
- International Multiple Sclerosis Genetics Consortium. MultipleMS Consortium Locus for Severity Implicates CNS Resilience in Progression of Multiple Sclerosis. Nature 2023, 619, 323–331. [Google Scholar] [CrossRef] [PubMed]
- Stern, Y. Cognitive Reserve and Alzheimer Disease. Alzheimer Dis. Assoc. Disord. 2006, 20, 112–117. [Google Scholar] [CrossRef] [PubMed]
- Vemuri, P.; Weigand, S.D.; Przybelski, S.A.; Knopman, D.S.; Smith, G.E.; Trojanowski, J.Q.; Shaw, L.M.; Decarli, C.S.; Carmichael, O.; Bernstein, M.A.; et al. Cognitive Reserve and Alzheimer’s Disease Biomarkers Are Independent Determinants of Cognition. Brain 2011, 134, 1479–1492. [Google Scholar] [CrossRef] [PubMed]
- Mountjoy, E.; Schmidt, E.M.; Carmona, M.; Schwartzentruber, J.; Peat, G.; Miranda, A.; Fumis, L.; Hayhurst, J.; Buniello, A.; Karim, M.A.; et al. An Open Approach to Systematically Prioritize Causal Variants and Genes at All Published Human GWAS Trait-Associated Loci. Nat. Genet. 2021, 53, 1527–1533. [Google Scholar] [CrossRef]
- Ghoussaini, M.; Mountjoy, E.; Carmona, M.; Peat, G.; Schmidt, E.M.; Hercules, A.; Fumis, L.; Miranda, A.; Carvalho-Silva, D.; Buniello, A.; et al. Open Targets Genetics: Systematic Identification of Trait-Associated Genes Using Large-Scale Genetics and Functional Genomics. Nucleic Acids Res. 2021, 49, D1311–D1320. [Google Scholar] [CrossRef]
- Karlsson, M.; Zhang, C.; Méar, L.; Zhong, W.; Digre, A.; Katona, B.; Sjöstedt, E.; Butler, L.; Odeberg, J.; Dusart, P.; et al. A Single-Cell Type Transcriptomics Map of Human Tissues. Sci. Adv. 2021, 7, eabh2169. [Google Scholar] [CrossRef]
- Uhlen, M.; Karlsson, M.J.; Zhong, W.; Tebani, A.; Pou, C.; Mikes, J.; Lakshmikanth, T.; Forsström, B.; Edfors, F.; Odeberg, J.; et al. A Genome-Wide Transcriptomic Analysis of Protein-Coding Genes in Human Blood Cells. Science 2019, 366, eaax9198. [Google Scholar] [CrossRef]
- Obayashi, T.; Kodate, S.; Hibara, H.; Kagaya, Y.; Kinoshita, K. COXPRESdb v8: An Animal Gene Coexpression Database Navigating from a Global View to Detailed Investigations. Nucleic Acids Res. 2023, 51, D80–D87. [Google Scholar] [CrossRef] [PubMed]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A Comprehensive Gene Set Enrichment Analysis Web Server 2016 Update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Bailey, A.; Kuleshov, M.V.; Clarke, D.J.B.; Evangelista, J.E.; Jenkins, S.L.; Lachmann, A.; Wojciechowicz, M.L.; Kropiwnicki, E.; Jagodnik, K.M.; et al. Gene Set Knowledge Discovery with Enrichr. Curr. Protoc. 2021, 1, e90. [Google Scholar] [CrossRef] [PubMed]
- Palasca, O.; Santos, A.; Stolte, C.; Gorodkin, J.; Jensen, L.J. TISSUES 2.0: An Integrative Web Resource on Mammalian Tissue Expression. Database 2018, 2018, bay003. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Jin, X.; Tsueng, G.; Afrasiabi, C.; Su, A.I. BioGPS: Building Your Own Mash-up of Gene Annotations and Expression Profiles. Nucleic Acids Res. 2016, 44, D313–D316. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Lan, Y.; Xu, J.; Quan, F.; Zhao, E.; Deng, C.; Luo, T.; Xu, L.; Liao, G.; Yan, M.; et al. CellMarker: A Manually Curated Resource of Cell Markers in Human and Mouse. Nucleic Acids Res. 2019, 47, D721–D728. [Google Scholar] [CrossRef] [PubMed]
