
Approach to the “Missing” Diarylsilylene: Formation, Characterization, and Intramolecular C–H Bond Activation of Blue Diarylsilylenes with Bulky Rind Groups †

 ,
,
Abstract
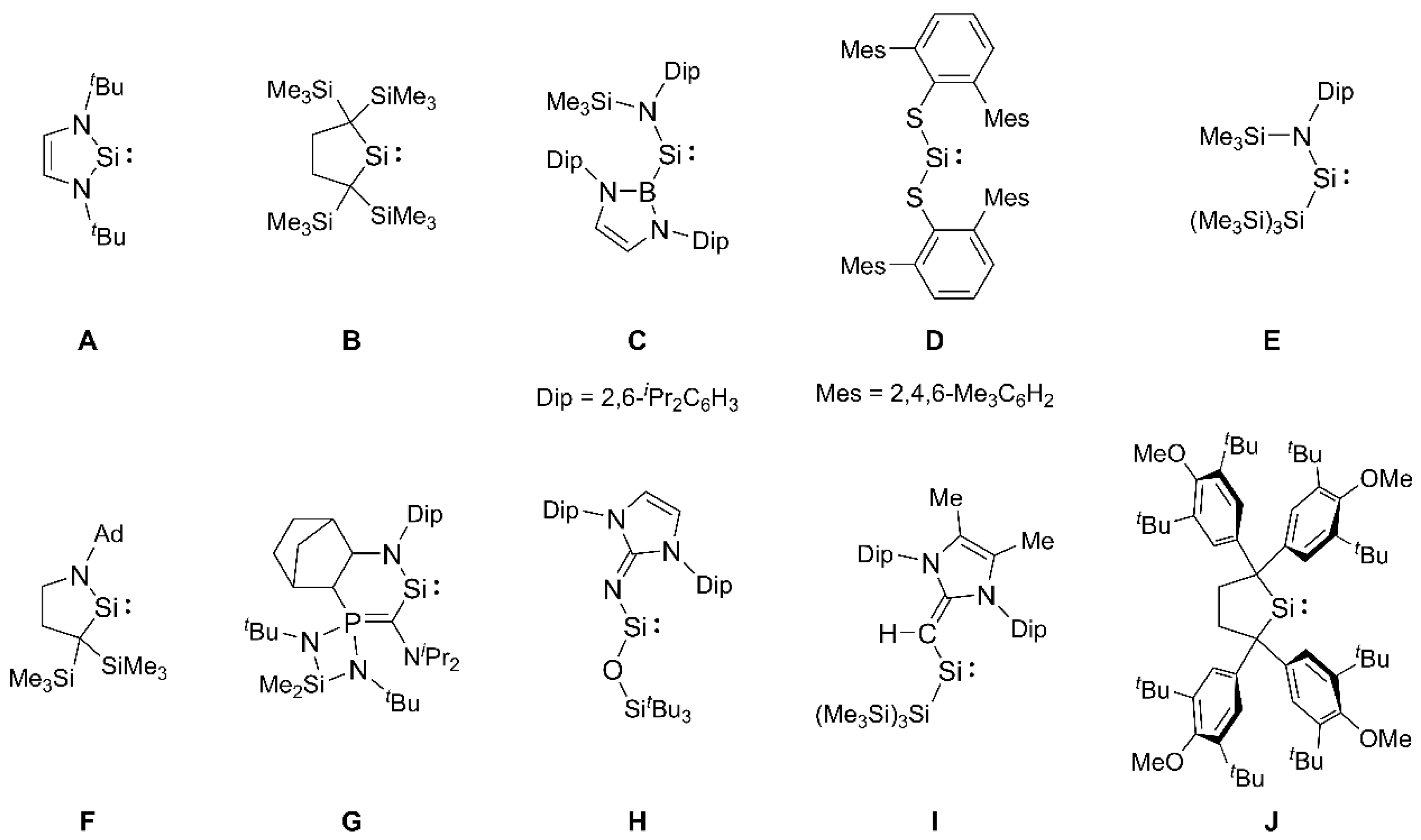
1. Introduction
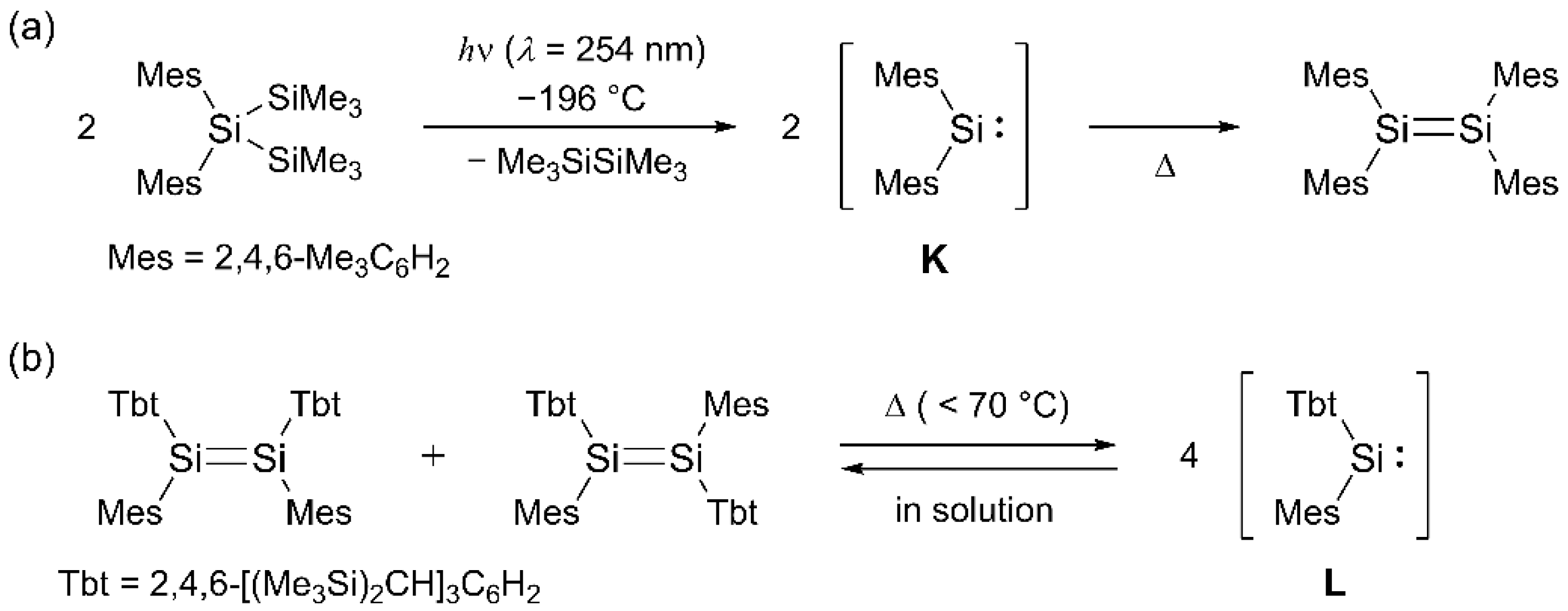
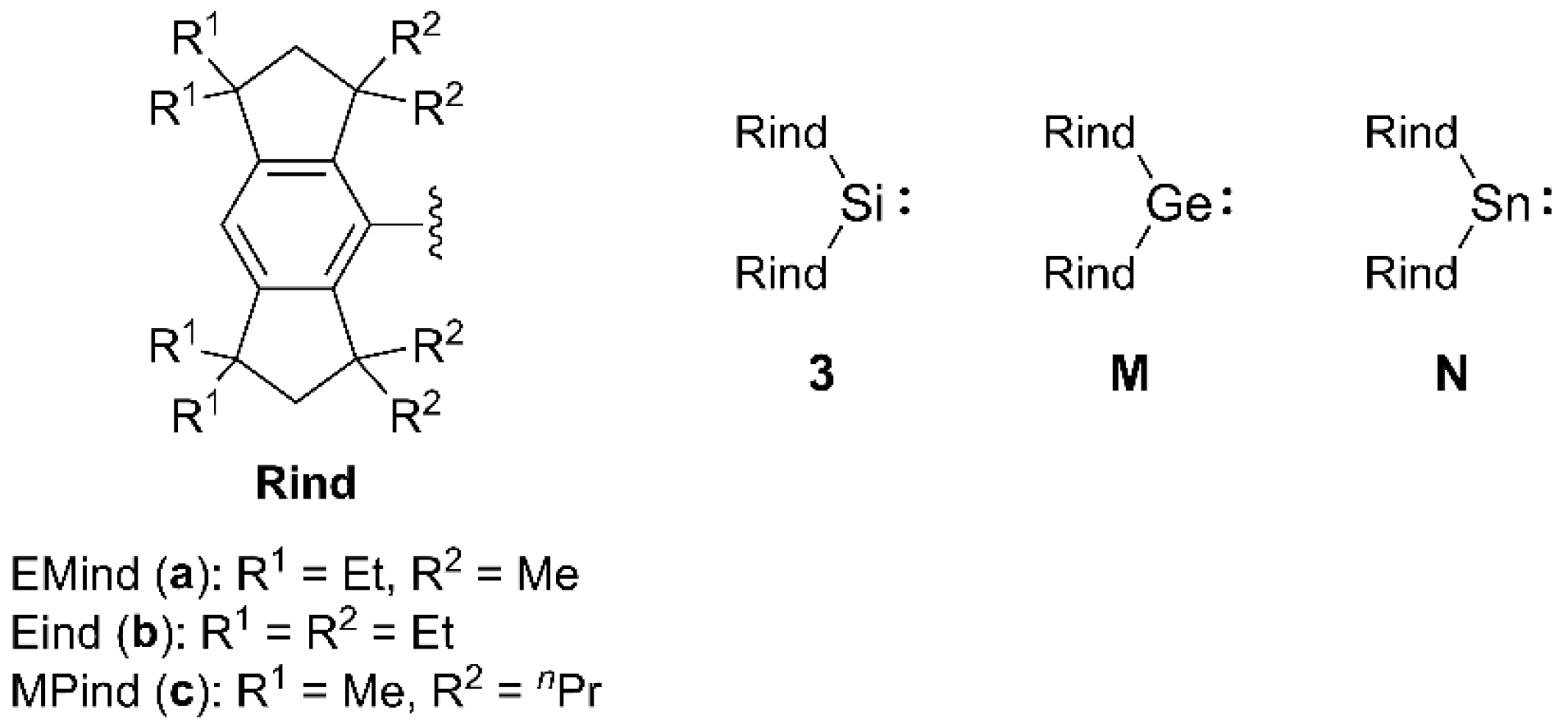
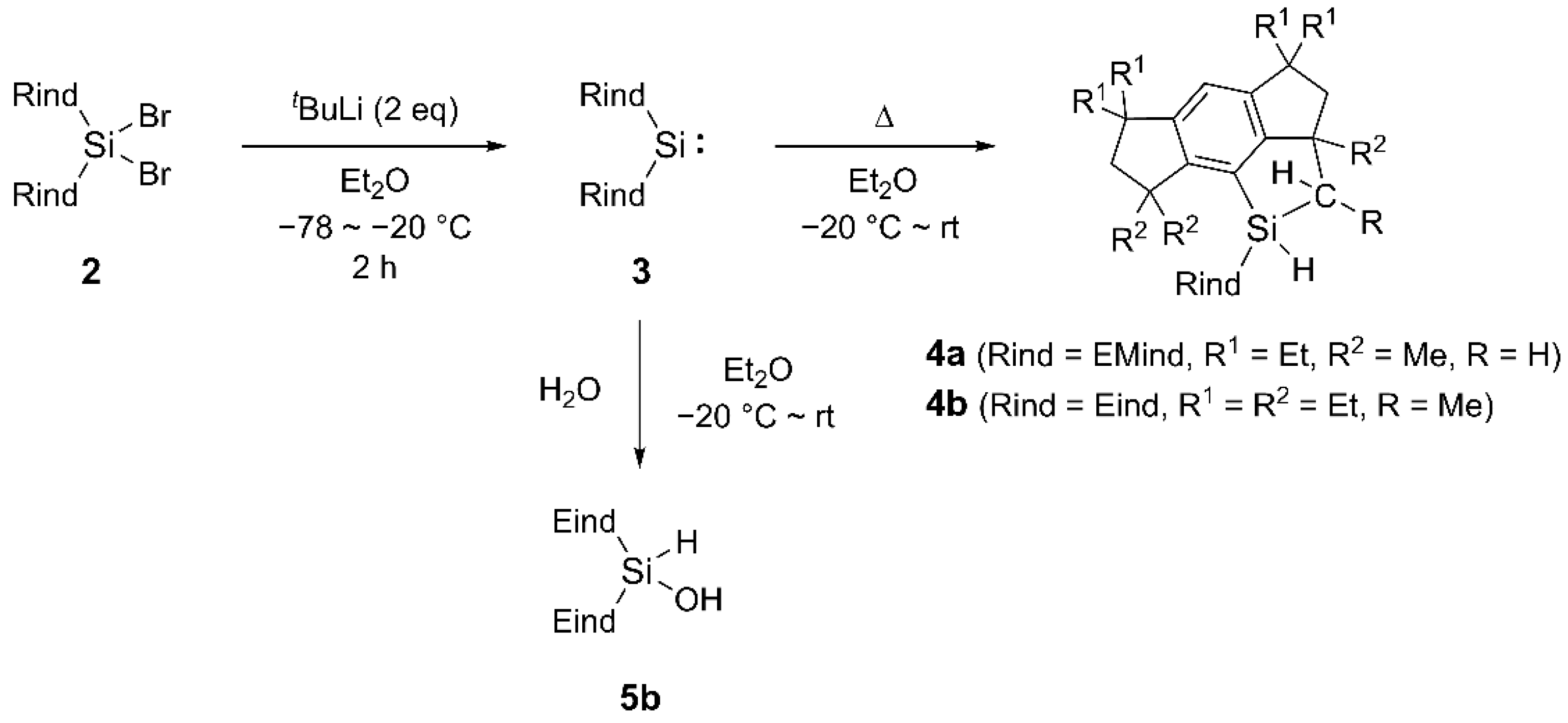
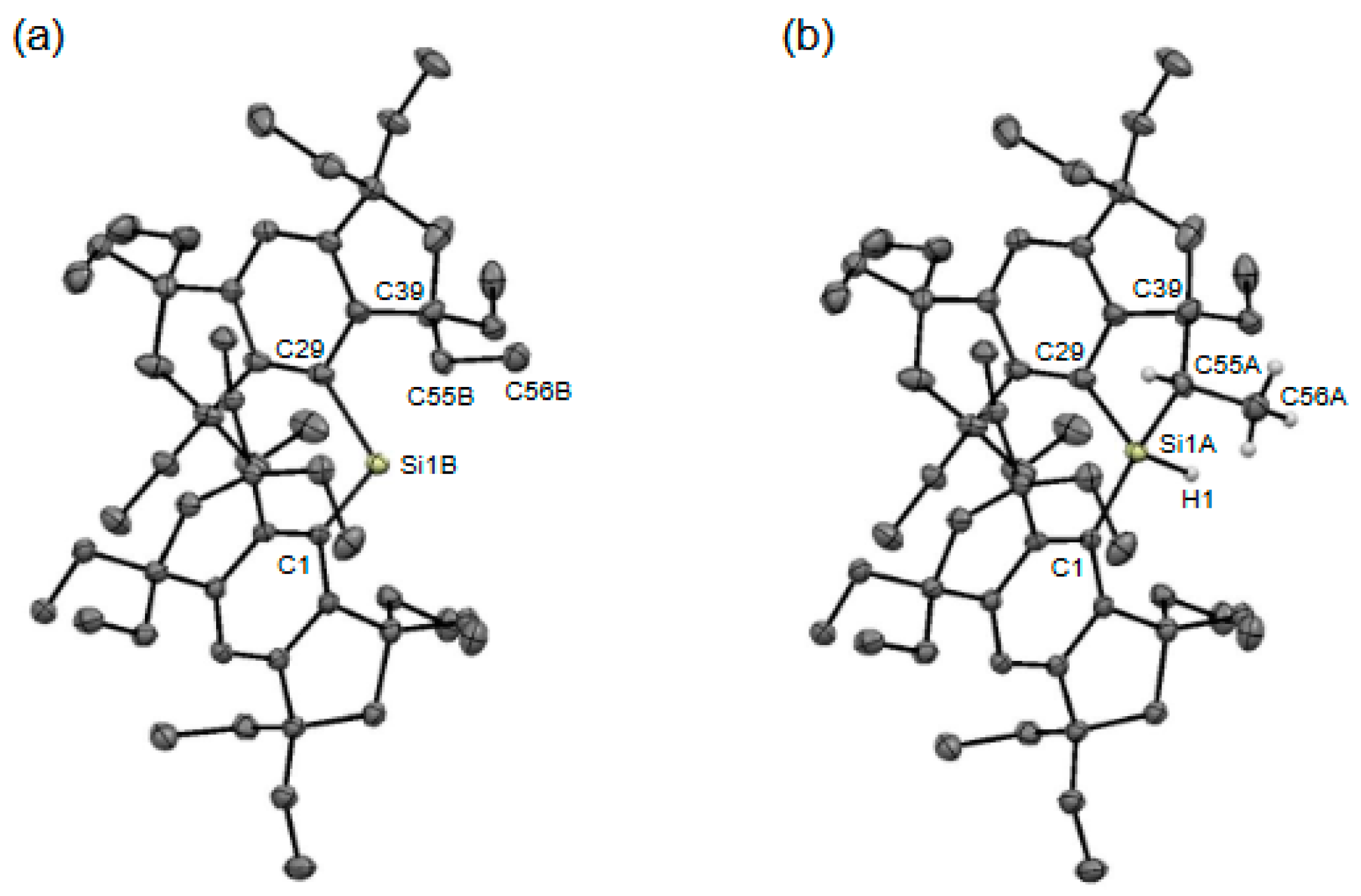
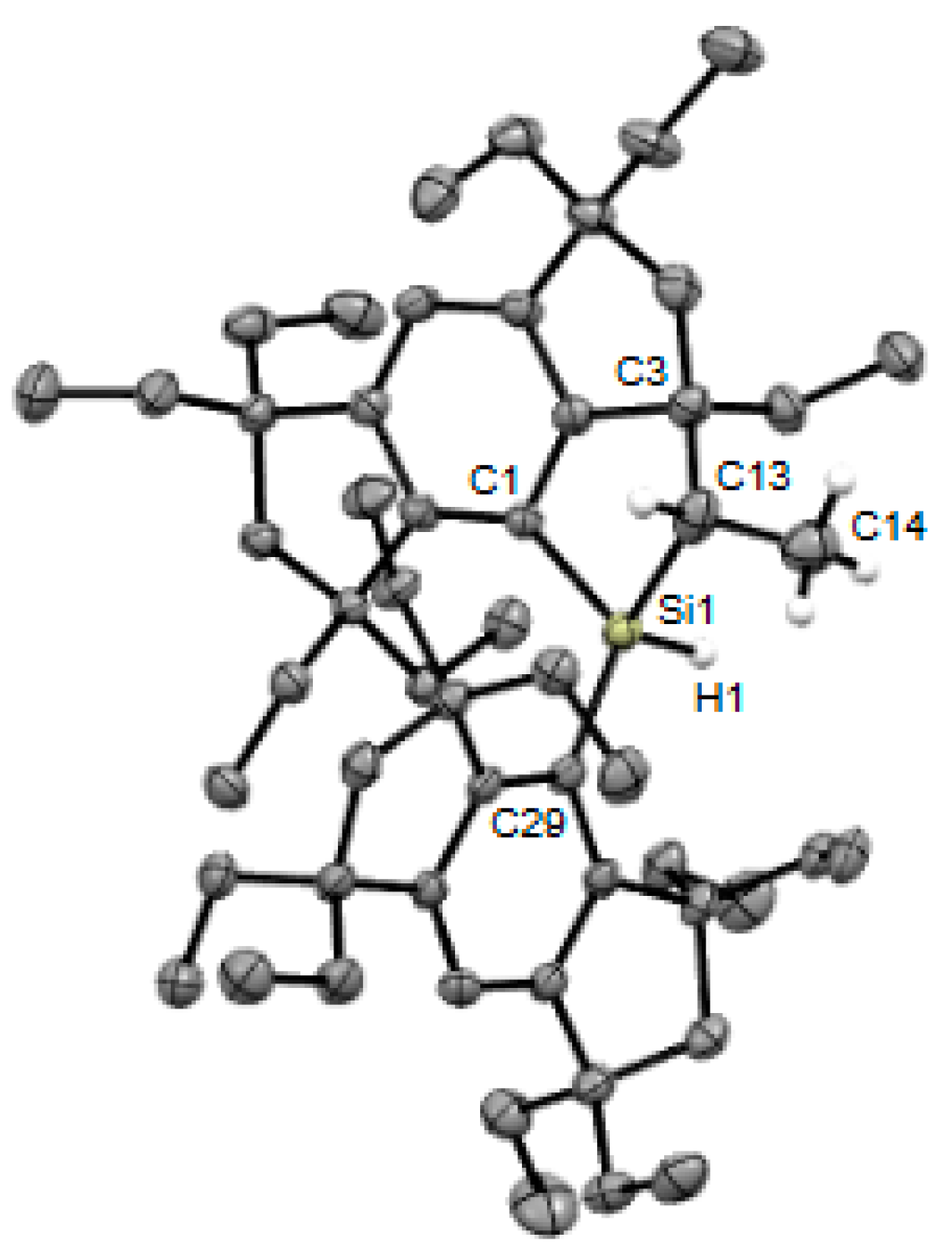
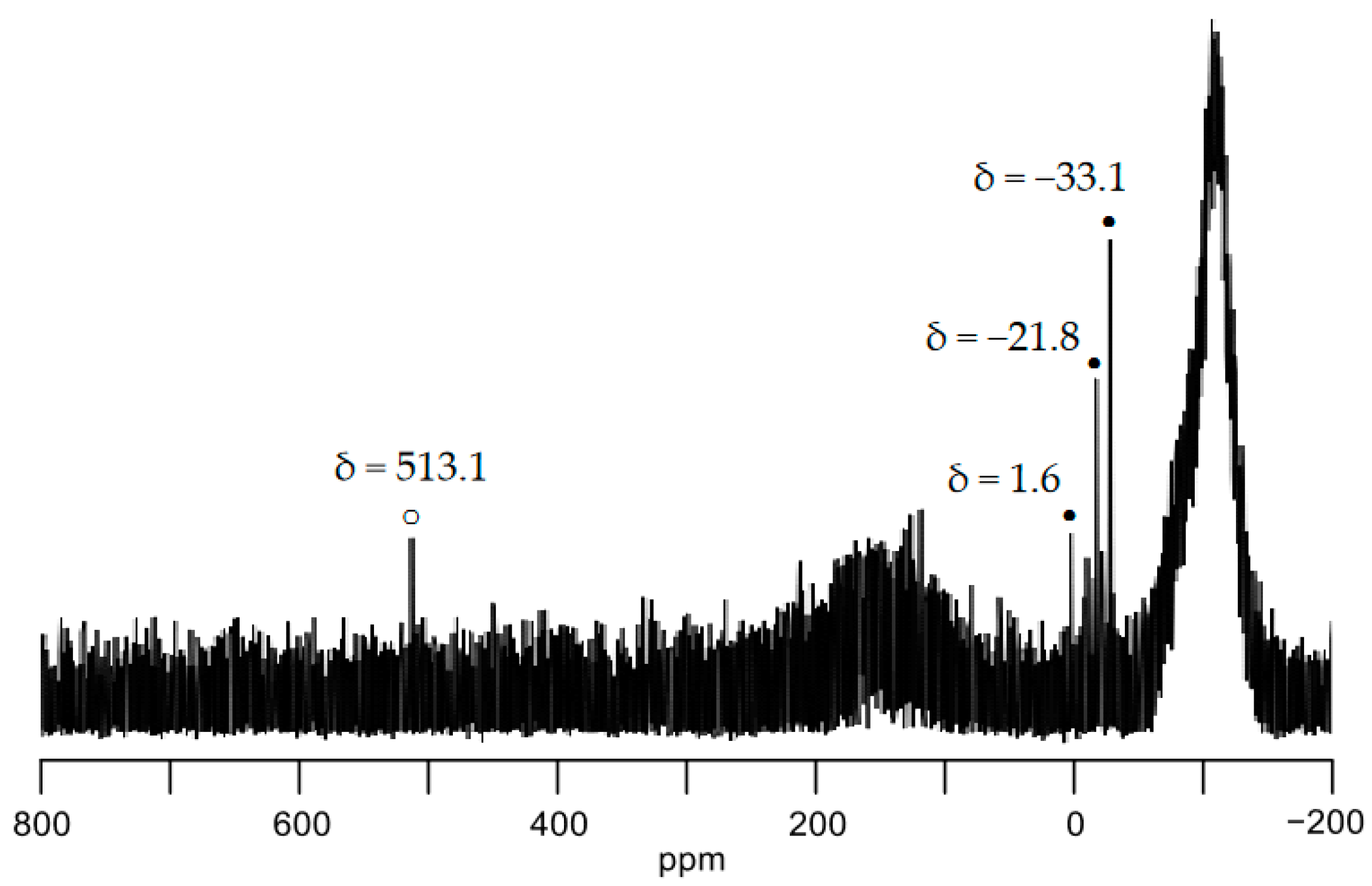
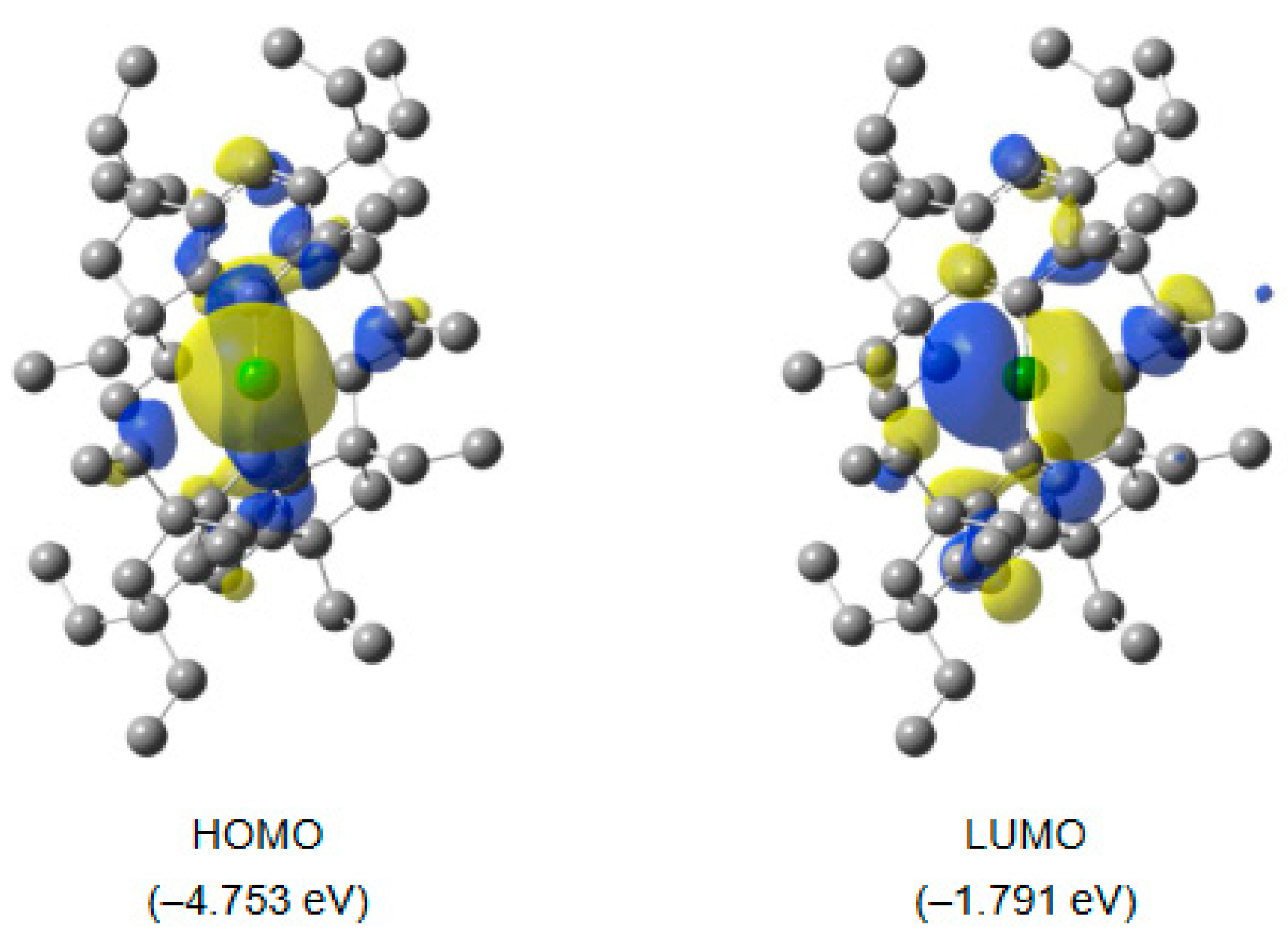
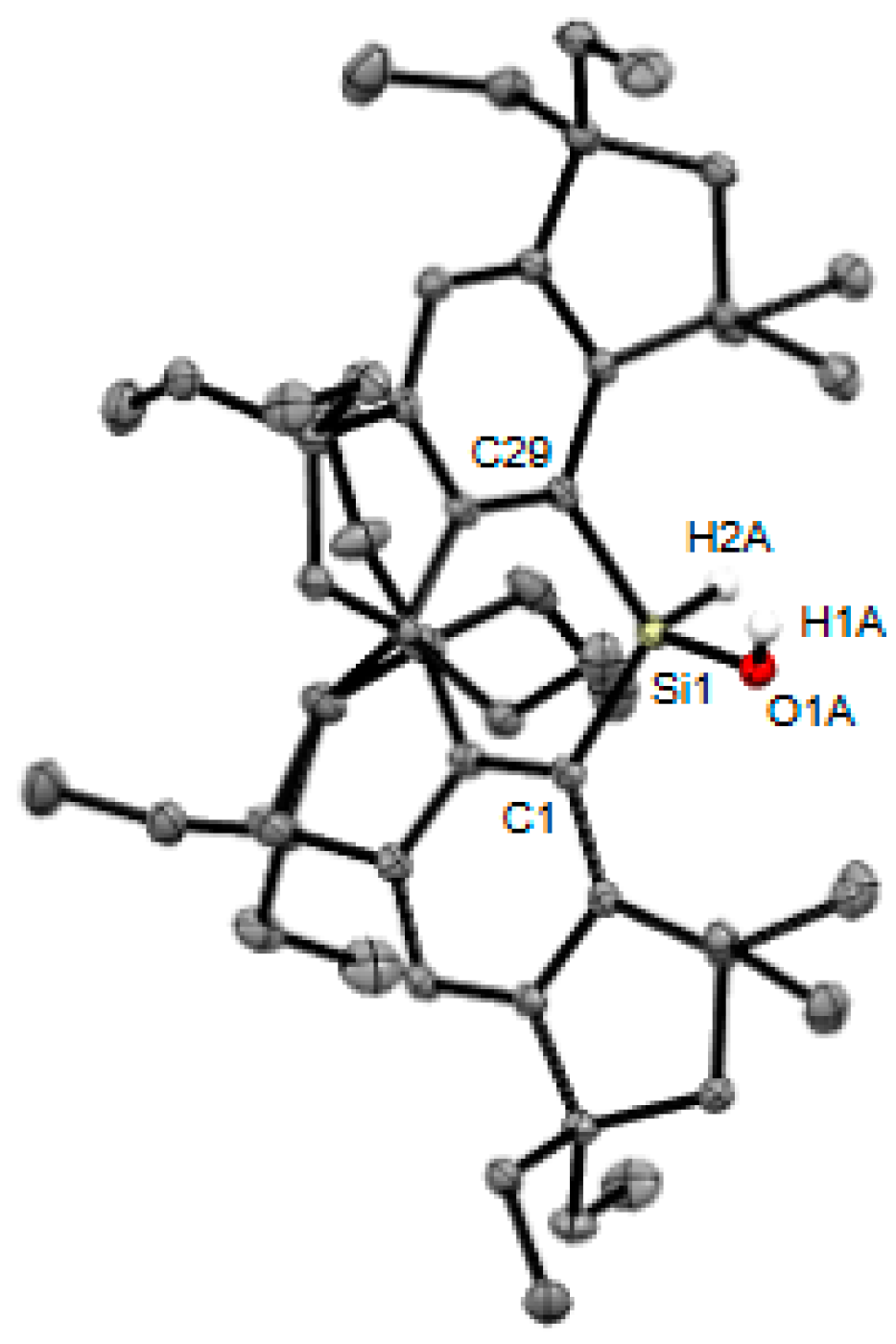
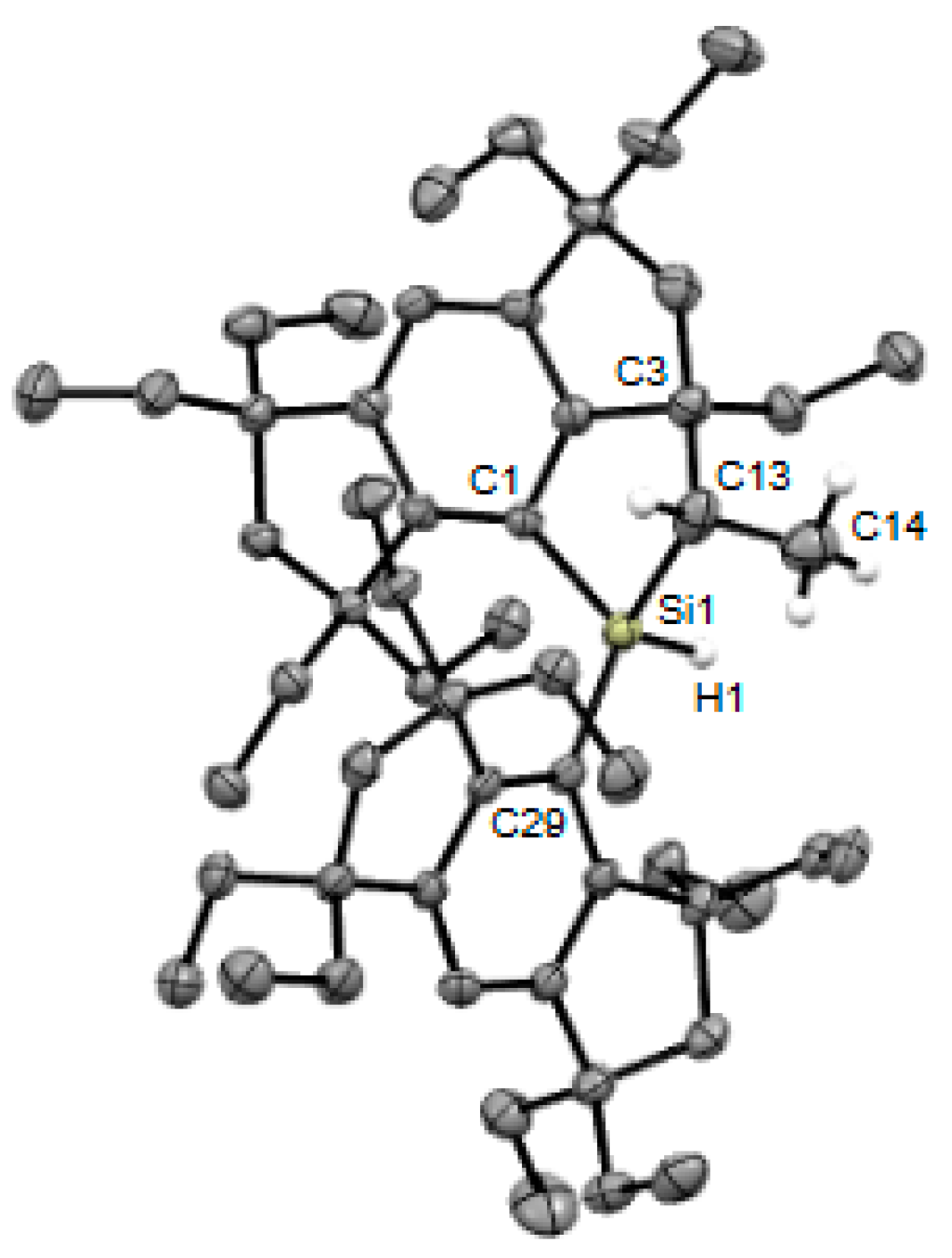
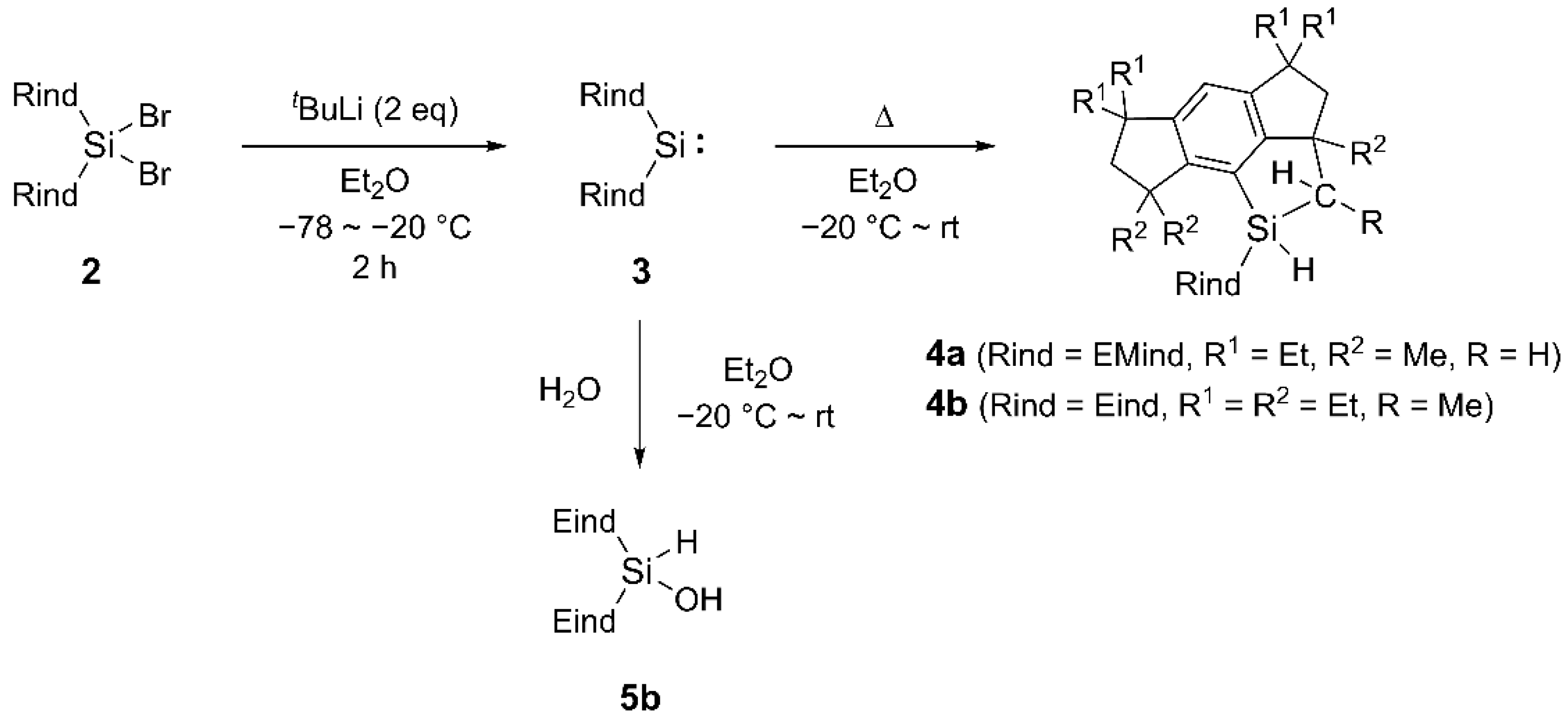
2. Results and Discussion
3. Materials and Methods
3.1. General Procedures
3.1.1. Synthesis of (Eind)2SiH2 (1b)
3.1.2. Synthesis of (Eind)2SiBr2 (2b)
3.1.3. Reduction of (EMind)2SiBr2 (2a) for UV-Vis Measurement of 3a
3.1.4. Reduction of (Eind)2SiBr2 (2b) for UV-Vis Measurement of 3b
3.1.5. Reduction of (Eind)2SiBr2 (2b) for XRD Measurement of Crystal-A
3.1.6. Reduction of (Eind)2SiBr2 (2b) for 29Si NMR Measurement of 3b
3.1.7. Reduction of (EMind)2SiBr2 (2a) for Thermal Reaction of 3a
3.1.8. Reduction of (Eind)2SiBr2 (2b) for Thermal Reaction of 3b
3.1.9. Hydrolysis of 3b for Synthesis of 5b
3.2. X-ray Crystallographic Studies of Crystal-A, Crystal-B, and 5b
3.2.1. Crystal-A (Co-Crystal of 3b and 4b)
3.2.2. Crystal-B (4b)
3.2.3. (Eind)2SiH(OH) (5b)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jutzi, P.; Holtmann, U.; Kanne, D.; Krüger, C.; Blom, R.; Gleiter, R.; Hyla-Kryspin, I. Decamethylsilicocene—The first stable silicon(II) compound: Synthesis, structure, and bonding. Chem. Ber. 1989, 122, 1629–1639. [Google Scholar] [CrossRef]
- Wang, L.; Li, Y.; Li, Z.; Kira, M. Isolable silylenes and their diverse reactivity. Coord. Chem. Rev. 2022, 457, 214413. [Google Scholar] [CrossRef]
