

Collagen Lattice Model, Populated with Heterogeneous Cancer-Associated Fibroblasts, Facilitates Advanced Reconstruction of Pancreatic Cancer Microenvironment

 , , , ,
, , , , 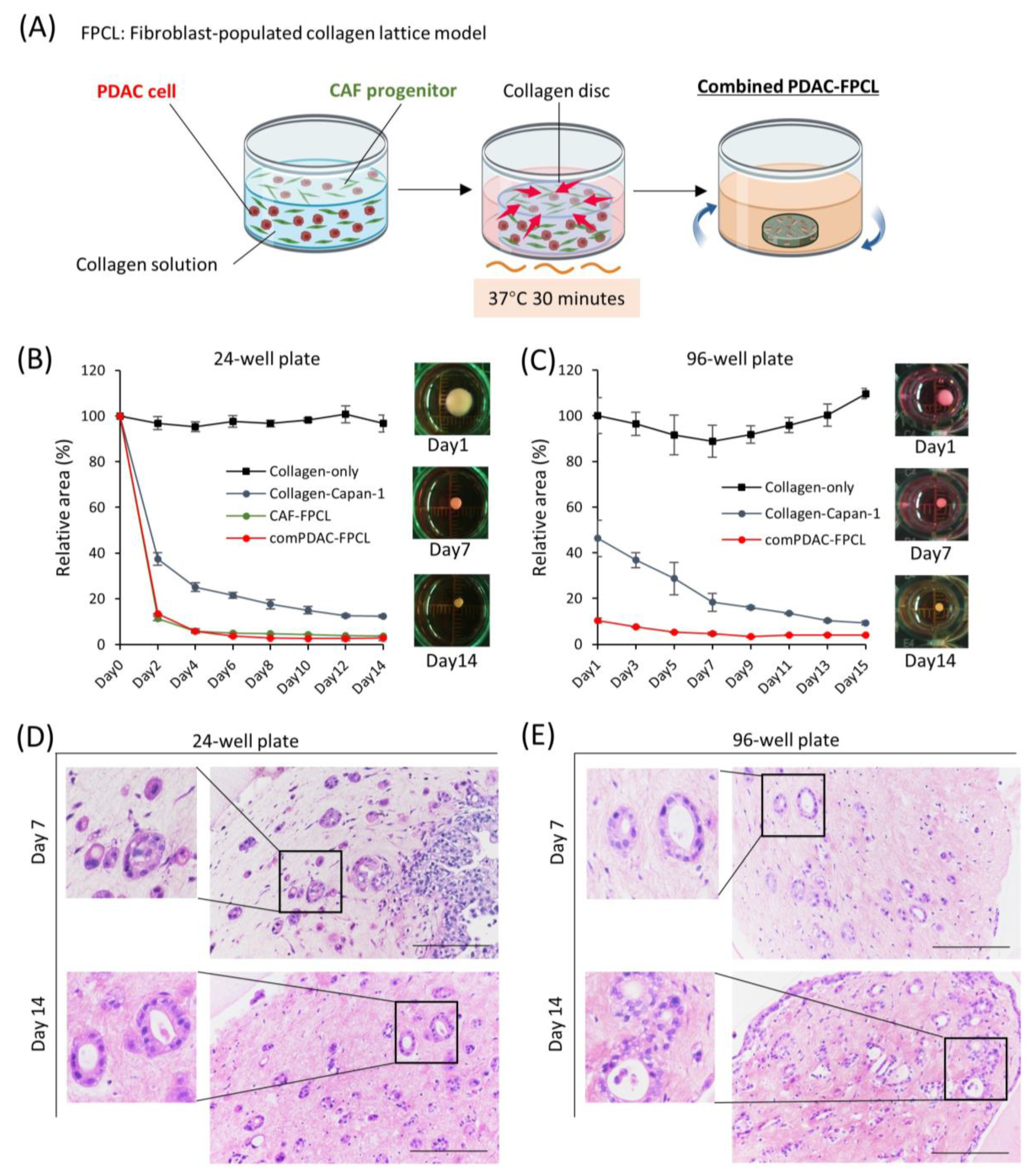{kind=link}
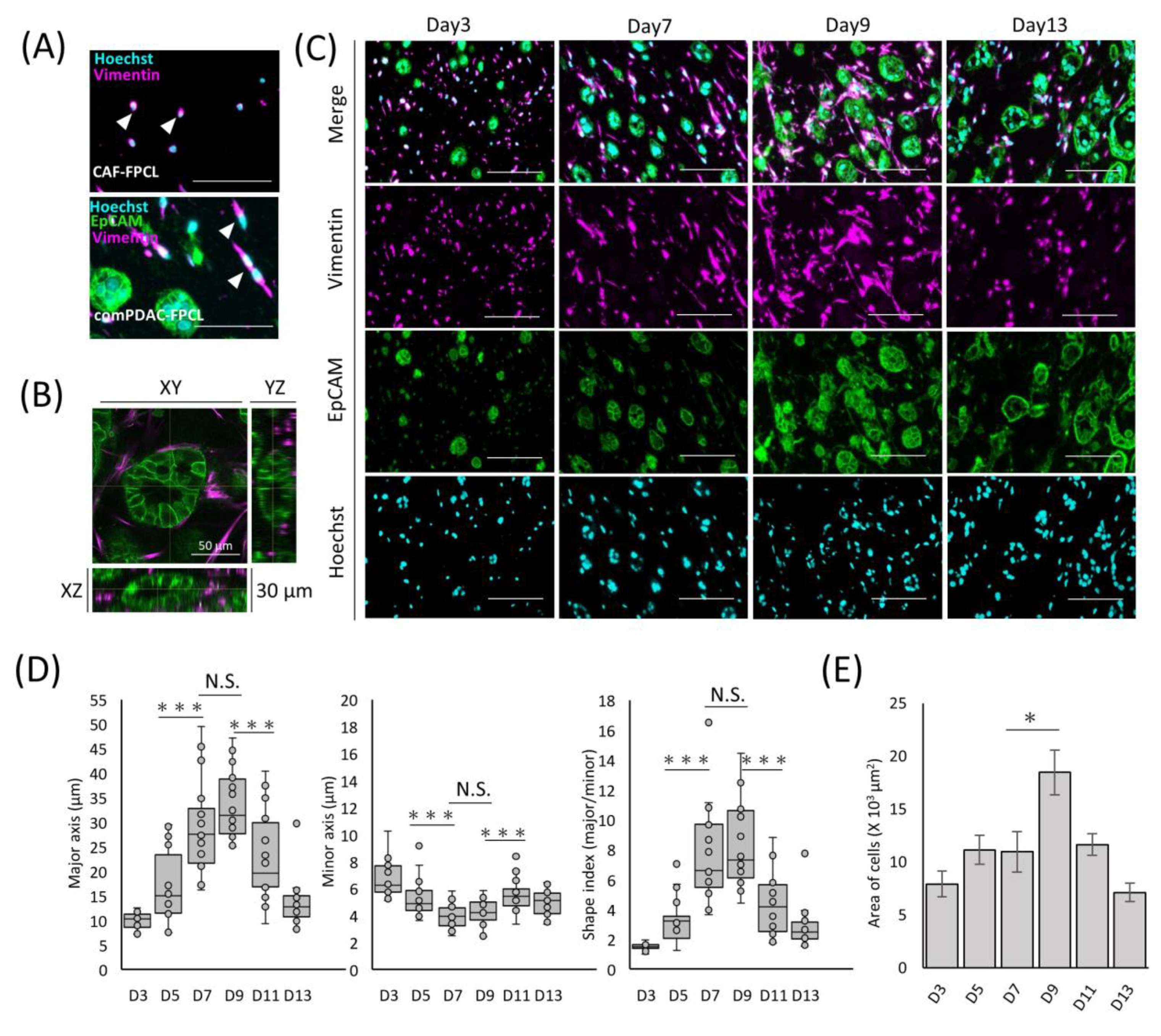{kind=link}
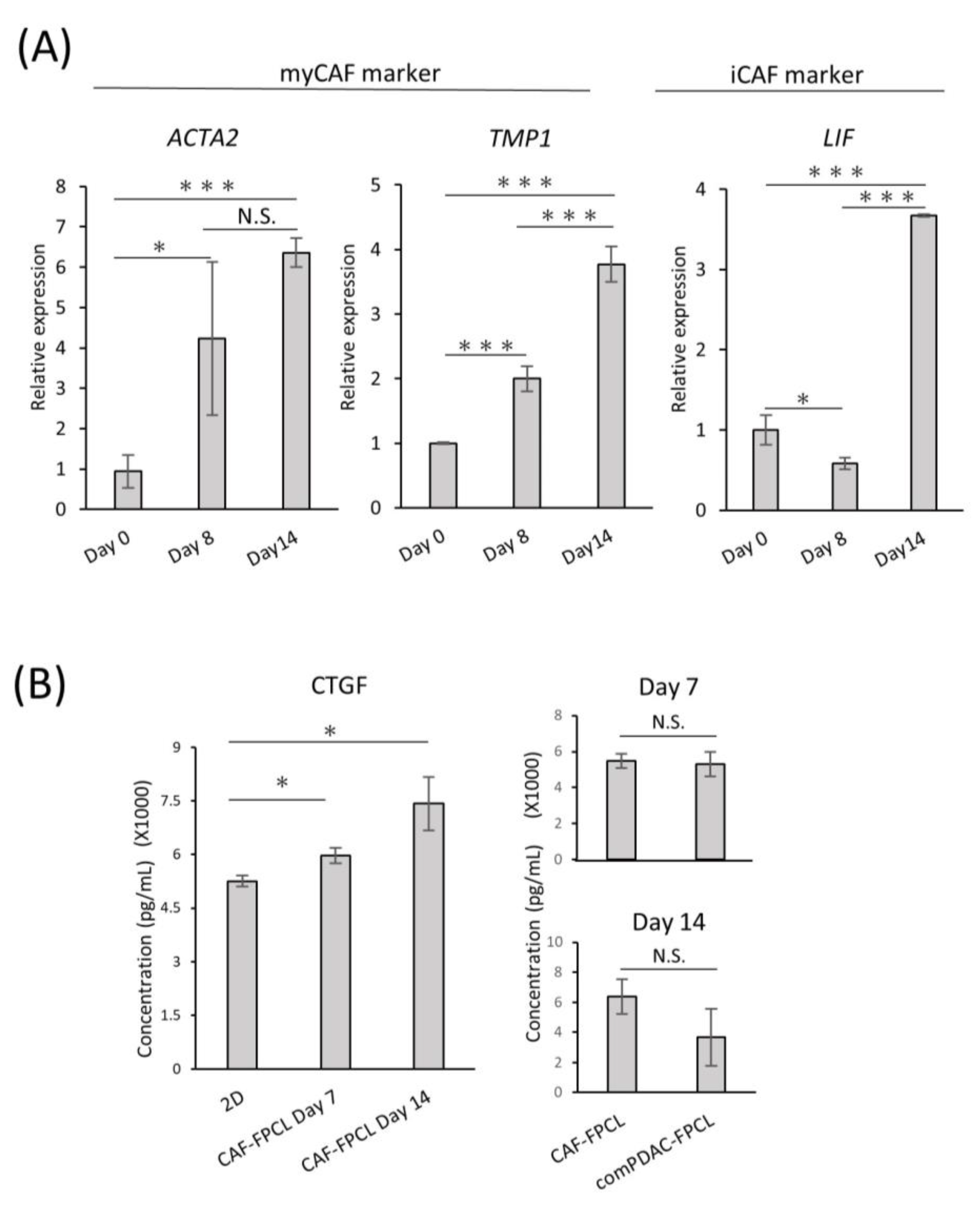{kind=link}
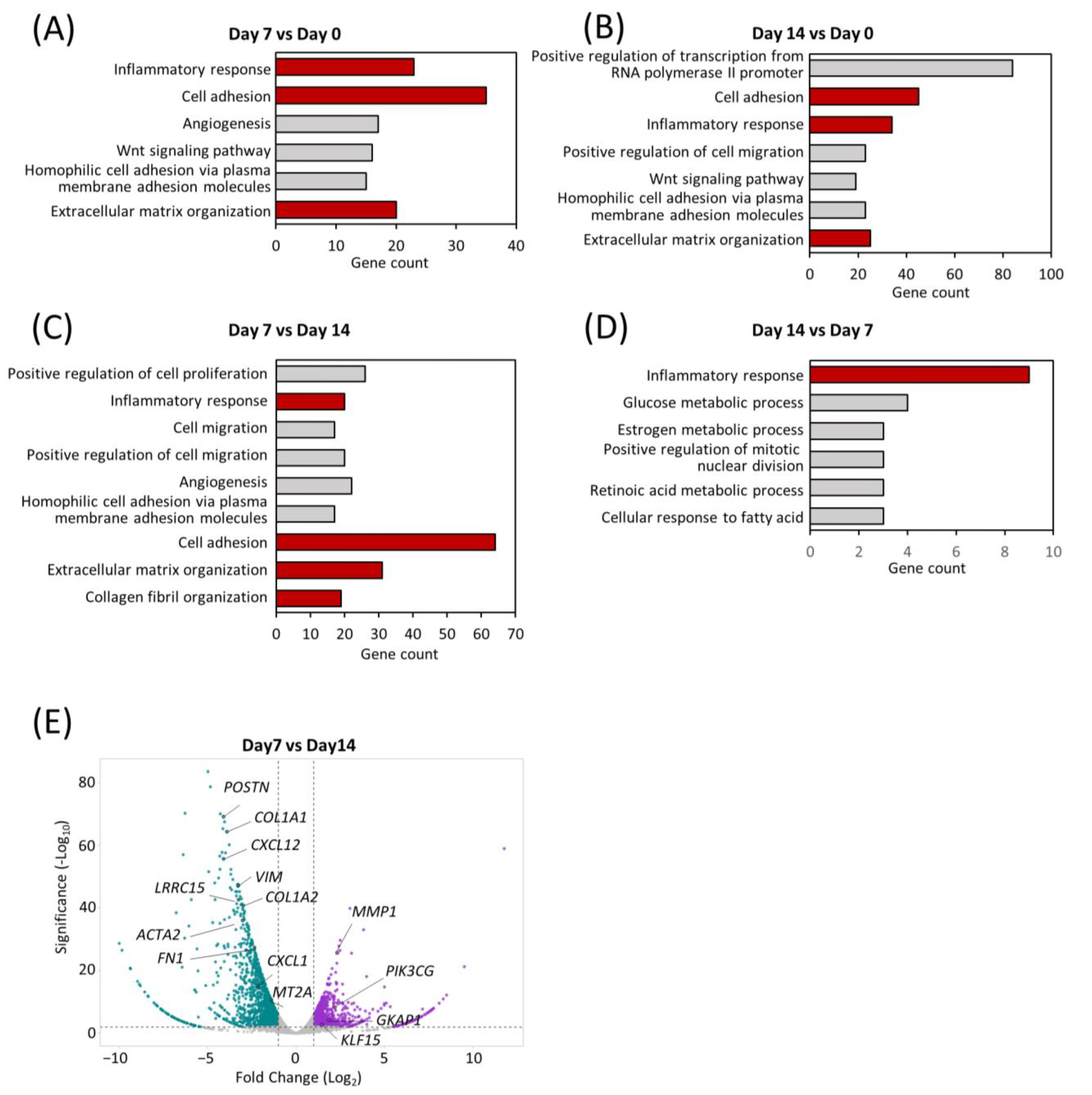{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
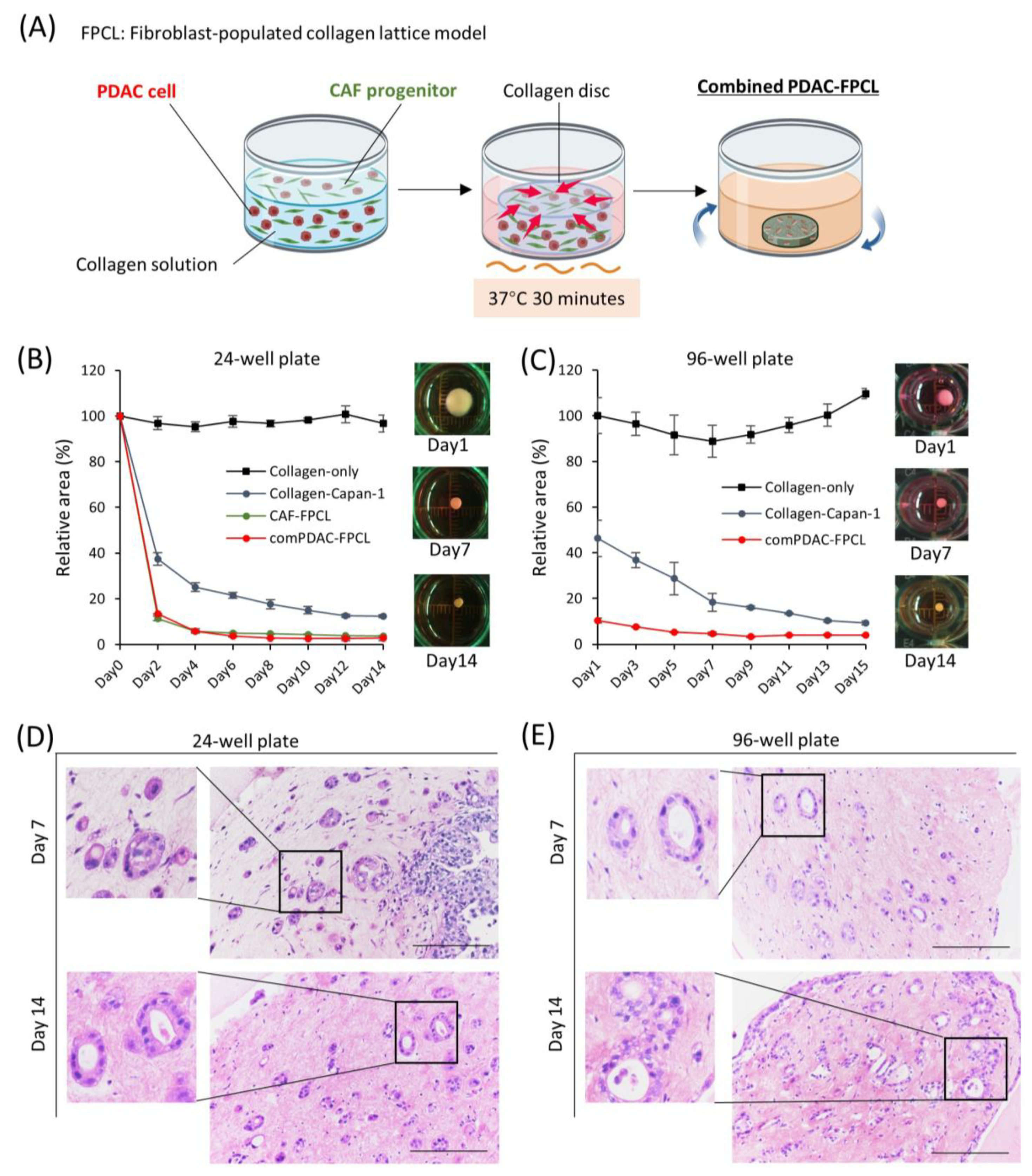
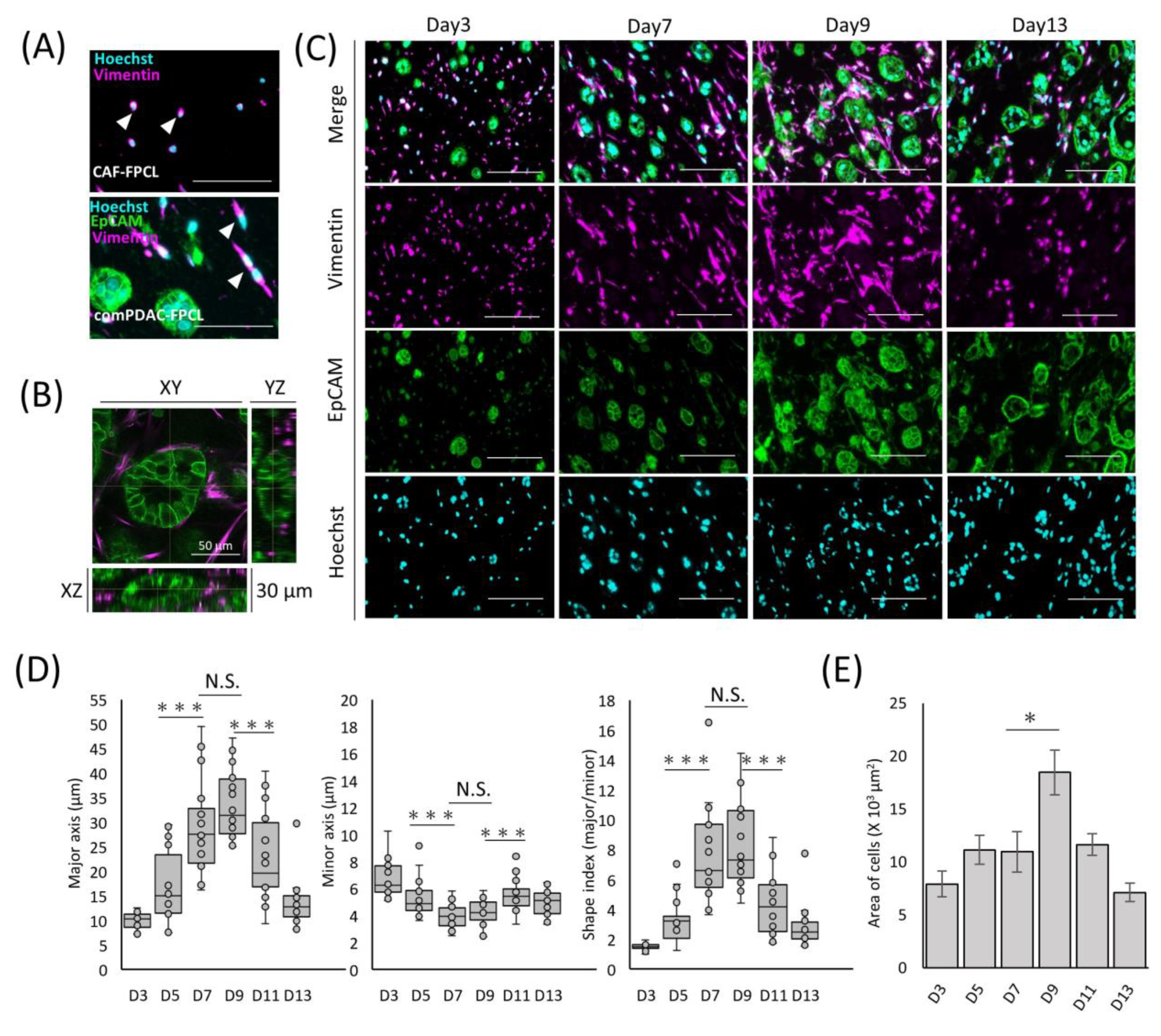
2.1. Morphological and Contractile Dynamics of PDAC-FPCL Models
2.2. Differential Morphological Dynamics of CAFs within PDAC-FPCL Models
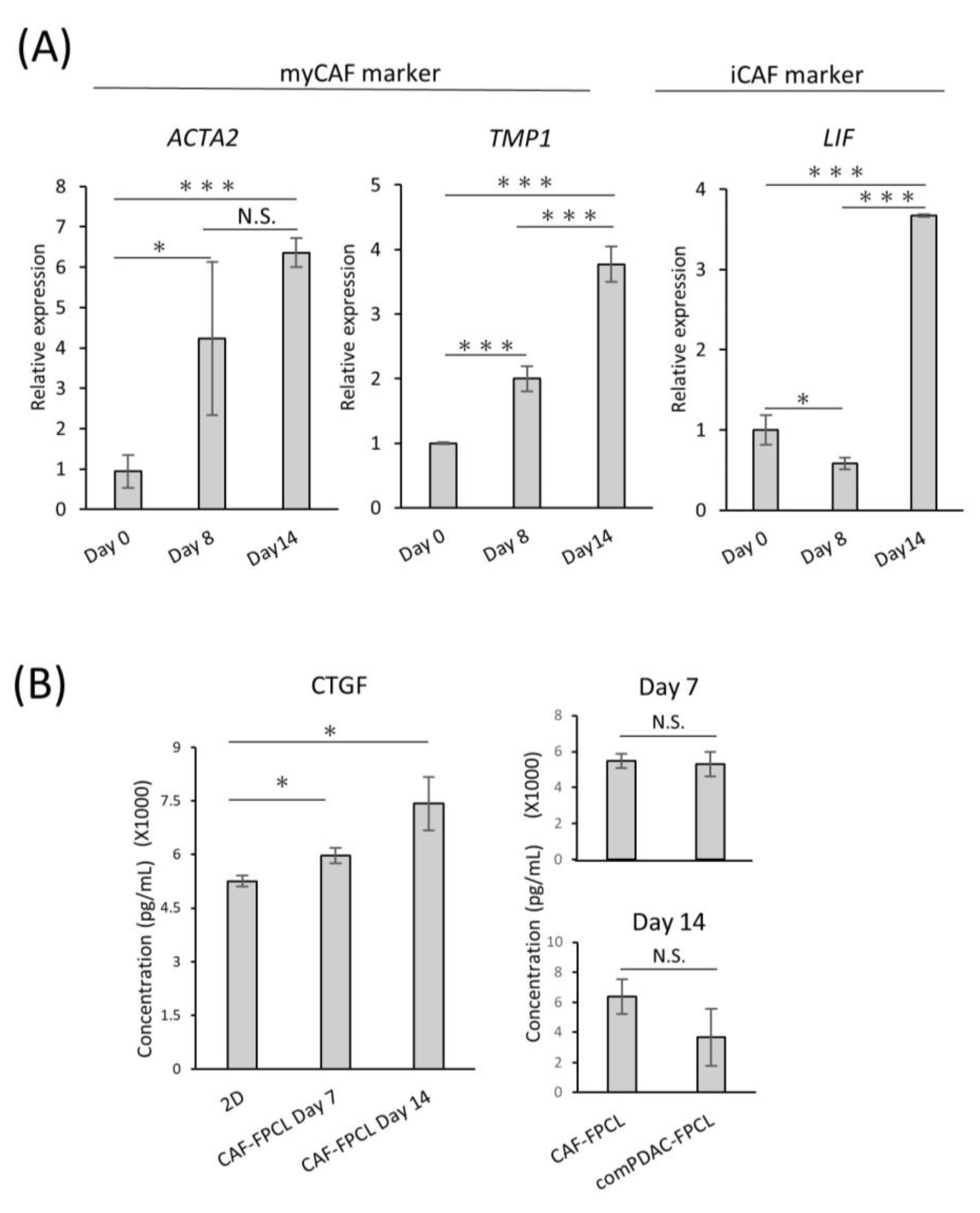
2.3. Differential Expression and Temporal Dynamics of CAF Subtypes
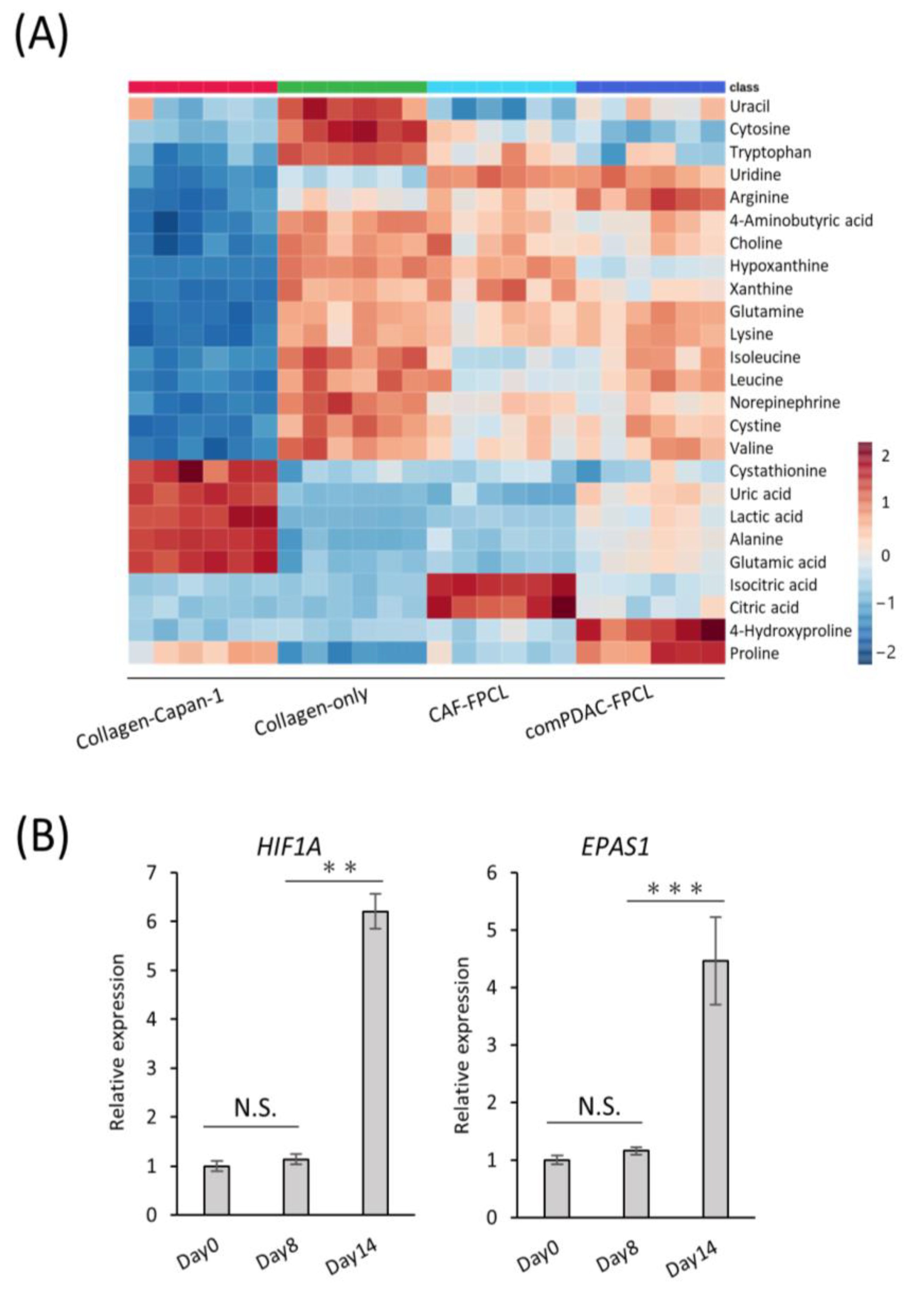
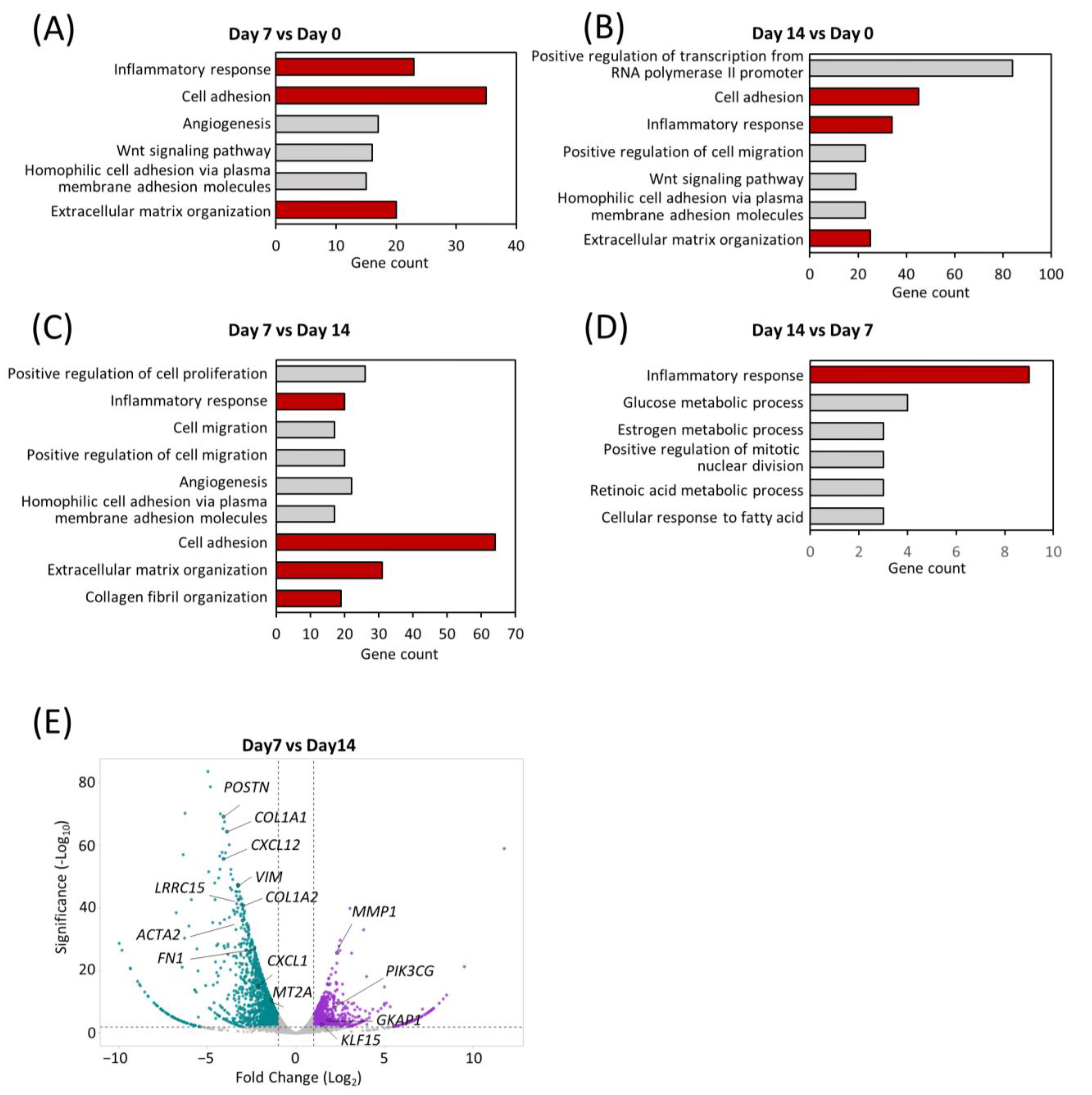
2.4. Transcriptomic Dynamics Show Structural and Metabolic Alterations
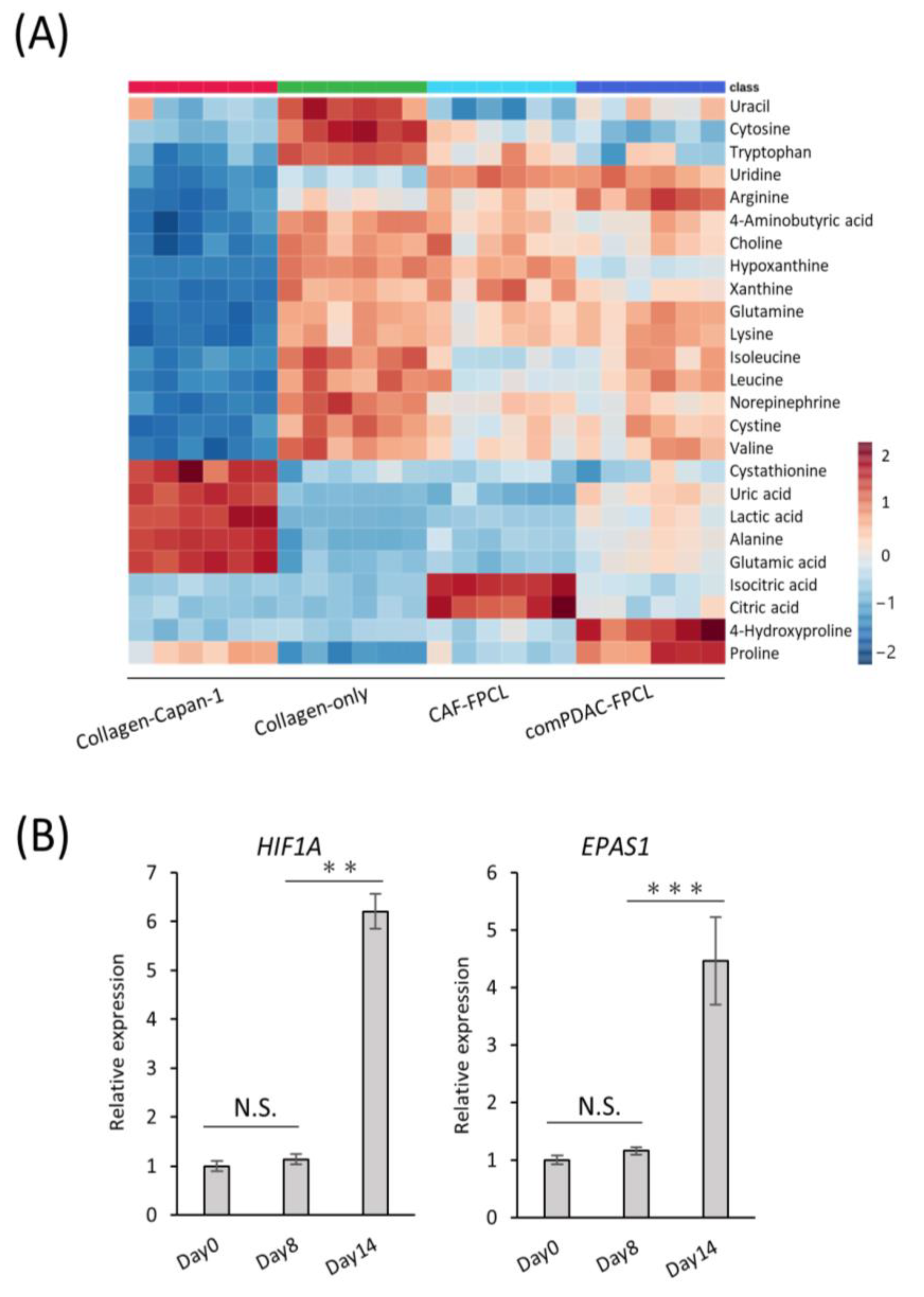
2.5. Characterization of Extracellular Metabolite Dynamics
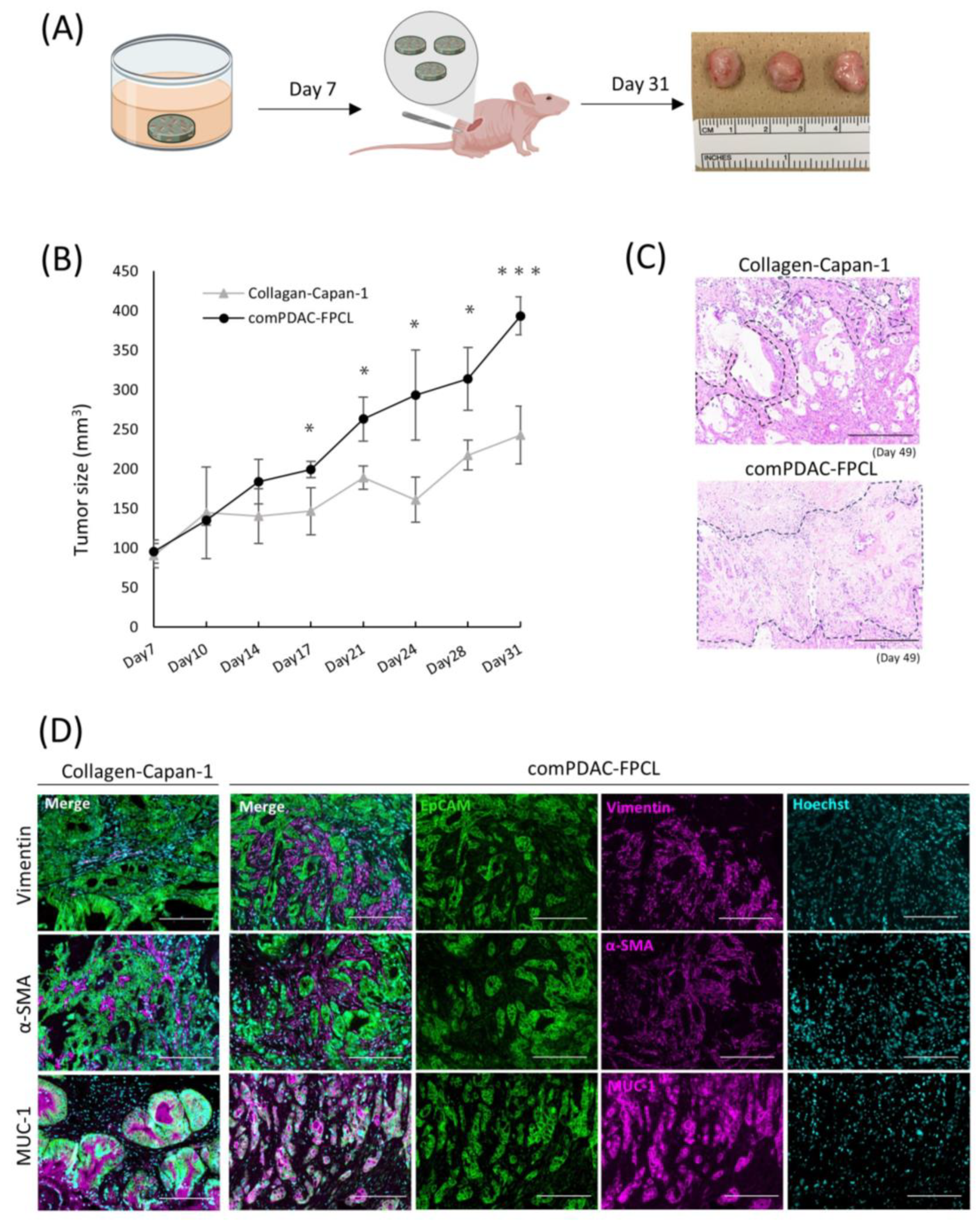
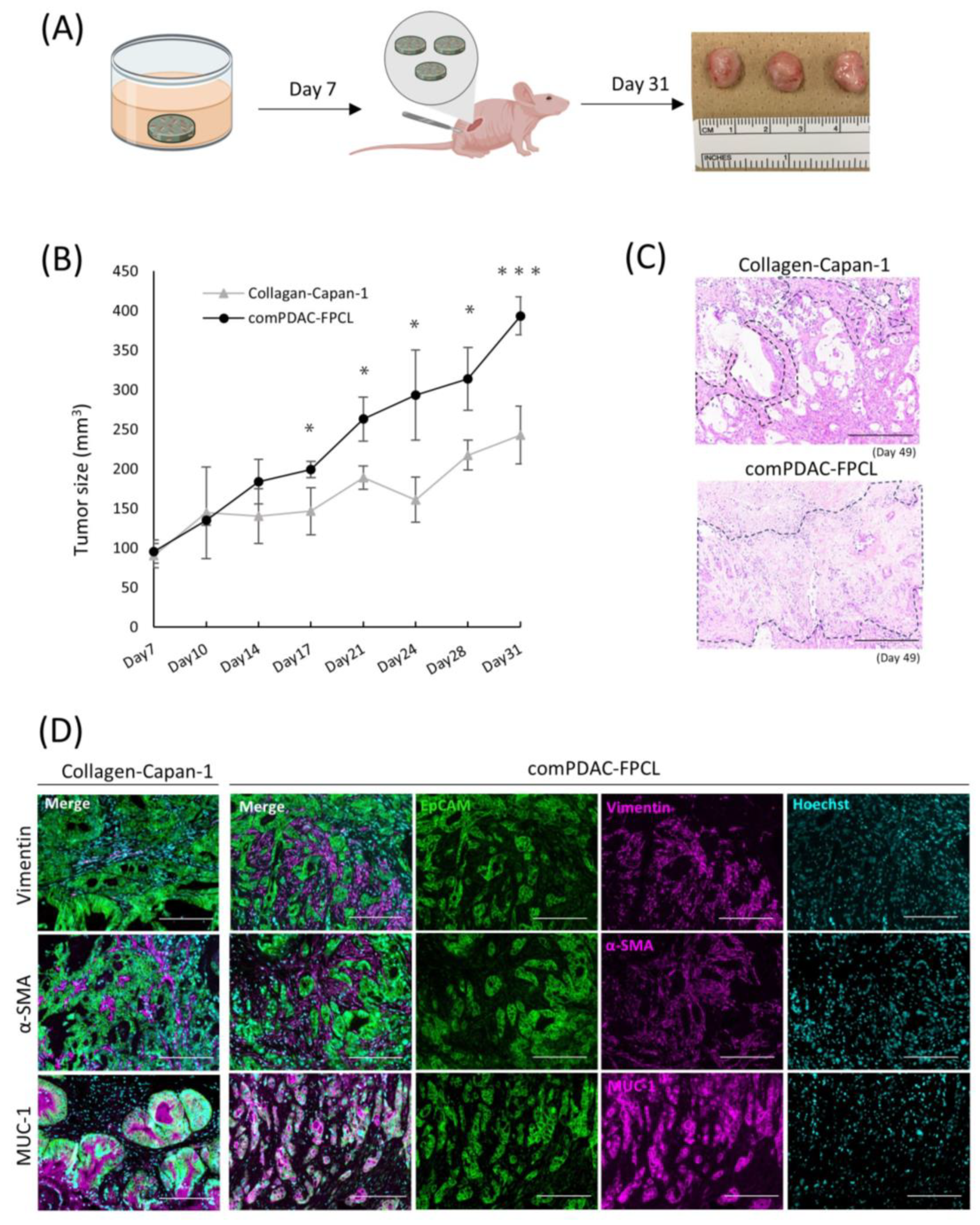
2.6. Validation of Tumorigenic Potential of 3D Tissues Generated by the comPDAC-FPCL Models In Vivo
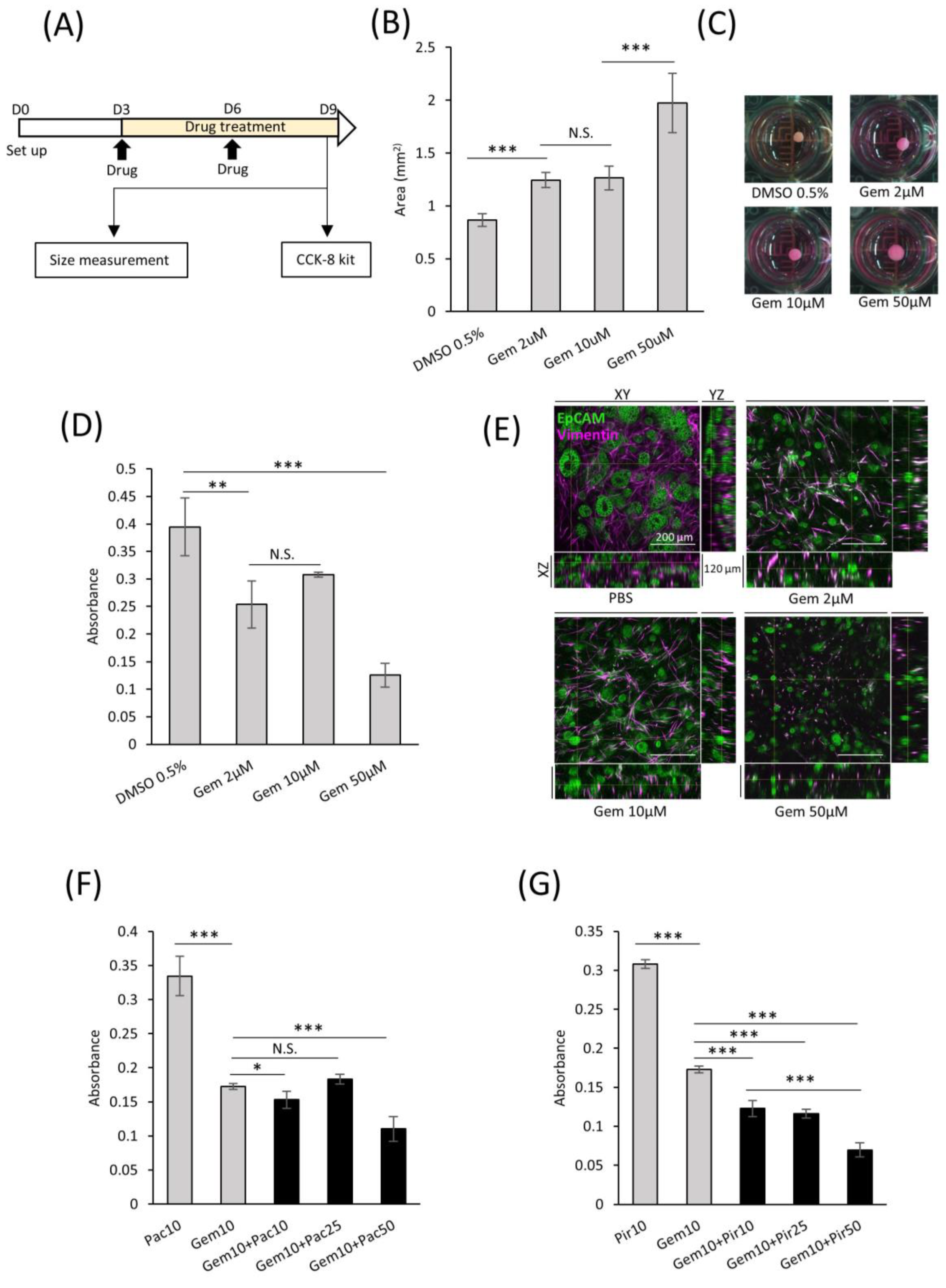
2.7. Applications of comPDAC-FPCL Models in Drug Screening
3. Discussion
4. Materials and Methods
4.1. Cell and Culture Conditions
4.2. Construction of 3D PDAC-FPCL Models
4.3. RNA Extraction and Quantitative Real-Time PCR (qPCR)
4.4. RNA-Seq Analysis
4.5. Conditioned Medium Collection and CTGF Analysis
4.6. Histological Analysis
4.7. 3D Reconstruction Imaging of the comPDAC-FPCL Model
4.8. Mouse Model and In Vivo Experiments
4.9. CCK-8 Assays
4.10. Metabolomic Analysis
4.11. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics, 2020. CA A Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting Cancer Incidence and Deaths to 2030: The Unexpected Burden of Thyroid, Liver, and Pancreas Cancers in the United States. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef] [PubMed]
- Ducreux, M.; Cuhna, A.S.; Caramella, C.; Hollebecque, A.; Burtin, P.; Goéré, D.; Seufferlein, T.; Haustermans, K.; Laethem, J.L.V.; Conroy, T.; et al. Cancer of the Pancreas: ESMO Clinical Practice Guidelines for Diagnosis, Treatment and Follow-Up. Ann. Oncol. 2015, 26, v56–v68. [Google Scholar] [CrossRef] [PubMed]