- Milacic, M.; Beavers, D.; Conley, P.; Gong, C.; Gillespie, M.; Griss, J.; Haw, R.; Jassal, B.; Matthews, L.; May, B.; et al. The Reactome Pathway Knowledgebase 2024. Nucleic Acids Res. 2024, 52, D672–D678. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.; Grishagin, I.; Wang, Y.; Zhao, T.; Greene, J.; Obenauer, J.C.; Ngan, D.; Nguyen, D.T.; Guha, R.; Jadhav, A.; et al. The NCATS BioPlanet—An Integrated Platform for Exploring the Universe of Cellular Signaling Pathways for Toxicology, Systems Biology, and Chemical Genomics. Front. Pharmacol. 2019, 10, 445. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Furumichi, M.; Sato, Y.; Ishiguro-Watanabe, M.; Tanabe, M. KEGG: Integrating Viruses and Cellular Organisms. Nucleic Acids Res. 2021, 49, D545–D551. [Google Scholar] [CrossRef]
- Mi, H.; Muruganujan, A.; Huang, X.; Ebert, D.; Mills, C.; Guo, X.; Thomas, P.D. Protocol Update for Large-Scale Genome and Gene Function Analysis with the PANTHER Classification System (v.14.0). Nat. Protoc. 2019, 14, 703–721. [Google Scholar] [CrossRef]
- Agrawal, A.; Balcı, H.; Hanspers, K.; Coort, S.L.; Martens, M.; Slenter, D.N.; Ehrhart, F.; Digles, D.; Waagmeester, A.; Wassink, I.; et al. WikiPathways 2024: Next Generation Pathway Database. Nucleic Acids Res. 2024, 52, D679–D689. [Google Scholar] [CrossRef] [PubMed]
- Oughtred, R.; Rust, J.; Chang, C.; Breitkreutz, B.J.; Stark, C.; Willems, A.; Boucher, L.; Leung, G.; Kolas, N.; Zhang, F.; et al. The BioGRID Database: A Comprehensive Biomedical Resource of Curated Protein, Genetic, and Chemical Interactions. Protein Sci. 2021, 30, 187–200. [Google Scholar] [CrossRef]
- Stafford, C.A.; Gassauer, A.M.; de Oliveira Mann, C.C.; Tanzer, M.C.; Fessler, E.; Wefers, B.; Nagl, D.; Kuut, G.; Sulek, K.; Vasilopoulou, C.; et al. Phosphorylation of Muramyl Peptides by NAGK Is Required for NOD2 Activation. Nature 2022, 609, 590–596. [Google Scholar] [CrossRef] [PubMed]
- Lessard, A.J.; LeBel, M.; Egarnes, B.; Préfontaine, P.; Thériault, P.; Droit, A.; Brunet, A.; Rivest, S.; Gosselin, J. Triggering of NOD2 Receptor Converts Inflammatory Ly6Chigh into Ly6Clow Monocytes with Patrolling Properties. Cell Rep. 2017, 20, 1830–1843. [Google Scholar] [CrossRef]
- Paneque, A.; Fortus, H.; Zheng, J.; Werlen, G.; Jacinto, E. The Hexosamine Biosynthesis Pathway: Regulation and Function. Genes 2023, 14, 933. [Google Scholar] [CrossRef]
- Neitzel, L.R.; Spencer, Z.T.; Nayak, A.; Cselenyi, C.S.; Benchabane, H.; Youngblood, C.A.Q.; Zouaoui, A.; Ng, V.; Stephens, L.; Hann, T.; et al. Developmental Regulation of Wnt Signaling by Nagk and the UDP-GlcNAc Salvage Pathway. Mech. Dev. 2019, 156, 20–31. [Google Scholar] [CrossRef]