- Shan, C.; Yao, S.; Driess, M. Where silylene–silicon centres matter in the activation of small molecules. Chem. Soc. Rev. 2020, 49, 6733–6754. [Google Scholar] [CrossRef]
- Fujimori, S.; Inoue, S. Small Molecule Activation by Two-Coordinate Silylenes. Chem. Eur. J. 2020, 2020, 3131–3142. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.-P.; Driess, M. Isolable Silylene Ligands Can Boost Efficiencies and Selectivities in Metal-Mediated Catalysis. Angew. Chem. Int. Ed. Engl. 2019, 58, 3715–3728. [Google Scholar] [CrossRef]
- Weetman, C.; Inoue, S. The Road Travelled: After Main-Group Elements as Transition Metals. ChemCatChem 2018, 10, 4213–4228. [Google Scholar] [CrossRef]
- Chu, T.; Nikonov, G.I. Oxidative Addition and Reductive Elimination at Main-Group Element Centers. Chem. Rev. 2018, 118, 3608–3680. [Google Scholar] [CrossRef]
- Hadlington, T.J.; Driess, M.; Jones, C. Low-valent group 14 element hydride chemistry: Towards catalysis. Chem. Soc. Rev. 2018, 47, 4176–4197. [Google Scholar] [CrossRef] [PubMed]
- Tacke, R.; Ribbeck, T. Bis(amidinato)- and bis(guanidinato)silylenes and silylenes with one sterically demanding amidinato or guanidinato ligand: Synthesis and reactivity. Dalton Trans. 2017, 46, 13628–13659. [Google Scholar] [CrossRef]
- Lee, V.Y. Organosilicon Compounds: Theory and Experiment (Synthesis); Elsevier: London, UK, 2017. [Google Scholar]
- Yadav, S.; Saha, S.; Sen, S.S. Compounds with Low-Valent p-Block Elements for Small Molecule Activation and Catalysis. ChemCatChem 2016, 8, 486–501. [Google Scholar] [CrossRef]
- Scheschkewitz, D. Functional Molecular Silicon Compounds II: Low Oxidation States; Springer: Basel, Switzerland, 2014. [Google Scholar]
