
Revealing the Interaction Mechanism between Mycobacterium tuberculosis GyrB and Novobiocin, SPR719 through Binding Thermodynamics and Dissociation Kinetics Analysis

Abstract
1. Introduction
2. Results
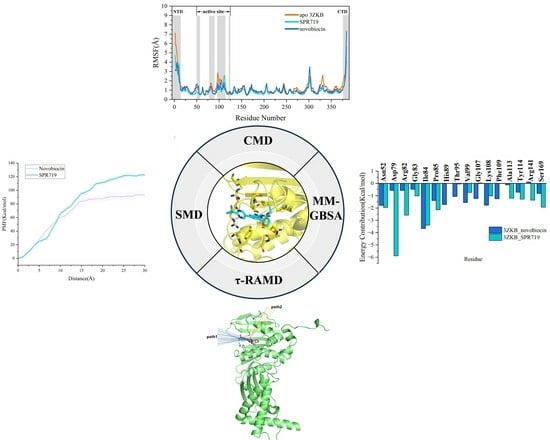
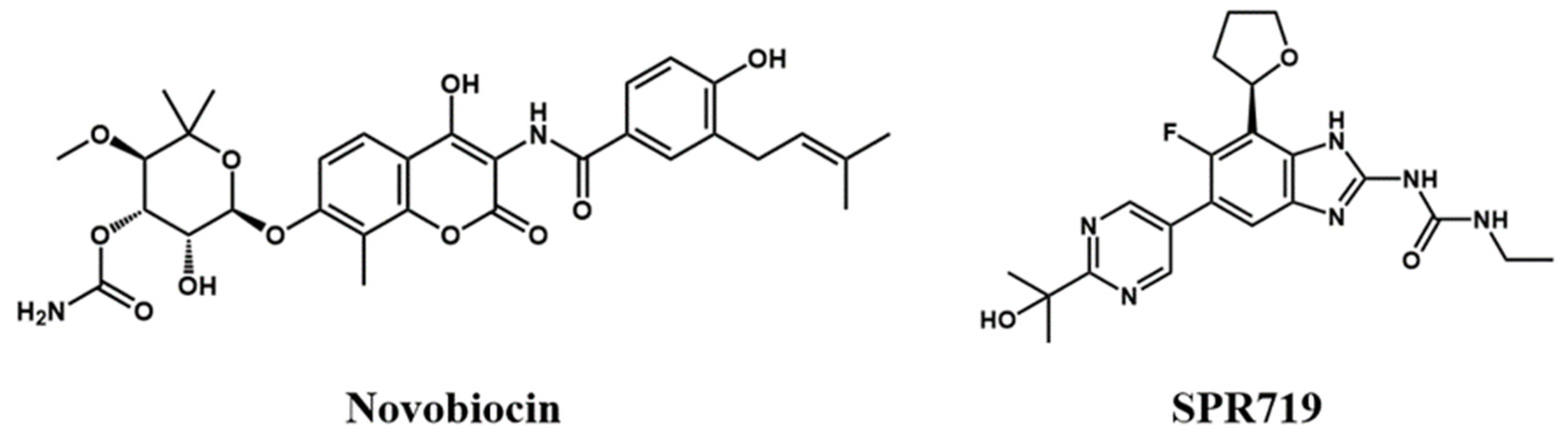
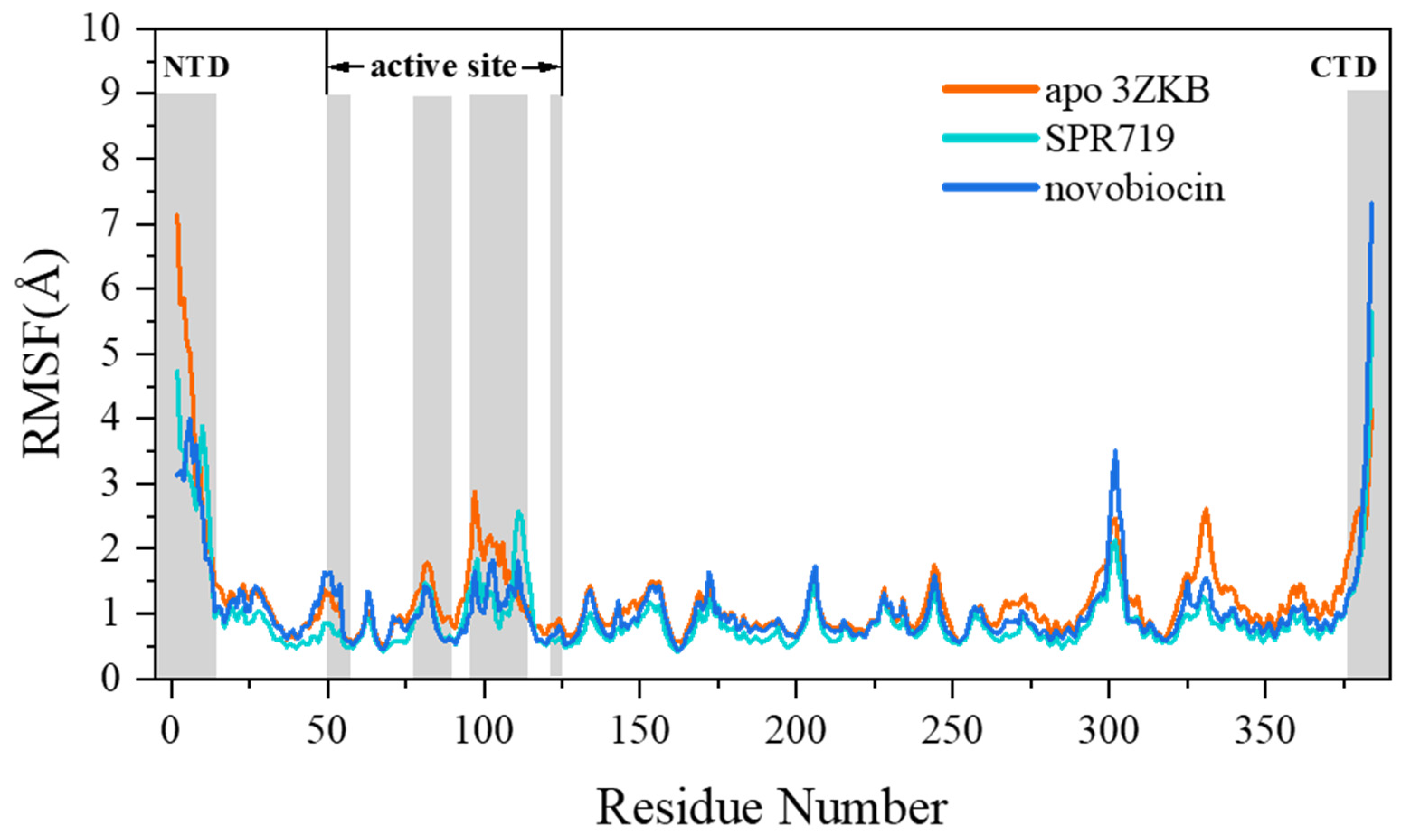
2.1. Electrostatic Interactions and van der Waals Interactions Are the Primary Driving Forces for the Binding of GyrB Inhibitors
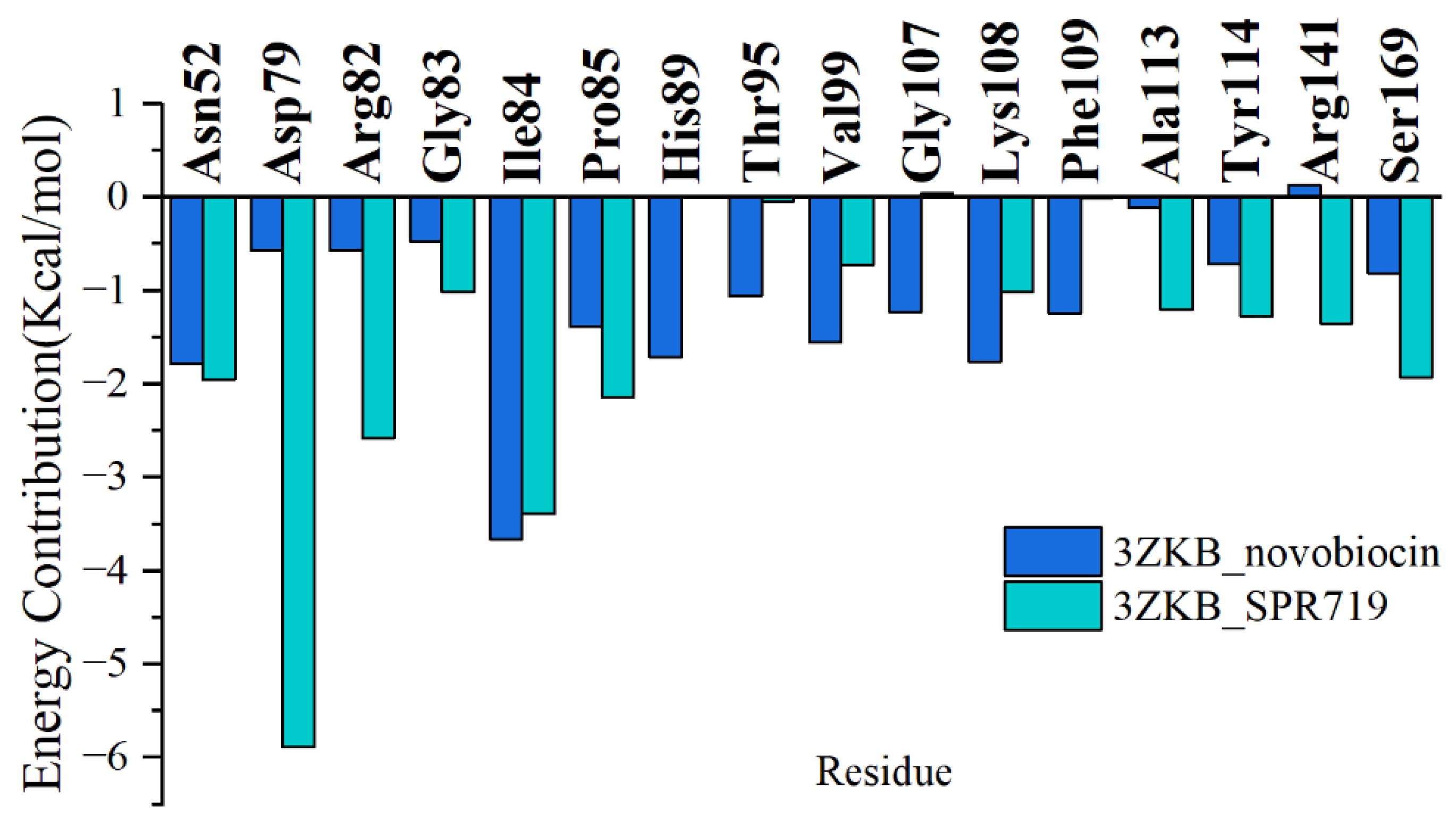
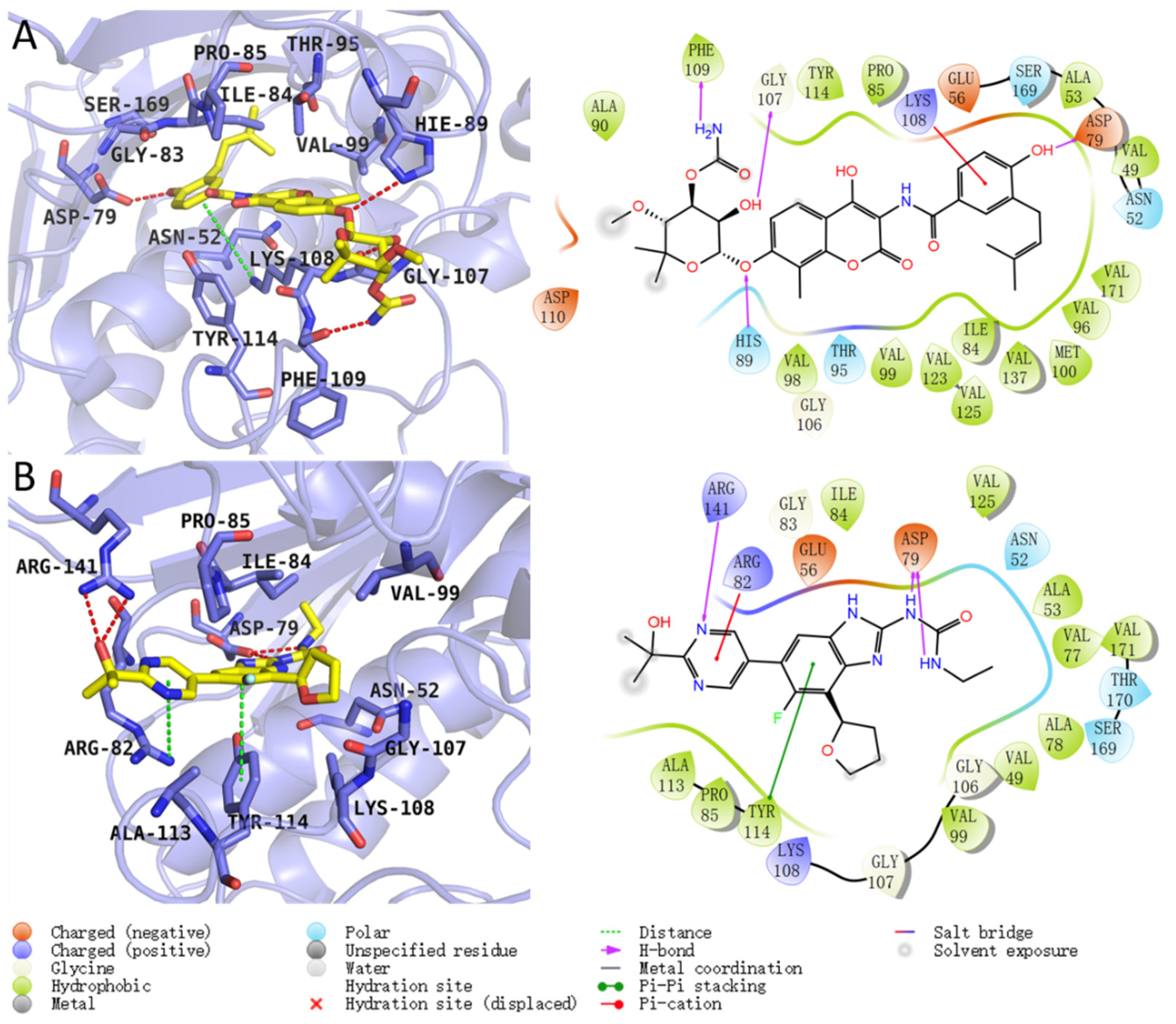
2.2. Residue Energy Decomposition Revealed the Key Residues for Inhibitor Binding to GyrB
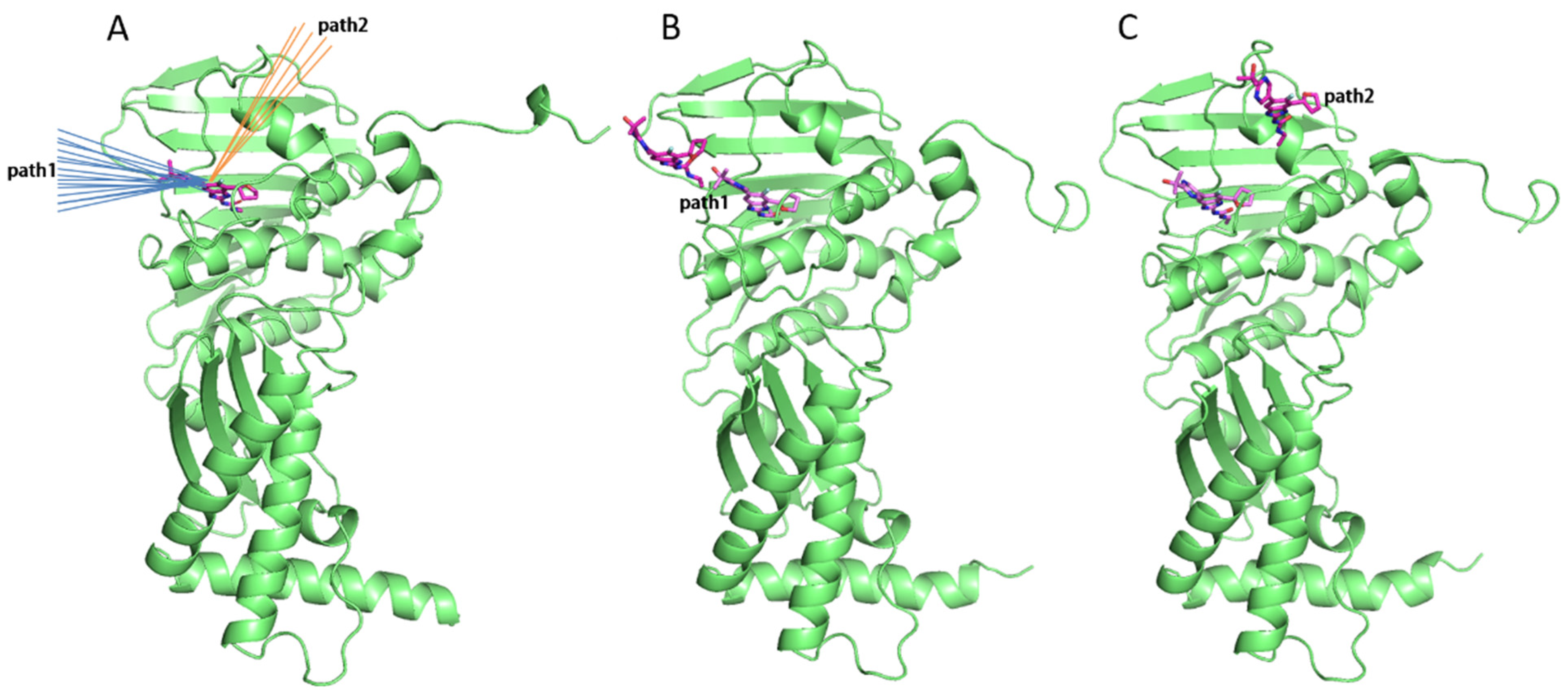
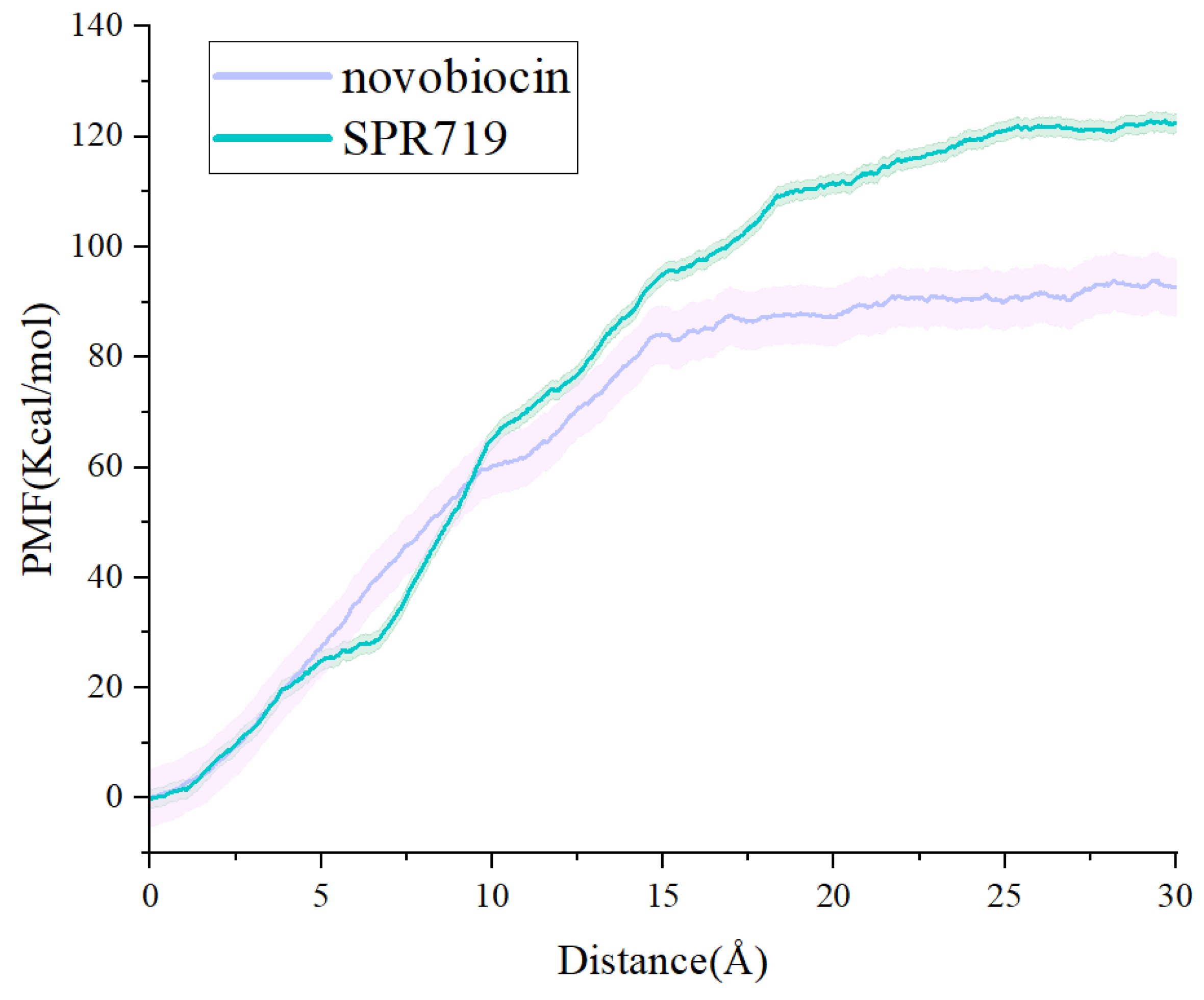
2.3. tau-RAMD Simulations Revealed That the Dissociation Pathways of Novobiocin and SPR719 Were Mainly from the ATP Channel of GyrB
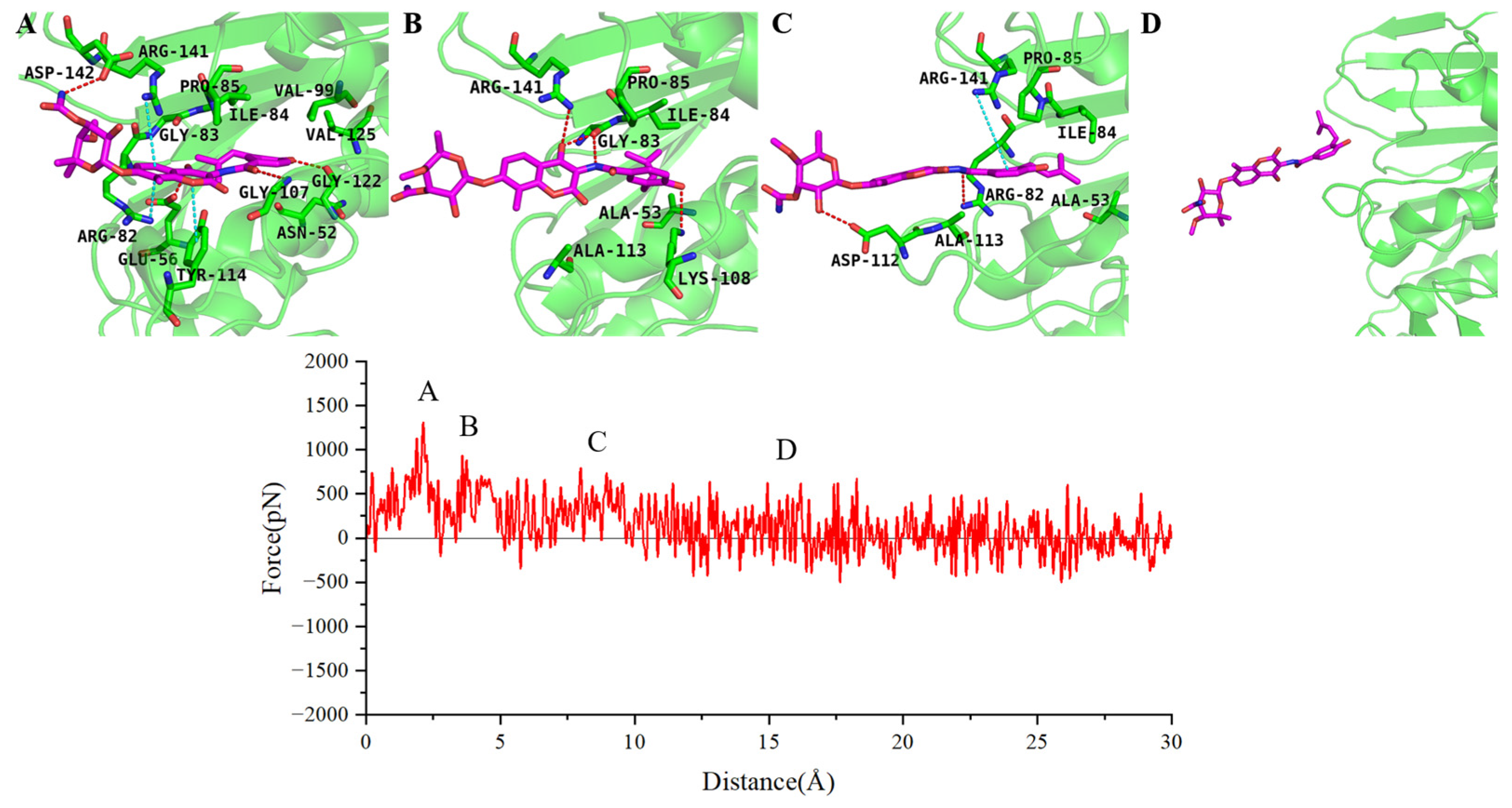
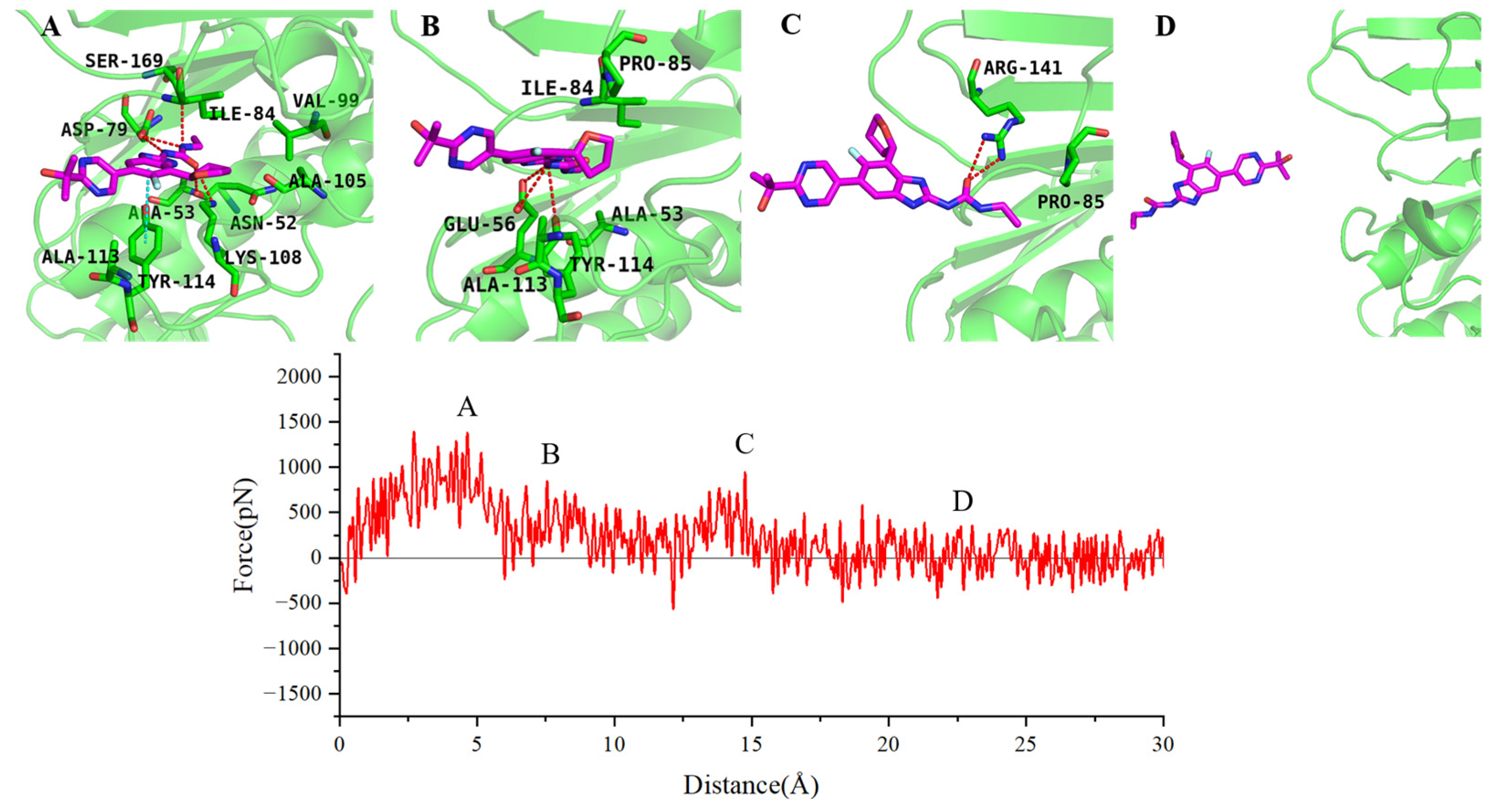
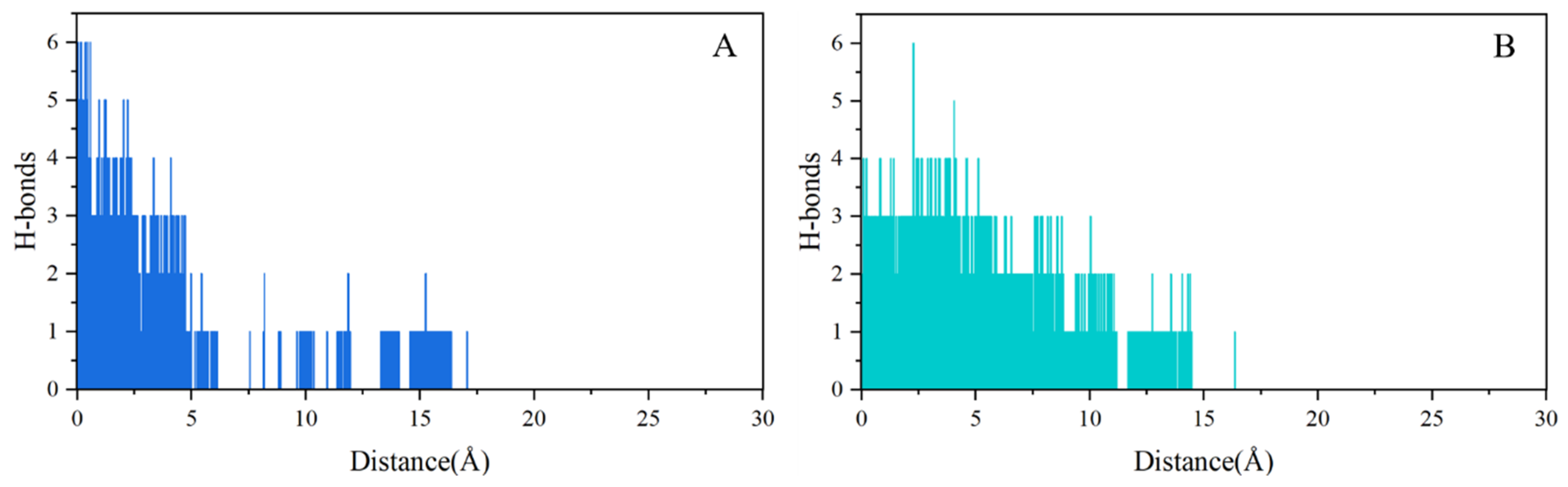
2.4. SMD Simulations Identified the Intermediate States in the Dissociation Process and the Key Amino Acids Involved
3. Discussion
4. Materials and Methods
4.1. Molecular Docking
4.2. System Preparation
4.3. Classical Molecular Dynamics Simulations
4.4. τ-Random Accelerated Molecular Dynamics (τ-RAMD) Simulation
4.5. Steered Molecular Dynamics Simulations
4.6. MM-GBSA (Molecular Mechanics-Generalized Born Surface Area) Calculations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- De Rycker, M.; Baragaña, B.; Duce, S.L.; Gilbert, I.H. Challenges and recent progress in drug discovery for tropical diseases. Nature 2018, 559, 498–506. [Google Scholar] [CrossRef] [PubMed]
- Simon, G.G. Impacts of neglected tropical disease on incidence and progression of HIV/AIDS, tuberculosis, and malaria: Scientific links. Int. J. Infect. Dis. 2016, 42, 54–57. [Google Scholar] [CrossRef]
- Bourzac, K. Infectious disease: Beating the big three. Nature 2014, 507, S4–S7. [Google Scholar] [CrossRef]
- World Health Organization. Global Tuberculosis Report 2023; World Health Organization: Geneva, Switzerland, 2023.
- Xu, G.; Liu, H.; Jia, X.; Wang, X.; Xu, P. Mechanisms and Detection Methods of Mycobacterium Tuberculosis Rifampicin Resistance: The Phenomenon of Drug Resistance Is Complex. Tuberculosis 2021, 128, 102083. [Google Scholar] [CrossRef] [PubMed]
- Pantel, A.; Petrella, S.; Veziris, N.; Brossier, F.; Bastian, S.; Jarlier, V.; Mayer, C.; Aubry, A. Extending the Definition of the GyrB Quinolone Resistance-Determining Region in Mycobacterium Tuberculosis DNA Gyrase for Assessing Fluoroquinolone Resistance in M. Tuberculosis. Antimicrob. Agents Chemother. 2012, 56, 1990–1996. [Google Scholar] [CrossRef]