- Campbell, S.; Mesaros, C.; Izzo, L.; Affronti, H.; Noji, M.; Schaffer, B.E.; Tsang, T.; Sun, K.; Trefely, S.; Kruijning, S.; et al. Glutamine Deprivation Triggers NAGK-Dependent Hexosamine Salvage. eLife 2021, 10, e62644. [Google Scholar] [CrossRef]
- Sun, S.; Hu, Y.; Ao, M.; Shah, P.; Chen, J.; Yang, W.; Jia, X.; Tian, Y.; Thomas, S.; Zhang, H. N-GlycositeAtlas: A Database Resource for Mass Spectrometry-Based Human N-Linked Glycoprotein and Glycosylation Site Mapping. Clin. Proteom. 2019, 16, 35. [Google Scholar] [CrossRef]
- Werneburg, S.; Jung, J.; Kunjamma, R.B.; Ha, S.K.; Luciano, N.J.; Willis, C.M.; Gao, G.; Biscola, N.P.; Havton, L.A.; Crocker, S.J.; et al. Targeted Complement Inhibition at Synapses Prevents Microglial Synaptic Engulfment and Synapse Loss in Demyelinating Disease. Immunity 2020, 52, 167–182.e7. [Google Scholar] [CrossRef] [PubMed]
- Hammond, J.W.; Bellizzi, M.J.; Ware, C.; Qiu, W.Q.; Saminathan, P.; Li, H.; Luo, S.; Ma, S.A.; Li, Y.; Gelbard, H.A. Complement-Dependent Synapse Loss and Microgliosis in a Mouse Model of Multiple Sclerosis. Brain Behav. Immun. 2020, 87, 739–750. [Google Scholar] [CrossRef]
- Michailidou, I.; Willems, J.G.P.; Kooi, E.J.; Van Eden, C.; Gold, S.M.; Geurts, J.J.G.; Baas, F.; Huitinga, I.; Ramaglia, V. Complement C1q-C3-Associated Synaptic Changes in Multiple Sclerosis Hippocampus. Ann. Neurol. 2015, 77, 1007–1026. [Google Scholar] [CrossRef] [PubMed]
- Ramaglia, V.; Hughes, T.R.; Donev, R.M.; Ruseva, M.M.; Wu, X.; Huitinga, I.; Baas, F.; Neal, J.W.; Morgan, B.P. C3-Dependent Mechanism of Microglial Priming Relevant to Multiple Sclerosis. Proc. Natl. Acad. Sci. USA 2012, 109, 965–970. [Google Scholar] [CrossRef]
- Ibañez-Vega, J.; Vilchez, C.; Jimenez, K.; Guevara, C.; Burgos, P.I.; Naves, R. Cellular and Molecular Regulation of the Programmed Death-1/Programmed Death Ligand System and Its Role in Multiple Sclerosis and Other Autoimmune Diseases. J. Autoimmun. 2021, 123, 102702. [Google Scholar] [CrossRef] [PubMed]
- Vogel, A.; Martin, K.; Soukup, K.; Halfmann, A.; Kerndl, M.; Brunner, J.S.; Hofmann, M.; Oberbichler, L.; Korosec, A.; Kuttke, M.; et al. JAK1 Signaling in Dendritic Cells Promotes Peripheral Tolerance in Autoimmunity through PD-L1-Mediated Regulatory T Cell Induction. Cell Rep. 2022, 38, 110420. [Google Scholar] [CrossRef]
- Sawcer, S.; Hellenthal, G.; Pirinen, M.; Spencer, C.C.A.; Patsopoulos, N.A.; Moutsianas, L.; Dilthey, A.; Su, Z.; Freeman, C.; Hunt, S.E.; et al. Genetic Risk and a Primary Role for Cell-Mediated Immune Mechanisms in Multiple Sclerosis. Nature 2011, 476, 214–219. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Jelcic, I.; Mühlenbruch, L.; Haunerdinger, V.; Toussaint, N.C.; Zhao, Y.; Cruciani, C.; Faigle, W.; Naghavian, R.; Foege, M.; et al. HLA-DR15 Molecules Jointly Shape an Autoreactive T Cell Repertoire in Multiple Sclerosis. Cell 2020, 183, 1264–1281.e20. [Google Scholar] [CrossRef]