- Asay, M.; Jones, C.; Driess, M. N-Heterocyclic Carbene Analogues with Low-Valent Group 13 and Group 14 Elements: Syntheses, Structures, and Reactivities of a New Generation of Multitalented Ligands. Chem. Rev. 2011, 111, 354–396. [Google Scholar] [CrossRef]
- Lee, V.Y.; Sekiguchi, A. Organometallic Compounds of Low-Coordinate Si, Ge, Sn and Pb; Wiley: West Sussex, UK, 2010. [Google Scholar]
- Kira, M. An isolable dialkylsilylene and its derivatives. A step toward comprehension of heavy unsaturated bonds. Chem. Commun. 2010, 46, 2893–2903. [Google Scholar] [CrossRef]
- Mizuhata, Y.; Sasamori, T.; Tokitoh, N. Stable heavier carbene analogues. Chem. Rev. 2009, 109, 3479–3511. [Google Scholar] [CrossRef]
- Denk, M.; Lennon, R.; Hayashi, R.; West, R.; Belyakov, A.V.; Verne, H.P.; Haaland, A.; Wagner, M.; Metzler, N. Synthesis and Structure of a Stable Silylene. J. Am. Chem. Soc. 1994, 116, 2691–2692. [Google Scholar] [CrossRef]
- Kira, M.; Ishida, S.; Iwamoto, T.; Kabuto, C. The First Isolable Dialkylsilylene. J. Am. Chem. Soc. 1999, 121, 9722–9723. [Google Scholar] [CrossRef]
- Protchenko, A.V.; Birjkumar, K.H.; Dange, D.; Schwarz, A.D.; Vidovic, D.; Jones, C.; Kaltsoyannis, N.; Mountford, P.; Aldridge, S. A Stable Two-Coordinate Acyclic Silylene. J. Am. Chem. Soc. 2012, 134, 6500–6503. [Google Scholar] [CrossRef]
- Rekken, B.D.; Brown, T.M.; Fettinger, J.C.; Tuononen, H.M.; Power, P.P. Isolation of a Stable, Acyclic, Two-Coordinate Silylene. J. Am. Chem. Soc. 2012, 134, 6504–6507. [Google Scholar] [CrossRef]
- Protchenko, A.V.; Schwarz, A.D.; Blake, M.P.; Jones, C.; Kaltsoyannis, N.; Mountford, P.; Aldridge, S. A Generic One-Pot Route to Acyclic Two-Coordinate Silylenes from Silicon(IV) Precursors: Synthesis and Structural Characterization of a Silylsilylene. Angew. Chem. Int. Ed. 2013, 52, 568–571. [Google Scholar] [CrossRef]