- Bailey, P.; Chang, D.K.; Nones, K.; Johns, A.L.; Patch, A.-M.; Gingras, M.-C.; Miller, D.K.; Christ, A.N.; Bruxner, T.J.C.; Quinn, M.C.; et al. Genomic Analyses Identify Molecular Subtypes of Pancreatic Cancer. Nature 2016, 531, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Brunner, M.; Wu, Z.; Krautz, C.; Pilarsky, C.; Grützmann, R.; Weber, G.F. Current Clinical Strategies of Pancreatic Cancer Treatment and Open Molecular Questions. Int. J. Mol. Sci. 2019, 20, 4543. [Google Scholar] [CrossRef]
- Conroy, T.; Desseigne, F.; Ychou, M.; Bouché, O.; Guimbaud, R.; Bécouarn, Y.; Adenis, A.; Raoul, J.-L.; Gourgou-Bourgade, S.; de la Fouchardière, C.; et al. FOLFIRINOX versus Gemcitabine for Metastatic Pancreatic Cancer. N. Engl. J. Med. 2011, 364, 1817–1825. [Google Scholar] [CrossRef]
- Rawla, P.; Sunkara, T.; Gaduputi, V. Epidemiology of Pancreatic Cancer: Global Trends, Etiology and Risk Factors. World J. Oncol. 2019, 10, 10–27. [Google Scholar] [CrossRef]
- Zhang, T.; Ren, Y.; Yang, P.; Wang, J.; Zhou, H. Cancer-Associated Fibroblasts in Pancreatic Ductal Adenocarcinoma. Cell Death Dis. 2022, 13, 897. [Google Scholar] [CrossRef]
- Geng, X.; Chen, H.; Zhao, L.; Hu, J.; Yang, W.; Li, G.; Cheng, C.; Zhao, Z.; Zhang, T.; Li, L.; et al. Cancer-Associated Fibroblast (CAF) Heterogeneity and Targeting Therapy of CAFs in Pancreatic Cancer. Front. Cell Dev. Biol. 2021, 9, 655152. [Google Scholar] [CrossRef]
- Pereira, B.A.; Vennin, C.; Papanicolaou, M.; Chambers, C.R.; Herrmann, D.; Morton, J.P.; Cox, T.R.; Timpson, P. CAF Subpopulations: A New Reservoir of Stromal Targets in Pancreatic Cancer. Trends Cancer 2019, 5, 724–741. [Google Scholar] [CrossRef]
- Garcıa-Manteiga, J.; Molina-Arcas, M.; Casado, F.J.; Mazo, A.; Pastor-Anglada, M. Nucleoside Transporter Profiles in Human Pancreatic Cancer Cells: Role of hCNT1 in 2′,2′-Difluorodeoxycytidine- Induced Cytotoxicity. Clin. Cancer Res. 2003, 9, 5000–5008. [Google Scholar]
- Mackey, J.R.; Yao, S.Y.M.; Smith, K.M.; Karpinski, E.; Baldwin, S.A.; Cass, C.E.; Young, J.D. Gemcitabine Transport in Xenopus Oocytes Expressing Recombinant Plasma Membrane Mammalian Nucleoside Transporters. JNCI J. Natl. Cancer Inst. 1999, 91, 1876–1881. [Google Scholar] [CrossRef]