- Hirano, K.; Takahashi, M.; Kazumi, Y.; Fukasawa, Y.; Abe, C. Mutation in pncA Is a Major Mechanism of Pyrazinamide Resistance in Mycobacterium Tuberculosis. Tuber. Lung Dis. 1998, 78, 117–122. [Google Scholar] [CrossRef]
- Stokes, S.S.; Vemula, R.; Pucci, M.J. Advancement of GyrB Inhibitors for Treatment of Infections Caused by Mycobacterium tuberculosis and Non-Tuberculous Mycobacteria. ACS Infect. Dis. 2020, 6, 1323–1331. [Google Scholar] [CrossRef]
- Kashyap, A.; Singh, P.K.; Silakari, O. Chemical Classes Targeting Energy Supplying GyrB Domain of Mycobacterium Tuberculosis. Tuberculosis 2018, 113, 43–54. [Google Scholar] [CrossRef]
- Barančoková, M.; Kikelj, D.; Ilaš, J. Recent Progress in the Discovery and Development of DNA Gyrase B Inhibitors. Future Med. Chem. 2018, 10, 1207–1227. [Google Scholar] [CrossRef]
- Tomioka, H.; Sato, K.; Shimizu, T.; Sano, C. Anti-Mycobacterium Tuberculosis Activities of New Fluoroquinolones in Combination with Other Antituberculous Drugs. J. Infect. 2002, 44, 160–165. [Google Scholar] [CrossRef] [PubMed]
- Hameed, H.M.A.; Tan, Y.; Islam, M.M.; Guo, L.; Chhotaray, C.; Wang, S.; Liu, Z.; Gao, Y.; Tan, S.; Yew, W.W.; et al. Phenotypic and Genotypic Characterization of Levofloxacin- and Moxifloxacin-Resistant Mycobacterium Tuberculosis Clinical Isolates in Southern China. J. Thorac. Dis. 2019, 11, 4613–4625. [Google Scholar] [CrossRef] [PubMed]
- Chopra, S.; Matsuyama, K.; Tran, T.; Malerich, J.P.; Wan, B.; Franzblau, S.G.; Lun, S.; Guo, H.; Maiga, M.C.; Bishai, W.R.; et al. Evaluation of Gyrase B as a Drug Target in Mycobacterium Tuberculosis. J. Antimicrob. Chemother. 2012, 67, 415–421. [Google Scholar] [CrossRef]
- Collin, F.; Karkare, S.; Maxwell, A. Exploiting Bacterial DNA Gyrase as a Drug Target: Current State and Perspectives. Appl. Microbiol. Biotechnol. 2011, 92, 479–497. [Google Scholar] [CrossRef] [PubMed]
- Sherer, B.A.; Hull, K.; Green, O.; Basarab, G.; Hauck, S.; Hill, P.; Loch, J.T.; Mullen, G.; Bist, S.; Bryant, J.; et al. Pyrrolamide DNA Gyrase Inhibitors: Optimization of Antibacterial Activity and Efficacy. Bioorganic Med. Chem. Lett. 2011, 21, 7416–7420. [Google Scholar] [CrossRef]
- Basarab, G.S.; Hill, P.J.; Garner, C.E.; Hull, K.; Green, O.; Sherer, B.A.; Dangel, P.B.; Manchester, J.I.; Bist, S.; Hauck, S.; et al. Optimization of Pyrrolamide Topoisomerase II Inhibitors Toward Identification of an Antibacterial Clinical Candidate (AZD5099). J. Med. Chem. 2014, 57, 6060–6082. [Google Scholar] [CrossRef]
- P, S.H.; Solapure, S.; Mukherjee, K.; Nandi, V.; Waterson, D.; Shandil, R.; Balganesh, M.; Sambandamurthy, V.K.; Raichurkar, A.K.; Deshpande, A.; et al. Optimization of Pyrrolamides as Mycobacterial GyrB ATPase Inhibitors: Structure-Activity Relationship and In Vivo Efficacy in a Mouse Model of Tuberculosis. Antimicrob. Agents Chemother. 2014, 58, 61–70. [Google Scholar] [CrossRef]