- Kosai, T.; Ishida, S.; Iwamoto, T. A Two-Coordinate Cyclic (Alkyl)(amino)silylene: Balancing Thermal Stability and Reactivity. Angew. Chem. Int. Ed. Engl. 2016, 55, 15554–15558. [Google Scholar] [CrossRef]
- Alvarado-Beltran, I.; Baceiredo, A.; Saffon-Merceron, N.; Branchadell, V.; Kato, T. Cyclic Amino(Ylide) Silylene: A Stable Heterocyclic Silylene with Strongly Electron-Donating Character. Angew. Chem. Int. Ed. Engl. 2016, 55, 16141–16144. [Google Scholar] [CrossRef]
- Wendel, D.; Reiter, D.; Porzelt, A.; Altmann, P.J.; Inoue, S.; Rieger, B. Silicon and Oxygen’s Bond of Affection: An Acyclic Three-Coordinate Silanone and Its Transformation to an Iminosiloxysilylene. J. Am. Chem. Soc. 2017, 139, 17193–17198. [Google Scholar] [CrossRef] [PubMed]
- Roy, M.M.D.; Ferguson, M.J.; McDonald, R.; Zhou, Y.; Rivard, E. A vinyl silylsilylene and its activation of strong homo- and heteroatomic bonds. Chem. Sci. 2019, 10, 6476–6481. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, R.; Ishida, S.; Iwamoto, T. An Isolable Silicon Analogue of a Ketone that Contains an Unperturbed Si=O Double Bond. Angew. Chem. Int. Ed. Engl. 2019, 58, 9425–9428. [Google Scholar] [CrossRef] [PubMed]
- West, R.; Fink, M.J.; Michl, J. Tetramesityldisilene, a Stable Compound Containing a Silicon-Silicon Double Bond. Science 1981, 214, 1343–1344. [Google Scholar] [CrossRef]
- Tokitoh, N.; Suzuki, H.; Okazaki, R. Synthesis, Structure, and Reactivity of Extremely Hindered Disilenes: The First Example of Thermal Dissociation of a Disilene into a Silylene. J. Am. Chem. Soc. 1993, 115, 10428–10429. [Google Scholar] [CrossRef]