- Ritzel, M.W.L.; Ng, A.M.L.; Yao, S.Y.M.; Graham, K.; Loewen, S.K.; Smith, K.M.; Hyde, R.J.; Karpinski, E.; Cass, C.E.; Baldwin, S.A.; et al. Recent Molecular Advances in Studies of the Concentrative Na+-Dependent Nucleoside Transporter (CNT) Family: Identification and Characterization of Novel Human and Mouse Proteins (hCNT3 and mCNT3) Broadly Selective for Purine and Pyrimidine Nucleosides (System cib). Molec. Membr. Biol. 2001, 18, 65–72. [Google Scholar] [CrossRef]
- Mackey, J.R.; Crawford, C.R.; Cass, C.E. Functional Nucleoside Transporters Are Required for Gemcitabine Influx and Manifestation of Toxicity in Cancer Cell Lines. Cancer Res. 1998, 58, 4349–4357. [Google Scholar]
- Nakano, Y.; Tanno, S.; Koizumi, K.; Nishikawa, T.; Nakamura, K.; Minoguchi, M.; Izawa, T.; Mizukami, Y.; Okumura, T.; Kohgo, Y. Gemcitabine Chemoresistance and Molecular Markers Associated with Gemcitabine Transport and Metabolism in Human Pancreatic Cancer Cells. Br. J. Cancer 2007, 96, 457–463. [Google Scholar] [CrossRef]
- Hosein, A.N.; Brekken, R.A.; Maitra, A. Pancreatic Cancer Stroma: An Update on Therapeutic Targeting Strategies. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 487–505. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Shi, Y.; Qian, F. Opportunities and Delusions Regarding Drug Delivery Targeting Pancreatic Cancer-Associated Fibroblasts. Adv. Drug Deliv. Rev. 2021, 172, 37–51. [Google Scholar] [CrossRef]
- Ene–Obong, A.; Clear, A.J.; Watt, J.; Wang, J.; Fatah, R.; Riches, J.C.; Marshall, J.F.; Chin–Aleong, J.; Chelala, C.; Gribben, J.G.; et al. Activated Pancreatic Stellate Cells Sequester CD8+ T Cells to Reduce Their Infiltration of the Juxtatumoral Compartment of Pancreatic Ductal Adenocarcinoma. Gastroenterology 2013, 145, 1121–1132. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.; Qian, Y.; Ye, Z.; Chen, H.; Xie, H.; Zhou, L.; Shen, Y.; Zheng, S. Cancer-Associated Fibroblasts Promote M2 Polarization of Macrophages in Pancreatic Ductal Adenocarcinoma. Cancer Med. 2017, 6, 463–470. [Google Scholar] [CrossRef]
- Eggermont, A.M.M.; Blank, C.U.; Mandala, M.; Long, G.V.; Atkinson, V.; Dalle, S.; Haydon, A.; Lichinitser, M.; Khattak, A.; Carlino, M.S.; et al. Adjuvant Pembrolizumab versus Placebo in Resected Stage III Melanoma. N. Engl. J. Med. 2018, 378, 1789–1801. [Google Scholar] [CrossRef]