- Mani, N.; Gross, C.H.; Parsons, J.D.; Hanzelka, B.; Müh, U.; Mullin, S.; Liao, Y.; Grillot, A.-L.; Stamos, D.; Charifson, P.S.; et al. In Vitro Characterization of the Antibacterial Spectrum of Novel Bacterial Type II Topoisomerase Inhibitors of the Aminobenzimidazole Class. Antimicrob. Agents Chemother. 2006, 50, 1228–1237. [Google Scholar] [CrossRef] [PubMed]
- Locher, C.P.; Jones, S.M.; Hanzelka, B.L.; Perola, E.; Shoen, C.M.; Cynamon, M.H.; Ngwane, A.H.; Wiid, I.J.; Van Helden, P.D.; Betoudji, F.; et al. A Novel Inhibitor of Gyrase B Is a Potent Drug Candidate for Treatment of Tuberculosis and Nontuberculosis Mycobacterial Infections. Antimicrob. Agents Chemother. 2015, 59, 1455–1465. [Google Scholar] [CrossRef] [PubMed]
- Kale, M.G.; Raichurkar, A.; P, S.H.; Waterson, D.; McKinney, D.; Manjunatha, M.R.; Kranthi, U.; Koushik, K.; Jena, L.K.; Shinde, V.; et al. Thiazolopyridine Ureas as Novel Antitubercular Agents Acting through Inhibition of DNA Gyrase B. J. Med. Chem. 2013, 56, 8834–8848. [Google Scholar] [CrossRef]
- Kale, R.R.; Kale, M.G.; Waterson, D.; Raichurkar, A.; Hameed, S.P.; Manjunatha, M.R.; Kishore Reddy, B.K.; Malolanarasimhan, K.; Shinde, V.; Koushik, K.; et al. Thiazolopyridone Ureas as DNA Gyrase B Inhibitors: Optimization of Antitubercular Activity and Efficacy. Bioorganic Med. Chem. Lett. 2014, 24, 870–879. [Google Scholar] [CrossRef]
- Manchester, J.I.; Dussault, D.D.; Rose, J.A.; Boriack-Sjodin, P.A.; Uria-Nickelsen, M.; Ioannidis, G.; Bist, S.; Fleming, P.; Hull, K.G. Discovery of a Novel Azaindole Class of Antibacterial Agents Targeting the ATPase Domains of DNA Gyrase and Topoisomerase IV. Bioorganic Med. Chem. Lett. 2012, 22, 5150–5156. [Google Scholar] [CrossRef]
- Jeankumar, V.U.; Renuka, J.; Santosh, P.; Soni, V.; Sridevi, J.P.; Suryadevara, P.; Yogeeswari, P.; Sriram, D. Thiazole–Aminopiperidine Hybrid Analogues: Design and Synthesis of Novel Mycobacterium Tuberculosis GyrB Inhibitors. Eur. J. Med. Chem. 2013, 70, 143–153. [Google Scholar] [CrossRef]
- Medapi, B.; Suryadevara, P.; Renuka, J.; Sridevi, J.P.; Yogeeswari, P.; Sriram, D. 4-Aminoquinoline Derivatives as Novel Mycobacterium Tuberculosis GyrB Inhibitors: Structural Optimization, Synthesis and Biological Evaluation. Eur. J. Med. Chem. 2015, 103, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Raad, I.I.; Hachem, R.Y.; Abi-Said, D.; Rolston, K.V.I.; Whimbey, E.; Buzaid, A.C.; Legha, S. A Prospective Crossover Randomized Trial of Novobiocin and Rifampin Prophylaxis for the Prevention of Intravascular Catheter Infections in Cancer Patients Treated with Interleukin-2. Cancer 1998, 82, 403–411. [Google Scholar] [CrossRef]