- Matsuo, T.; Suzuki, K.; Fukawa, T.; Li, B.; Ito, M.; Shoji, Y.; Otani, T.; Li, L.; Kobayashi, M.; Hachiya, M.; et al. Synthesis and Structures of a Series of Bulky “Rind-Br” Based on a Rigid Fused-Ring s-Hydrindacene Skeleton. Bull. Chem. Soc. Jpn. 2011, 84, 1178–1191. [Google Scholar] [CrossRef]
- Ohno, R.; Numata, Y.; Konaka, S.; Yagura, S.; Kuroda, A.; Harada, M.; Fujita, N.; Hayakawa, N.; Nakai, H.; Rosas-Sánchez, A.; et al. Synthesis and Characterization of a Series of Diarylgermylene and Dihalodigermenes Having Fused-Ring Bulky “Rind” Groups. Bull. Chem. Soc. Jpn. 2021, 94, 1931–1939. [Google Scholar] [CrossRef]
- Suzuki, K.; Numata, Y.; Fujita, N.; Hayakawa, N.; Tanikawa, T.; Hashizume, D.; Tamao, K.; Fueno, H.; Tanaka, K.; Matsuo, T. A stable free tetragermacyclobutadiene incorporating fused-ring bulky EMind groups. Chem. Commun. 2018, 54, 2200–2203. [Google Scholar] [CrossRef]
- Li, L.; Fukawa, T.; Matsuo, T.; Hashizume, D.; Fueno, H.; Tanaka, K.; Tamao, K. A stable germanone as the first isolated heavy ketone with a terminal oxygen atom. Nat. Chem. 2012, 4, 361–365. [Google Scholar]
- Kuroda, A.; Fujita, N.; Horita, T.; Ota, K.; Rosas-Sánchez, A.; Hoshino, M.; Hashizume, D.; Matsuo, T. Formation and Reactions of Ge=O Double-bonded Species Bearing EMind Groups. Chem. Lett. 2022, 51, 828–831. [Google Scholar] [CrossRef]
- Numata, Y.; Nishikawa, Y.; Inoue, K.; Ohnishi, H.; Konaka, S.; Tanikawa, T.; Hashizume, D.; Matsuo, T. A Series of Room-Temperature Thermally Stable Bromostannylenes Bearing the Bulky Rind Group: Synthesis, Characterization, and Crystal Structures. Organometallics 2021, 40, 1956–1965. [Google Scholar] [CrossRef]