- Horn, L.; Mansfield, A.S.; Szczęsna, A.; Havel, L.; Krzakowski, M.; Hochmair, M.J.; Huemer, F.; Losonczy, G.; Johnson, M.L.; Nishio, M.; et al. First-Line Atezolizumab plus Chemotherapy in Extensive-Stage Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 379, 2220–2229. [Google Scholar] [CrossRef] [PubMed]
- Schmid, P.; Adams, S.; Rugo, H.S.; Schneeweiss, A.; Barrios, C.H.; Iwata, H.; Diéras, V.; Hegg, R.; Im, S.-A.; Shaw Wright, G.; et al. Atezolizumab and Nab-Paclitaxel in Advanced Triple-Negative Breast Cancer. N. Engl. J. Med. 2018, 379, 2108–2121. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, Y.; Oda, T.; Mori, N.; Kida, Y.S. Adipose-Derived Mesenchymal Stem Cells Differentiate into Pancreatic Cancer-Associated Fibroblasts in Vitro. FEBS Open Bio. 2020, 10, 2268–2281. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.A.; Park, H.; Lim, E.H.; Lee, K.W. Exosomes from Breast Cancer Cells Can Convert Adipose Tissue-Derived Mesenchymal Stem Cells into Myofibroblast-like Cells. Int. J. Oncol. 2012, 40, 130–138. [Google Scholar] [CrossRef]
- Lin, R.; Wang, S.; Zhao, R.C. Exosomes from Human Adipose-Derived Mesenchymal Stem Cells Promote Migration through Wnt Signaling Pathway in a Breast Cancer Cell Model. Mol. Cell Biochem. 2013, 383, 13–20. [Google Scholar] [CrossRef]
- Cho, J.A.; Park, H.; Lim, E.H.; Kim, K.H.; Choi, J.S.; Lee, J.H.; Shin, J.W.; Lee, K.W. Exosomes from Ovarian Cancer Cells Induce Adipose Tissue-Derived Mesenchymal Stem Cells to Acquire the Physical and Functional Characteristics of Tumor-Supporting Myofibroblasts. Gynecol. Oncol. 2011, 123, 379–386. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, Y.; Oda, T.; Inagaki, Y.; Kushige, H.; Saito, Y.; Mori, N.; Takayama, Y.; Kumagai, Y.; Mitsuyama, T.; Kida, Y.S. Adipose-Derived Mesenchymal Stem Cells Differentiate into Heterogeneous Cancer-Associated Fibroblasts in a Stroma-Rich Xenograft Model. Sci. Rep. 2021, 11, 4690. [Google Scholar] [CrossRef] [PubMed]
- Pampaloni, F.; Reynaud, E.G.; Stelzer, E.H.K. The Third Dimension Bridges the Gap between Cell Culture and Live Tissue. Nat. Rev. Mol. Cell Biol. 2007, 8, 839–845. [Google Scholar] [CrossRef] [PubMed]