- Gjorgjieva, M.; Tomašič, T.; Barančokova, M.; Katsamakas, S.; Ilaš, J.; Tammela, P.; Peterlin Mašič, L.; Kikelj, D. Discovery of Benzothiazole Scaffold-Based DNA Gyrase B Inhibitors. J. Med. Chem. 2016, 59, 8941–8954. [Google Scholar] [CrossRef] [PubMed]
- Aragaw, W.W.; Cotroneo, N.; Stokes, S.; Pucci, M.; Critchley, I.; Gengenbacher, M.; Dick, T. In Vitro Resistance against DNA Gyrase Inhibitor SPR719 in Mycobacterium Avium and Mycobacterium Abscessus. Microbiol. Spectr. 2022, 10, e01321-21. [Google Scholar] [CrossRef]
- Talley, A.K.; Thurston, A.; Moore, G.; Gupta, V.K.; Satterfield, M.; Manyak, E.; Stokes, S.; Dane, A.; Melnick, D. First-in-Human Evaluation of the Safety, Tolerability, and Pharmacokinetics of SPR720, a Novel Oral Bacterial DNA Gyrase (GyrB) Inhibitor for Mycobacterial Infections. Antimicrob. Agents Chemother. 2021, 65, e01208-21. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, A.; Roué, M.; Spitzfaden, C.; Petrella, S.; Aubry, A.; Hann, M.; Bax, B.; Mayer, C. Mycobacterium tuberculosis DNA Gyrase ATPase Domain Structures Suggest a Dissociative Mechanism That Explains How ATP Hydrolysis Is Coupled to Domain Motion. Biochem. J. 2013, 456, 263–273. [Google Scholar] [CrossRef]
- Santos Nascimento, J.d.I.; Aquino, M.d.T.; Silva-Júnior, F.d.E. Repurposing FDA-approved Drugs Targeting SARS-CoV2 3CLpro: A Study by Applying Virtual Screening, Molecular Dynamics, MM-PBSA Calculations and Covalent Docking. Lett. Drug Des. Discov. 2022, 19, 637–653. [Google Scholar] [CrossRef]
- Zhang, Q.; Han, J.; Zhu, Y.; Tan, S.; Liu, H. Binding Thermodynamics and Dissociation Kinetics Analysis Uncover the Key Structural Motifs of Phenoxyphenol Derivatives as the Direct InhA Inhibitors and the Hotspot Residues of InhA. Int. J. Mol. Sci. 2022, 23, 10102. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.; Zhang, Q.; Wang, J.; Gao, P.; Xie, G.; Liu, H.; Yao, X. Molecular Modeling Study on the Interaction Mechanism between the LRRK2 G2019S Mutant and Type I Inhibitors by Integrating Molecular Dynamics Simulation, Binding Free Energy Calculations, and Pharmacophore Modeling. ACS Chem. Neurosci. 2022, 13, 599–612. [Google Scholar] [CrossRef] [PubMed]
- Grillot, A.-L.; Tiran, A.L.; Shannon, D.; Krueger, E.; Liao, Y.; O’Dowd, H.; Tang, Q.; Ronkin, S.; Wang, T.; Waal, N.; et al. Second-Generation Antibacterial Benzimidazole Ureas: Discovery of a Preclinical Candidate with Reduced Metabolic Liability. J. Med. Chem. 2014, 57, 8792–8816. [Google Scholar] [CrossRef] [PubMed]