- Wang, D.; Zhai, C.; Chen, Y.; He, Y.; Chen, X.; Wang, S.; Zhao, L.; Frenking, G.; Wang, X.; Tan, G. An isolable germylene radical with a one-coordinate germanium atom. Nat. Chem. 2023, 15, 200–205. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Chen, W.; Zhai, C.; Zhao, L.; Ye, S.; Tan, G. Monosubstituted Doublet Sn(I) Radical Featuring Substantial Unquenched Orbital Angular Momentum. J. Am. Chem. Soc. 2023, 145, 6914–6920. [Google Scholar] [CrossRef] [PubMed]
- Hayakawa, N.; Morimoto, T.; Takagi, A.; Tanikawa, T.; Hashizume, D.; Matsuo, T. Synthesis and Structures of Sterically Congested Diarylsilanes Bearing Two Bulky Rind Groups. Chem. Lett. 2016, 45, 409–411. [Google Scholar] [CrossRef]
- Sasamori, T.; Hironaka, K.; Sugiyama, Y.; Takagi, N.; Nagase, S.; Hosoi, Y.; Furukawa, Y.; Tokitoh, N. Synthesis and Reactions of a Stable 1,2-Diaryl-1,2-dibromodisilene: A Precursor for Substituted Disilenes and a 1,2-Diaryldisilyne. J. Am. Chem. Soc. 2008, 130, 13856–13857. [Google Scholar] [CrossRef]
- Michalczyk, M.J.; Fink, M.J.; DeYoung, D.J.; Carlson, C.W.; Welsh, K.M.; West, R.; Michl, J. Organosilylenes and Their Dimerization to Disilenes. Silicon Germanium Tin Lead Compd. 1986, 9, 75–83. [Google Scholar]
- Conlin, R.T.; Netto-Ferreira, J.C.; Zhang, S.; Scaiano, J.C. Kinetic study of dimesitylsilylene by laser flash photolysis. Organometallics 1990, 9, 1332–1334. [Google Scholar] [CrossRef]
- Moiseev, A.G.; Leigh, W.J. Diphenylsilylene. J. Am. Chem. Soc. 2006, 128, 14442–14443. [Google Scholar] [CrossRef]
- Hayakawa, N.; Sugahara, T.; Numata, Y.; Kawaai, H.; Yamatani, K.; Nishimura, S.; Goda, S.; Suzuki, Y.; Tanikawa, T.; Nakai, H.; et al. 1,2-Dihalodigermenes bearing bulky Eind groups: Synthesis, characterization, and conversion to halogermylenoids. Dalton Trans. 2018, 47, 814–822. [Google Scholar] [CrossRef]