- Cukierman, E.; Pankov, R.; Stevens, D.R.; Yamada, K.M. Taking Cell-Matrix Adhesions to the Third Dimension. Science 2001, 294, 1708–1712. [Google Scholar] [CrossRef]
- Weiswald, L.-B.; Bellet, D.; Dangles-Marie, V. Spherical Cancer Models in Tumor Biology. Neoplasia 2015, 17, 1–15. [Google Scholar] [CrossRef]
- Ben-David, U.; Ha, G.; Tseng, Y.-Y.; Greenwald, N.F.; Oh, C.; Shih, J.; McFarland, J.M.; Wong, B.; Boehm, J.S.; Beroukhim, R.; et al. Patient-Derived Xenografts Undergo Mouse-Specific Tumor Evolution. Nat. Genet. 2017, 49, 1567–1575. [Google Scholar] [CrossRef] [PubMed]
- Barros, A.S.; Costa, E.C.; Nunes, A.S.; de Melo-Diogo, D.; Correia, I.J. Comparative Study of the Therapeutic Effect of Doxorubicin and Resveratrol Combination on 2D and 3D (Spheroids) Cell Culture Models. Int. J. Pharm. 2018, 551, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Alemany-Ribes, M.; Semino, C.E. Bioengineering 3D Environments for Cancer Models. Adv. Drug Deliv. Rev. 2014, 79–80, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Jubelin, C.; Muñoz-Garcia, J.; Griscom, L.; Cochonneau, D.; Ollivier, E.; Heymann, M.-F.; Vallette, F.M.; Oliver, L.; Heymann, D. Three-Dimensional in Vitro Culture Models in Oncology Research. Cell Biosci. 2022, 12, 155. [Google Scholar] [CrossRef] [PubMed]
- Bell, E.; Ivarsson, B.; Merrill, C. Production of a Tissue-like Structure by Contraction of Collagen Lattices by Human Fibroblasts of Different Proliferative Potential in Vitro. Proc. Natl. Acad. Sci. USA 1979, 76, 1274–1278. [Google Scholar] [CrossRef] [PubMed]
- Stylianopoulos, T.; Martin, J.D.; Chauhan, V.P.; Jain, S.R.; Diop-Frimpong, B.; Bardeesy, N.; Smith, B.L.; Ferrone, C.R.; Hornicek, F.J.; Boucher, Y.; et al. Causes, Consequences, and Remedies for Growth-Induced Solid Stress in Murine and Human Tumors. Proc. Natl. Acad. Sci. USA 2012, 109, 15101–15108. [Google Scholar] [CrossRef] [PubMed]
- Schuth, S.; Le Blanc, S.; Krieger, T.G.; Jabs, J.; Schenk, M.; Giese, N.A.; Büchler, M.W.; Eils, R.; Conrad, C.; Strobel, O. Patient-Specific Modeling of Stroma-Mediated Chemoresistance of Pancreatic Cancer Using a Three-Dimensional Organoid-Fibroblast Co-Culture System. J. Exp. Clin. Cancer Res. 2022, 41, 312. [Google Scholar] [CrossRef] [PubMed]