- Tambe, P.M.; Bhowmick, S.; Chaudhary, S.K.; Khan, M.R.; Wabaidur, S.M.; Muddassir, M.; Patil, P.C.; Islam, M.A. Structure-Based Screening of DNA GyraseB Inhibitors for Therapeutic Applications in Tuberculosis: A Pharmacoinformatics Study. Appl. Biochem. Biotechnol. 2020, 192, 1107–1123. [Google Scholar] [CrossRef]
- Kamsri, P.; Punkvang, A.; Hannongbua, S.; Suttisintong, K.; Kittakoop, P.; Spencer, J.; Mulholland, A.J.; Pungpo, P. In silico study directed towards identification of the key structural features of GyrB inhibitors targeting MTB DNA gyrase: HQSAR, CoMSIA and molecular dynamics simulations. SAR QSAR Environ. Res. 2019, 30, 775–800. [Google Scholar] [CrossRef]
- Gl, B.; Rajput, R.; Gupta, M.; Dahiya, P.; Thakur, J.K.; Bhatnagar, R.; Grover, A. Structure-based drug repurposing to inhibit the DNA gyrase of Mycobacterium tuberculosis. Biochem. J. 2020, 477, 4167–4190. [Google Scholar] [CrossRef]
- Arévalo, J.M.C.; Amorim, J.C. Virtual screening, optimization and molecular dynamics analyses highlighting a pyrrolo[1,2-a]quinazoline derivative as a potential inhibitor of DNA gyrase B of Mycobacterium tuberculosis. Sci. Rep. 2022, 12, 4742. [Google Scholar] [CrossRef]
- Bayly, C.I.; Cieplak, P.; Cornell, W.; Kollman, P.A. A Well-Behaved Electrostatic Potential Based Method Using Charge Restraints for Deriving Atomic Charges: The RESP Model. J. Phys. Chem. 1993, 97, 10269–10280. [Google Scholar] [CrossRef]
- Kristyán, S.; Ruzsinszky, A.; Csonka, G.I. Accurate Thermochemistry from Corrected Hartree-Fock Results: Rapid Estimation of Nearly Experimental Quality Total Energy Using the Small 6-31G(d) Basis Set. Theor. Chem. Acc. 2001, 106, 319–328. [Google Scholar] [CrossRef]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and Testing of a General Amber Force Field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Liu, S.; Lee, T.-S.; Ji, B.; Man, V.H.; York, D.M.; Wang, J. Fast, Accurate, and Reliable Protocols for Routine Calculations of Protein–Ligand Binding Affinities in Drug Design Projects Using AMBER GPU-TI with ff14SB/GAFF. ACS Omega 2020, 5, 4611–4619. [Google Scholar] [CrossRef]
- Götz, A.W.; Williamson, M.J.; Xu, D.; Poole, D.; Le Grand, S.; Walker, R.C. Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 1. Generalized Born. J. Chem. Theory Comput. 2012, 8, 1542–1555. [Google Scholar] [CrossRef]
- Andersen, H.C. Molecular Dynamics Simulations at Constant Pressure and/or Temperature. J. Chem. Phys. 1980, 72, 2384–2393. [Google Scholar] [CrossRef]
- Martyna, G.J.; Hughes, A.; Tuckerman, M.E. Molecular Dynamics Algorithms for Path Integrals at Constant Pressure. J. Chem. Phys. 1999, 110, 3275–3290. [Google Scholar] [CrossRef]