- Ando, W.; Hamada, Y.; Sekiguchi, A. Reactions of Oxasilacyclopropane. Generation of Silanediyl by Photo and Thermal Induced Cycloelimination. J. Chem. Soc. Chem. Commun. 1983, 17, 952–954. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Rev. D.01; Gaussian, Inc.: Wallingford, CT, USA, 2010. [Google Scholar]
- Kosa, M.; Karni, M.; Apeloig, Y. Were Reactions of Triplet Silylenes Observed? J. Am. Chem. Soc. 2013, 135, 9032–9040. [Google Scholar] [CrossRef] [PubMed]
- Guthardt, R.; Jacob, H.L.; Bruhn, C.; Siemeling, U. A complete series of N-heterocyclic tetrylenes (Si–Pb) with a 1,1′-ferrocenediyl backbone enabled by 1,3,2-diazaborolyl N-substituents. Dalton Trans. 2023, 52, 14380–14389. [Google Scholar] [CrossRef]
- Ishida, S.; Abe, T.; Hirakawa, F.; Kosai, T.; Sato, K.; Kira, M.; Iwamoto, T. Persistent Dialkylsilanone Generated by Dehydrobromination of Dialkylbromosilanol. Chem. Eur. J. 2015, 21, 15100–15103. [Google Scholar] [CrossRef] [PubMed]
- Krempner, C.; Martens, K.; Reinke, H. Synthesis and structure of silyl acetonitriles. J. Organomet. Chem. 2007, 692, 5799–5803. [Google Scholar] [CrossRef]
- CrystalClear; Rigaku/MSC. Inc.: The Woodlands, TX, USA, 2005.
- CrysAlisPro; Agilent Technologies Ltd.: Oxfordshire, UK, 2014.
- Burla, M.C.; Caliandro, R.; Camalli, M.; Carrozzini, B.; Cascarano, G.L.; Caro, L.D.; Giacovazzo, C.; Polidori, G.; Siliqi, D.; Spagna, R. IL MILIONE: A suite of computer programs for crystal structure solution of proteins. J. Appl. Cryst. 2007, 40, 609–613. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A 2015, A71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C 2015, C71, 3–8. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | Si–C (Å) | C(sp2)–Si–C(sp2) (°) |
|---|---|---|
| experimental | ||
| 1b [37] | 1.950(4), 1.938(5) | 123.60(17) |
| 2b [37] | 1.934 (3) | 116.60(13) |
| 3b (Crystal-A) | 1.941(5), 2.004(5) | 112.3(2) |