- Aden, N.; Nuttall, A.; Shiwen, X.; de Winter, P.; Leask, A.; Black, C.M.; Denton, C.P.; Abraham, D.J.; Stratton, R.J. Epithelial Cells Promote Fibroblast Activation via IL-1α in Systemic Sclerosis. J. Investig. Dermatol. 2010, 130, 2191–2200. [Google Scholar] [CrossRef] [PubMed]
- Schild, C.; Trueb, B. Mechanical Stress Is Required for High-Level Expression of Connective Tissue Growth Factor. Exp. Cell Res. 2002, 274, 83–91. [Google Scholar] [CrossRef]
- Miyazaki, Y.; Mori, N.; Akagi, Y.; Oda, T.; Kida, Y.S. Potential Metabolite Markers for Pancreatic Cancer Identified by Metabolomic Analysis of Induced Cancer-Associated Fibroblasts. Cancers 2022, 14, 1375. [Google Scholar] [CrossRef]
- Nihashi, Y.; Song, X.; Yamamoto, M.; Setoyama, D.; Kida, Y.S. Decoding Metabolic Symbiosis between Pancreatic Cancer Cells and Cancer-Associated Fibroblasts Using Cultured Tumor Microenvironment. Int. J. Mol. Sci. 2023, 24, 11015. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.; Ren, S.; Li, C.; Guo, K.; Lu, Z.; Tian, L.; He, J.; Zhang, K.; Cao, Y.; Liu, S.; et al. Biomarkers for Pancreatic Cancer Based on Tissue and Serum Metabolomics Analysis in a Multicenter Study. Cancer Med. 2023, 12, 5158–5171. [Google Scholar] [CrossRef] [PubMed]
- Elyada, E.; Bolisetty, M.; Laise, P.; Flynn, W.F.; Courtois, E.T.; Burkhart, R.A.; Teinor, J.A.; Belleau, P.; Biffi, G.; Lucito, M.S.; et al. Cross-Species Single-Cell Analysis of Pancreatic Ductal Adenocarcinoma Reveals Antigen-Presenting Cancer-Associated Fibroblasts. Cancer Discov. 2019, 9, 1102–1123. [Google Scholar] [CrossRef] [PubMed]
- Rice, A.J.; Cortes, E.; Lachowski, D.; Cheung, B.C.H.; Karim, S.A.; Morton, J.P.; del Río Hernández, A. Matrix Stiffness Induces Epithelial–Mesenchymal Transition and Promotes Chemoresistance in Pancreatic Cancer Cells. Oncogenesis 2017, 6, e352. [Google Scholar] [CrossRef] [PubMed]
- Dupont, S.; Morsut, L.; Aragona, M.; Enzo, E.; Giulitti, S.; Cordenonsi, M.; Zanconato, F.; Le Digabel, J.; Forcato, M.; Bicciato, S.; et al. Role of YAP/TAZ in Mechanotransduction. Nature 2011, 474, 179–183. [Google Scholar] [CrossRef] [PubMed]
- Osuna de la Peña, D.; Trabulo, S.M.D.; Collin, E.; Liu, Y.; Sharma, S.; Tatari, M.; Behrens, D.; Erkan, M.; Lawlor, R.T.; Scarpa, A.; et al. Bioengineered 3D Models of Human Pancreatic Cancer Recapitulate In Vivo Tumour Biology. Nat. Commun. 2021, 12, 5623. [Google Scholar] [CrossRef] [PubMed]