- York, D.M.; Darden, T.A.; Pedersen, L.G. The Effect of Long-Range Electrostatic Interactions in Simulations of Macromolecular Crystals: A Comparison of the Ewald and Truncated List Methods. J. Chem. Phys. 1993, 99, 8345–8348. [Google Scholar] [CrossRef]
- Ryckaert, J.-P.; Ciccotti, G.; Berendsen, H.J.C. Numerical Integration of the Cartesian Equations of Motion of a System with Constraints: Molecular Dynamics of n-Alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef]
- Muvva, C.; Murugan, N.A.; Kumar Choutipalli, V.S.; Subramanian, V. Unraveling the Unbinding Pathways of Products Formed in Catalytic Reactions Involved in SIRT1–3: A Random Acceleration Molecular Dynamics Simulation Study. J. Chem. Inf. Model. 2019, 59, 4100–4115. [Google Scholar] [CrossRef] [PubMed]
- Kokh, D.B.; Wade, R.C. G Protein-Coupled Receptor–Ligand Dissociation Rates and Mechanisms from τRAMD Simulations. J. Chem. Theory Comput. 2021, 17, 6610–6623. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Zhao, N.; Meng, X.; Yu, F.; Yao, X.; Liu, H. The prediction of protein-ligand unbinding for modern drug discovery. Expert Opin. Drug Discov. 2022, 17, 191–205. [Google Scholar] [CrossRef]
- Kokh, D.B.; Amaral, M.; Bomke, J.; Grädler, U.; Musil, D.; Buchstaller, H.-P.; Dreyer, M.K.; Frech, M.; Lowinski, M.; Vallee, F.; et al. Estimation of Drug-Target Residence Times by τ-Random Acceleration Molecular Dynamics Simulations. J. Chem. Theory Comput. 2018, 14, 3859–3869. [Google Scholar] [CrossRef]
- Phillips, J.C.; Hardy, D.J.; Maia, J.D.C.; Stone, J.E.; Ribeiro, J.V.; Bernardi, R.C.; Buch, R.; Fiorin, G.; Hénin, J.; Jiang, W.; et al. Scalable Molecular Dynamics on CPU and GPU Architectures with NAMD. J. Chem. Phys. 2020, 153, 044130. [Google Scholar] [CrossRef] [PubMed]
- Izrailev, S.; Stepaniants, S.; Isralewitz, B.; Kosztin, D.; Lu, H.; Molnar, F.; Wriggers, W.; Schulten, K. Steered Molecular Dynamics. In Computational Molecular Dynamics: Challenges, Methods, Ideas; Deuflhard, P., Hermans, J., Leimkuhler, B., Mark, A.E., Reich, S., Skeel, R.D., Eds.; Lecture Notes in Computational Science and Engineering; Springer: Berlin/Heidelberg, Germany, 1999; Volume 4, pp. 39–65. [Google Scholar]
- Shen, L.; Shen, J.; Luo, X.; Cheng, F.; Xu, Y.; Chen, K.; Arnold, E.; Ding, J.; Jiang, H. Steered Molecular Dynamics Simulation on the Binding of NNRTI to HIV-1 RT. Biophys. J. 2003, 84, 3547–3563. [Google Scholar] [CrossRef]
- Liu, X.; Xu, Y.; Wang, X.; Barrantes, F.J.; Jiang, H. Unbinding of Nicotine from the Acetylcholine Binding Protein: Steered Molecular Dynamics Simulations. J. Phys. Chem. B 2008, 112, 4087–4093. [Google Scholar] [CrossRef]