- Gasperi, C.; Wiltgen, T.; McGinnis, J.; Cerri, S.; Moridi, T.; Ouellette, R.; Pukaj, A.; Voon, C.; Bafligil, C.; Lauerer, M.; et al. A Genetic Risk Variant for Multiple Sclerosis Severity Is Associated with Brain Atrophy. Ann. Neurol. 2023, 94, 1080–1085. [Google Scholar] [CrossRef] [PubMed]
- Grosveld, F.; Van Staalduinen, J.; Stadhouders, R. Transcriptional Regulation by (Super)Enhancers: From Discovery to Mechanisms. Annu. Rev. Genom. Hum. Genet. 2021, 22, 127–146. [Google Scholar] [CrossRef]
- Giacopuzzi, E.; Popitsch, N.; Taylor, J.C. GREEN-DB: A Framework for the Annotation and Prioritization of Non-Coding Regulatory Variants from Whole-Genome Sequencing Data. Nucleic Acids Res. 2022, 50, 2522–2535. [Google Scholar] [CrossRef]
- Brodie, A.; Azaria, J.R.; Ofran, Y. How Far from the SNP May the Causative Genes Be? Nucleic Acids Res. 2016, 44, 6046–6054. [Google Scholar] [CrossRef]
- Zhu, Z.; Zhang, F.; Hu, H.; Bakshi, A.; Robinson, M.R.; Powell, J.E.; Montgomery, G.W.; Goddard, M.E.; Wray, N.R.; Visscher, P.M.; et al. Integration of Summary Data from GWAS and EQTL Studies Predicts Complex Trait Gene Targets. Nat. Genet. 2016, 48, 481–487. [Google Scholar] [CrossRef]
- Savage, J.E.; Jansen, P.R.; Stringer, S.; Watanabe, K.; Bryois, J.; De Leeuw, C.A.; Nagel, M.; Awasthi, S.; Barr, P.B.; Coleman, J.R.I.; et al. Genome-Wide Association Meta-Analysis in 269,867 Individuals Identifies New Genetic and Functional Links to Intelligence. Nat. Genet. 2018, 50, 912–919. [Google Scholar] [CrossRef]
- Fadista, J.; Manning, A.K.; Florez, J.C.; Groop, L. The (in)Famous GWAS P-Value Threshold Revisited and Updated for Low-Frequency Variants. Eur. J. Hum. Genet. 2016, 24, 1202–1205. [Google Scholar] [CrossRef] [PubMed]
- Vasileiou, E.S.; Filippatou, A.G.; Pimentel Maldonado, D.; Kalaitzidis, G.; Ehrhardt, H.; Lambe, J.; Saidha, S.; Sotirchos, E.S.; Mowry, E.M.; Calabresi, P.A.; et al. Socioeconomic Disparity Is Associated with Faster Retinal Neurodegeneration in Multiple Sclerosis. Brain 2021, 144, 3664–3673. [Google Scholar] [CrossRef]
- Mao, L.; Dhar, A.; Meng, G.; Fuss, I.; Montgomery-Recht, K.; Yang, Z.; Xu, Q.; Kitani, A.; Strober, W. Blau Syndrome NOD2 Mutations Result in Loss of NOD2 Cross-Regulatory Function. Front. Immunol. 2022, 13, 988862. [Google Scholar] [CrossRef]
- Miceli-Richard, C.; Lesage, S.; Rybojad, M.; Prieur, A.M.; Manouvrier-Hanu, S.; Häfner, R.; Chamaillard, M.; Zouali, H.; Thomas, G.; Hugot, J.P. CARD15 Mutations in Blau Syndrome. Nat. Genet. 2001, 29, 19–20. [Google Scholar] [CrossRef]
- Nayar, S.; Morrison, J.K.; Giri, M.; Gettler, K.; Chuang, L.-s.; Walker, L.A.; Ko, H.M.; Kenigsberg, E.; Kugathasan, S.; Merad, M.; et al. A Myeloid-Stromal Niche and Gp130 Rescue in NOD2-Driven Crohn’s Disease. Nature 2021, 593, 275–281. [Google Scholar] [CrossRef] [PubMed]
- Hugot, J.P.; Chamaillard, M.; Zouali, H.; Lesage, S.; Cézard, J.P.; Belaiche, J.; Almer, S.; Tysk, C.; O’morain, C.A.; Gassull, M.; et al. Association of NOD2 Leucine-Rich Repeat Variants with Susceptibility to Crohn’s Disease. Nature 2001, 411, 599–603. [Google Scholar] [CrossRef]