| 4b (Crystal-A) | 1.919(2), 1.873(3), | 119.57(12) |
| 1.904(4) | ||
| 4b (Crystal-B) | 1.899(3), 1.909(2), | 122.11(11) |
| 1.964(3) | ||
| 5b | 1.9090(11), 1.9228(12) | 122.05(5) |
| calculations | ||
| 3b (singlet) | 1.9470, 1.9489 | 112.00 |
| 3b (triplet) | 1.8980, 1.8926 | 127.95 |
| 4b | 1.9099, 1.9012, | 119.50 |
| 1.9358 | ||
| 5b | 1.9294, 1.9174 | 121.32 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mochihara, K.; Morimoto, T.; Ota, K.; Marumoto, S.; Hashizume, D.; Matsuo, T. Approach to the “Missing” Diarylsilylene: Formation, Characterization, and Intramolecular C–H Bond Activation of Blue Diarylsilylenes with Bulky Rind Groups. Int. J. Mol. Sci. 2024, 25, 3761. https://doi.org/10.3390/ijms25073761
Mochihara K, Morimoto T, Ota K, Marumoto S, Hashizume D, Matsuo T. Approach to the “Missing” Diarylsilylene: Formation, Characterization, and Intramolecular C–H Bond Activation of Blue Diarylsilylenes with Bulky Rind Groups. International Journal of Molecular Sciences. 2024; 25(7):3761. https://doi.org/10.3390/ijms25073761
Chicago/Turabian StyleMochihara, Kazuki, Tatsuto Morimoto, Kei Ota, Shinsuke Marumoto, Daisuke Hashizume, and Tsukasa Matsuo. 2024. "Approach to the “Missing” Diarylsilylene: Formation, Characterization, and Intramolecular C–H Bond Activation of Blue Diarylsilylenes with Bulky Rind Groups" International Journal of Molecular Sciences 25, no. 7: 3761. https://doi.org/10.3390/ijms25073761
APA StyleMochihara, K., Morimoto, T., Ota, K., Marumoto, S., Hashizume, D., & Matsuo, T. (2024). Approach to the “Missing” Diarylsilylene: Formation, Characterization, and Intramolecular C–H Bond Activation of Blue Diarylsilylenes with Bulky Rind Groups. International Journal of Molecular Sciences, 25(7), 3761. https://doi.org/10.3390/ijms25073761