- Patel, J.S.; Berteotti, A.; Ronsisvalle, S.; Rocchia, W.; Cavalli, A. Steered Molecular Dynamics Simulations for Studying Protein–Ligand Interaction in Cyclin-Dependent Kinase 5. J. Chem. Inf. Model. 2014, 54, 470–480. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.; Uzelac, I.; Gottfries, J.; Eriksson, L.A. Exploration of Multiple Sortase A Protein Conformations in Virtual Screening. Sci. Rep. 2016, 6, 20413. [Google Scholar] [CrossRef]
- Jin, H.; Zhu, J.; Dong, Y.; Han, W. Exploring the Different Ligand Escape Pathways in Acylaminoacyl Peptidase by Random Acceleration and Steered Molecular Dynamics Simulations. RSC Adv. 2016, 6, 10987–10996. [Google Scholar] [CrossRef]
- Wang, E.; Sun, H.; Wang, J.; Wang, Z.; Liu, H.; Zhang, J.Z.H.; Hou, T. End-Point Binding Free Energy Calculation with MM/PBSA and MM/GBSA: Strategies and Applications in Drug Design. Chem. Rev. 2019, 119, 9478–9508. [Google Scholar] [CrossRef]
- Onufriev, A.; Bashford, D.; Case, D.A. Modification of the Generalized Born Model Suitable for Macromolecules. J. Phys. Chem. B 2000, 104, 3712–3720. [Google Scholar] [CrossRef]
- Forouzesh, N.; Mishra, N. An Effective MM/GBSA Protocol for Absolute Binding Free Energy Calculations: A Case Study on SARS-CoV-2 Spike Protein and the Human ACE2 Receptor. Molecules 2021, 26, 2383. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Sun, H.; Wang, J.; Zhu, F.; Liu, H.; Wang, Z.; Lei, T.; Li, Y.; Hou, T. Assessing the Performance of MM/PBSA and MM/GBSA Methods. 8. Predicting Binding Free Energies and Poses of Protein–RNA Complexes. RNA 2018, 24, 1183–1194. [Google Scholar] [CrossRef]
- Chen, J.; Wang, J.; Zhang, Q.; Chen, K.; Zhu, W. Probing Origin of Binding Difference of Inhibitors to MDM2 and MDMX by Polarizable Molecular Dynamics Simulation and QM/MM-GBSA Calculation. Sci. Rep. 2015, 5, 17421. [Google Scholar] [CrossRef] [PubMed]
- Ren, X.; Zeng, R.; Tortorella, M.; Wang, J.; Wang, C. Structural Insight into Inhibition of CsrA-RNA Interaction Revealed by Docking, Molecular Dynamics and Free Energy Calculations. Sci. Rep. 2017, 7, 14934. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, N.; Aparoy, P. Application of Per-Residue Energy Decomposition to Identify the Set of Amino Acids Critical for in Silico Prediction of COX-2 Inhibitory Activity. Heliyon 2020, 6, e04944. [Google Scholar] [CrossRef] [PubMed]
- Tian, M.; Li, H.; Yan, X.; Gu, J.; Zheng, P.; Luo, S.; Zhangsun, D.; Chen, Q.; Ouyang, Q. Application of Per-Residue Energy Decomposition to Design Peptide Inhibitors of PSD95 GK Domain. Front. Mol. Biosci. 2022, 9, 848353. [Google Scholar] [CrossRef]
- Lu, B.; Zhang, D.; McCammon, J.A. Computation of Electrostatic Forces between Solvated Molecules Determined by the Poisson–Boltzmann Equation Using a Boundary Element Method. J. Chem. Phys. 2005, 122, 214102. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Energy | 3ZKB-Novobiovin | 3ZKB-SPR719 |