- Ogura, Y.; Bonen, D.K.; Inohara, N.; Nicolae, D.L.; Chen, F.F.; Ramos, R.; Britton, H.; Moran, T.; Karaliuskas, R.; Duerr, R.H.; et al. A Frameshift Mutation in NOD2 Associated with Susceptibility to Crohn’s Disease. Nature 2001, 411, 603–606. [Google Scholar] [CrossRef] [PubMed]
- Fani Maleki, A.; Cisbani, G.; Laflamme, N.; Prefontaine, P.; Plante, M.M.; Baillargeon, J.; Rangachari, M.; Gosselin, J.; Rivest, S. Selective Immunomodulatory and Neuroprotective Effects of a NOD2 Receptor Agonist on Mouse Models of Multiple Sclerosis. Neurotherapeutics 2021, 18, 889–904. [Google Scholar] [CrossRef] [PubMed]
- Chauvin, C.; Alvarez-Simon, D.; Radulovic, K.; Boulard, O.; Laine, W.; Delacre, M.; Waldschmitt, N.; Segura, E.; Kluza, J.; Chamaillard, M.; et al. NOD2 in Monocytes Negatively Regulates Macrophage Development through TNFalpha. Front. Immunol. 2023, 14, 1181823. [Google Scholar] [CrossRef]
- Hedl, M.; Li, J.; Cho, J.H.; Abraham, C. Chronic Stimulation of Nod2 Mediates Tolerance to Bacterial Products. Proc. Natl. Acad. Sci. USA 2007, 104, 19440–19445. [Google Scholar] [CrossRef]
- Prescott, D.; Maisonneuve, C.; Yadav, J.; Rubino, S.J.; Girardin, S.E.; Philpott, D.J. NOD2 Modulates Immune Tolerance via the GM-CSF-Dependent Generation of CD103+ Dendritic Cells. Proc. Natl. Acad. Sci. USA 2020, 117, 10946–10957. [Google Scholar] [CrossRef]
- Yamasaki, R.; Lu, H.; Butovsky, O.; Ohno, N.; Rietsch, A.M.; Cialic, R.; Wu, P.M.; Doykan, C.E.; Lin, J.; Cotleur, A.C.; et al. Differential Roles of Microglia and Monocytes in the Inflamed Central Nervous System. J. Exp. Med. 2014, 211, 1533–1549. [Google Scholar] [CrossRef]
- Paré, A.; Mailhot, B.; Lévesque, S.A.; Juzwik, C.; Doss, P.M.I.A.; Lécuyer, M.A.; Prat, A.; Rangachari, M.; Fournier, A.; Lacroix, S. IL-1β Enables CNS Access to CCR2hi Monocytes and the Generation of Pathogenic Cells through GM-CSF Released by CNS Endothelial Cells. Proc. Natl. Acad. Sci. USA 2018, 115, E1194–E1203. [Google Scholar] [CrossRef]
- Ajami, B.; Bennett, J.L.; Krieger, C.; McNagny, K.M.; Rossi, F.M.V. Infiltrating Monocytes Trigger EAE Progression, but Do Not Contribute to the Resident Microglia Pool. Nat. Neurosci. 2011, 14, 1142–1150. [Google Scholar] [CrossRef]
- Fischer, H.J.; Finck, T.L.K.; Pellkofer, H.L.; Reichardt, H.M.; Lühder, F. Glucocorticoid Therapy of Multiple Sclerosis Patients Induces Anti-Inflammatory Polarization and Increased Chemotaxis of Monocytes. Front. Immunol. 2019, 10, 1200. [Google Scholar] [CrossRef]
- Zahoor, I.; Rui, B.; Khan, J.; Datta, I.; Giri, S. An Emerging Potential of Metabolomics in Multiple Sclerosis: A Comprehensive Overview. Cell Mol. Life Sci. 2021, 78, 3181–3203. [Google Scholar] [CrossRef]
- Brandt, A.U.; Sy, M.; Bellmann-Strobl, J.; Newton, B.L.; Pawling, J.; Zimmermann, H.G.; Yu, Z.; Chien, C.; Dörr, J.; Wuerfel, J.T.; et al. Association of a Marker of N-Acetylglucosamine With Progressive Multiple Sclerosis and Neurodegeneration. JAMA Neurol. 2021, 78, 842–852. [Google Scholar] [CrossRef]