- Elia, I.; Broekaert, D.; Christen, S.; Boon, R.; Radaelli, E.; Orth, M.F.; Verfaillie, C.; Grünewald, T.G.P.; Fendt, S.-M. Proline Metabolism Supports Metastasis Formation and Could Be Inhibited to Selectively Target Metastasizing Cancer Cells. Nat. Commun. 2017, 8, 15267. [Google Scholar] [CrossRef] [PubMed]
- Scott, G.K.; Yau, C.; Becker, B.C.; Khateeb, S.; Mahoney, S.; Jensen, M.B.; Hann, B.; Cowen, B.J.; Pegan, S.D.; Benz, C.C. Targeting Mitochondrial Proline Dehydrogenase with a Suicide Inhibitor to Exploit Synthetic Lethal Interactions with P53 Upregulation and Glutaminase Inhibition. Mol. Cancer Ther. 2019, 18, 1374–1385. [Google Scholar] [CrossRef] [PubMed]
- Mello, A.M.; Ngodup, T.; Lee, Y.; Donahue, K.L.; Li, J.; Rao, A.; Carpenter, E.S.; Crawford, H.C.; Pasca di Magliano, M.; Lee, K.E. Hypoxia Promotes an Inflammatory Phenotype of Fibroblasts in Pancreatic Cancer. Oncogenesis 2022, 11, 56. [Google Scholar] [CrossRef]
- Awaji, M.; Singh, R.K. Cancer-Associated Fibroblasts’ Functional Heterogeneity in Pancreatic Ductal Adenocarcinoma. Cancers 2019, 11, 290. [Google Scholar] [CrossRef]
- Schwörer, S.; Cimino, F.V.; Ros, M.; Tsanov, K.M.; Ng, C.; Lowe, S.W.; Carmona-Fontaine, C.; Thompson, C.B. Hypoxia Potentiates the Inflammatory Fibroblast Phenotype Promoted by Pancreatic Cancer Cell-Derived Cytokines. Cancer Res. 2023, 83, 1596–1610. [Google Scholar] [CrossRef]
- Chen, Y.; Yang, S.; Tavormina, J.; Tampe, D.; Zeisberg, M.; Wang, H.; Mahadevan, K.K.; Wu, C.-J.; Sugimoto, H.; Chang, C.-C.; et al. Oncogenic Collagen I Homotrimers from Cancer Cells Bind to A3β1 Integrin and Impact Tumor Microbiome and Immunity to Promote Pancreatic Cancer. Cancer Cell 2022, 40, 818–834.e9. [Google Scholar] [CrossRef] [PubMed]
- Tian, C.; Huang, Y.; Clauser, K.R.; Rickelt, S.; Lau, A.N.; Carr, S.A.; Vander Heiden, M.G.; Hynes, R.O. Suppression of Pancreatic Ductal Adenocarcinoma Growth and Metastasis by Fibrillar Collagens Produced Selectively by Tumor Cells. Nat. Commun. 2021, 12, 2328. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Nihashi, Y.; Yamamoto, M.; Setoyama, D.; Kunisaki, Y.; Kida, Y.S. Exploring the Role of Desmoplastic Physical Stroma in Pancreatic Cancer Progression Using a Three-Dimensional Collagen Matrix Model. Bioengineering 2023, 10, 1437. [Google Scholar] [CrossRef] [PubMed]