|---|---|---|
| ΔEele | −48.72 | −62.04 |
| ΔEvdw | −61.13 | −59.87 |
| ΔEMM a | −109.85 | −121.91 |
| ΔGSA | −8.28 | −6.53 |
| ΔGGB | 72.26 | 68.51 |
| ΔGsol b | 63.98 | 61.98 |
| ΔGpolar c | 23.54 | 6.47 |
| ΔGnonpolar d | −69.41 | −66.4 |
| ΔHbind | −52.5 | −59.95 |
| −TΔS | 28.16 | 33.14 |
| ΔGbind | −24.34 | −26.81 |
| Ki (nM) | 13 | 9 |
| ΔGexp e | −10.82 | −11.04 |
| Complex | Donor | Acceptor | Distance (Å) | Angel (°) | Occupancy (%) |
|---|---|---|---|---|---|
| 3ZKB_novobiocin | ligand@O11-H30 | ASP_79@OD1 | 2.5876 | 168.1364 | 99.91 |
| ligand@O5-H11 | GLY_107@O1479 | 2.6696 | 165.8257 | 99.22 | |
| ligand@N1-H17 | PHE_109@O1521 | 2.8626 | 161.4976 | 63.76 | |
| HIE_89@NE2-HE2 | ligand@O5 | 2.8912 | 154.5943 | 25.39 | |
| ASN_52@ND2-HD2 | ligand@O9 | 2.8714 | 160.222 | 13.12 | |
| GLY_107@NH | ligand@O6 | 2.8935 | 143.3523 | 11.15 | |
| 3ZKB_SPR719 | ligand@N6-H20 | ASP_79@OD1 | 2.782 | 158.5813 | 76.02 |
| ligand@N3-H12 | ASP_79@OD1 | 2.7976 | 159.6323 | 74.85 | |
| ARG_141@NH1-HH12 | ligand@N4 | 2.8998 | 148.7218 | 35.25 | |
| ARG_141@NH2-HH22 | ligand@O2 | 2.8837 | 162.1836 | 27.17 | |
| ligand@N6-H20 | ASP_79@OD2 | 2.7771 | 158.7187 | 20.58 | |
| ligand@N3-H12 | ASP_79@OD2 | 2.7982 | 159.7471 | 20.52 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qiu, X.; Zhang, Q.; Li, Z.; Zhang, J.; Liu, H. Revealing the Interaction Mechanism between Mycobacterium tuberculosis GyrB and Novobiocin, SPR719 through Binding Thermodynamics and Dissociation Kinetics Analysis. Int. J. Mol. Sci. 2024, 25, 3764. https://doi.org/10.3390/ijms25073764
Qiu X, Zhang Q, Li Z, Zhang J, Liu H. Revealing the Interaction Mechanism between Mycobacterium tuberculosis GyrB and Novobiocin, SPR719 through Binding Thermodynamics and Dissociation Kinetics Analysis. International Journal of Molecular Sciences. 2024; 25(7):3764. https://doi.org/10.3390/ijms25073764
Chicago/Turabian StyleQiu, Xiaofei, Qianqian Zhang, Zhaoguo Li, Juan Zhang, and Huanxiang Liu. 2024. "Revealing the Interaction Mechanism between Mycobacterium tuberculosis GyrB and Novobiocin, SPR719 through Binding Thermodynamics and Dissociation Kinetics Analysis" International Journal of Molecular Sciences 25, no. 7: 3764. https://doi.org/10.3390/ijms25073764
APA StyleQiu, X., Zhang, Q., Li, Z., Zhang, J., & Liu, H. (2024). Revealing the Interaction Mechanism between Mycobacterium tuberculosis GyrB and Novobiocin, SPR719 through Binding Thermodynamics and Dissociation Kinetics Analysis. International Journal of Molecular Sciences, 25(7), 3764. https://doi.org/10.3390/ijms25073764