- Sy, M.; Brandt, A.U.; Lee, S.U.; Newton, B.L.; Pawling, J.; Golzar, A.; Rahman, A.M.A.; Yu, Z.; Cooper, G.; Scheel, M.; et al. N-Acetylglucosamine Drives Myelination by Triggering Oligodendrocyte Precursor Cell Differentiation. J. Biol. Chem. 2020, 295, 17413–17424. [Google Scholar] [CrossRef]
- Grigorian, A.; Araujo, L.; Naidu, N.N.; Place, D.J.; Choudhury, B.; Demetriou, M. N-Acetylglucosamine Inhibits T-Helper 1 (Th1)/T-Helper 17 (Th17) Cell Responses and Treats Experimental Autoimmune Encephalomyelitis. J. Biol. Chem. 2011, 286, 40133–40141. [Google Scholar] [CrossRef] [PubMed]
- Sy, M.; Newton, B.L.; Pawling, J.; Hayama, K.L.; Cordon, A.; Yu, Z.; Kuhle, J.; Dennis, J.W.; Brandt, A.U.; Demetriou, M. N-Acetylglucosamine Inhibits Inflammation and Neurodegeneration Markers in Multiple Sclerosis: A Mechanistic Trial. J. Neuroinflam. 2023, 20, 209. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Tissue or Cell Type | Regulated Gene | Distance from TSS | Beta Value | p-Value |
|---|---|---|---|---|
| blood cells | NAGK | 385,525 bp | −0.316 | 6.7 × 10−101 |
| ZNF638 | 173,308 bp | +0.166 | 3.2 × 10−51 | |
| MCEE | 319,630 bp | −0.123 | 3.6 × 10−34 | |
| CLEC4F | >500 Kb | +0.0573 | 1.4 × 10−8 | |
| MPHOSPH10 | 319,559 bp | −0.0511 | 4.2 × 10−7 | |
| IFN-γ-stimulated monocytes | NAGK | 385,525 bp | −0.0972 | 1.9 × 10−6 |
| testis | DYSF | 3692 bp | −0.290 | 2.3 × 10−6 |
| induced pluripotent stem cells | DYSF | 3692 bp | −0.263 | 5.0 × 10−9 |
| Gene (Gene Symbol and HPA URL) | Mean Normalized TPM in Immune Cells | Mean Normalized TPM in Neural Cells |
|---|---|---|
| NAGK https://www.proteinatlas.org/ENSG00000124357-NAGK/single+cell+type (accessed on 12 February 2024) | dendritic cells: 119.6 monocytes: 85.4 macrophages: 104.9 T-cells: 25.4 B-cells: 44.2 | excitatory neurons: 11.1 inhibitory neurons: 14.5 astrocytes: 14.2 microglia: 25 oligodendrocytes: 22.3 |
| ZNF638 https://www.proteinatlas.org/ENSG00000075292-ZNF638/single+cell+type (accessed on 12 February 2024) | dendritic cells: 48.7 monocytes: 53.8 macrophages: 52.5 T-cells: 60.4 B-cells: 61.9 | excitatory neurons: 277.2 inhibitory neurons: 257 astrocytes: 216.1 microglia: 215.6 oligodendrocytes: 576.6 |
| MCEE https://www.proteinatlas.org/ENSG00000124370-MCEE/single+cell+type (accessed on 12 February 2024) | dendritic cells: 12.1 monocytes: 5.9 macrophages: 10.1 T-cells: 9.7 B-cells: 7.8 | excitatory neurons: 12.8 inhibitory neurons: 13.9 astrocytes: 13 microglia: 13.4 oligodendrocytes: 19.4 |
| CLEC4F https://www.proteinatlas.org/ENSG00000152672-CLEC4F/single+cell+type (accessed on 12 February 2024) | dendritic cells: 0 monocytes: 13.9 macrophages: 3.6 T-cells: 0.1 B-cells: 0 | excitatory neurons: 0.2 inhibitory neurons: 0.1 astrocytes: 0 microglia: 0 oligodendrocytes: 0.1 |