- Imai, Y.; Mori, N.; Nihashi, Y.; Kumagai, Y.; Shibuya, Y.; Oshima, J.; Sasaki, M.; Sasaki, K.; Aihara, Y.; Sekido, M.; et al. Therapeutic Potential of Adipose Stem Cell-Derived Conditioned Medium on Scar Contraction Model. Biomedicines 2022, 10, 2388. [Google Scholar] [CrossRef]
- Mori, N.; Akagi, Y.; Imai, Y.; Takayama, Y.; Kida, Y.S. Fabrication of Perfusable Vascular Channels and Capillaries in 3D Liver-like Tissue. Sci. Rep. 2020, 10, 5646. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, X.; Nihashi, Y.; Imai, Y.; Mori, N.; Kagaya, N.; Suenaga, H.; Shin-ya, K.; Yamamoto, M.; Setoyama, D.; Kunisaki, Y.; et al. Collagen Lattice Model, Populated with Heterogeneous Cancer-Associated Fibroblasts, Facilitates Advanced Reconstruction of Pancreatic Cancer Microenvironment. Int. J. Mol. Sci. 2024, 25, 3740. https://doi.org/10.3390/ijms25073740
Song X, Nihashi Y, Imai Y, Mori N, Kagaya N, Suenaga H, Shin-ya K, Yamamoto M, Setoyama D, Kunisaki Y, et al. Collagen Lattice Model, Populated with Heterogeneous Cancer-Associated Fibroblasts, Facilitates Advanced Reconstruction of Pancreatic Cancer Microenvironment. International Journal of Molecular Sciences. 2024; 25(7):3740. https://doi.org/10.3390/ijms25073740
Chicago/Turabian StyleSong, Xiaoyu, Yuma Nihashi, Yukiko Imai, Nobuhito Mori, Noritaka Kagaya, Hikaru Suenaga, Kazuo Shin-ya, Masamichi Yamamoto, Daiki Setoyama, Yuya Kunisaki, and et al. 2024. "Collagen Lattice Model, Populated with Heterogeneous Cancer-Associated Fibroblasts, Facilitates Advanced Reconstruction of Pancreatic Cancer Microenvironment" International Journal of Molecular Sciences 25, no. 7: 3740. https://doi.org/10.3390/ijms25073740
APA StyleSong, X., Nihashi, Y., Imai, Y., Mori, N., Kagaya, N., Suenaga, H., Shin-ya, K., Yamamoto, M., Setoyama, D., Kunisaki, Y., & Kida, Y. S. (2024). Collagen Lattice Model, Populated with Heterogeneous Cancer-Associated Fibroblasts, Facilitates Advanced Reconstruction of Pancreatic Cancer Microenvironment. International Journal of Molecular Sciences, 25(7), 3740. https://doi.org/10.3390/ijms25073740