| MPHOSPH10 https://www.proteinatlas.org/ENSG00000124383-MPHOSPH10/single+cell+type (accessed on 12 February 2024) | dendritic cells: 42 monocytes: 27.8 macrophages: 21.1 T-cells: 43.6 B-cells: 35.2 | excitatory neurons: 9.5 inhibitory neurons: 10.9 astrocytes: 7.7 microglia: 14.3 oligodendrocytes: 14.8 |
| DYSF https://www.proteinatlas.org/ENSG00000135636-DYSF/single+cell+type (accessed on 12 February 2024) | dendritic cells: 0.4 monocytes: 3.5 macrophages: 10 T-cells: 0.7 B-cells: 1.3 | excitatory neurons: 33.6 inhibitory neurons: 12.5 astrocytes: 0.9 microglia: 16.1 oligodendrocytes: 58.4 |
| PMID | Assigned Cell Types |
|---|---|
| 27864467 | Blood pre-conventional dendritic cells |
| 29808007 | Monocytes and dendritic cells |
| 31597962 | Fetal liver hematopoietic cells |
| 30692988 | Tonsils dendritic cells |
| 30402542 | Decidual macrophages and dendritic cells |
| 31594933 | Blood monocytes |
| 29352091 | Effector memory CD4+ T lymphocytes |
| 33352111 | Neonatal liver monocytes |
| 32351704 | Blood, liver or spleen dendritic cells |
| 30737144 | Skin regulatory T-cells |
| Library | Cell Type | Adjusted p-Value |
|---|---|---|
| Jensen Tissues | Monocyte | 6.205 × 10−17 |
| Intestine | 3.604 × 10−14 | |
| Natural killer cell | 8.235 × 10−14 | |
| Blood | 2.823 × 10−13 | |
| Blood platelet | 5.428 × 10−13 | |
| Human Gene Atlas | CD33+ myeloid cell | 0.001403 |
| CD56+ natural killer cell | 0.007790 | |
| CD14+ monocytes | 0.009458 | |
| Whole blood | 0.01149 | |
| CellMarker Augmented 2021 | Pancreatic Islet cell | 1.508 × 10−7 |
| Basal cell | 0.0004272 | |
| Monocyte from feta kidney | 0.0008851 | |
| Liver regulatory T-cell | 0.0008851 |
| Gene | Classical Monocytes | Non-Classical Monocytes |
|---|---|---|
| NAGK | 1.53 | 1.28 |
| PKM | 1.53 | 1.33 |
| PGK1 | 1.86 | 1.66 |
| ALDOA | 1.70 | 1.33 |
| LDHB | −0.8 | NS |
| PGAM1 | 1.6 | 1.5 |
| TPI1 | NS | 1.25 |
| MDH1 | 1.5 | NS |
| ENO1 | 1.68 | 1.58 |
| GAPDH | 2.4 | 1.65 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nataf, S.; Guillen, M.; Pays, L. The Immunometabolic Gene N-Acetylglucosamine Kinase Is Uniquely Involved in the Heritability of Multiple Sclerosis Severity. Int. J. Mol. Sci. 2024, 25, 3803. https://doi.org/10.3390/ijms25073803
Nataf S, Guillen M, Pays L. The Immunometabolic Gene N-Acetylglucosamine Kinase Is Uniquely Involved in the Heritability of Multiple Sclerosis Severity. International Journal of Molecular Sciences. 2024; 25(7):3803. https://doi.org/10.3390/ijms25073803
Chicago/Turabian StyleNataf, Serge, Marine Guillen, and Laurent Pays. 2024. "The Immunometabolic Gene N-Acetylglucosamine Kinase Is Uniquely Involved in the Heritability of Multiple Sclerosis Severity" International Journal of Molecular Sciences 25, no. 7: 3803. https://doi.org/10.3390/ijms25073803
APA StyleNataf, S., Guillen, M., & Pays, L. (2024). The Immunometabolic Gene N-Acetylglucosamine Kinase Is Uniquely Involved in the Heritability of Multiple Sclerosis Severity. International Journal of Molecular Sciences, 25(7), 3803. https://doi.org/10.3390/ijms25073803