

Ferroptosis: Frenemy of Radiotherapy



Abstract
1. Introduction
2. Ferroptosis
2.1. Terms and Conditions for Ferroptosis Execution
2.2. Involvement of Lipid Metabolism in Ferroptosis
2.3. The Role of Iron Metabolism during Ferroptosis
2.4. Mitochondrial Involvement in Ferroptosis
2.5. Ferroptosis Defense Systems
2.5.1. SLC7A11-GSH-GPX4 Axis
2.5.2. BH4 Axis
2.5.3. The Ubiquinol (CoQH2) Axis

3. Ferroptosis and RT
3.1. Interplay between Ferroptosis and RT
3.2. Effect of Hypoxia on Ferroptosis

4. Ferroptosis Inducers and the Role in RT Effectiveness
4.1. Drug Repurposing for Radiosensitizing Properties via Ferroptosis
4.2. Drugs Containing Radiosensitizing Properties Linked to Ferroptosis Induction
- 1.
- Sulfasalazine
- 2.
- Disulfiram
- 3.
- Sorafenib
4.3. Drugs with Ferroptosis Inducing Capacities and Radiosensitizing Properties
- 4.
- Auranofin
- 5.
- Metformin
- 6.
- Artesunate
- 7.
- Statins
- 8.
- Vorinostat
- 9.
- Olaparib
- 10.
- Tyrosine kinase inhibitors (Lapatinib and Neratinib)

{kind=link}
{kind=link}
{kind=link}
| Drug | FDA Approved Condition | FIN in Cancer Types | Molecular Mechanism |
|---|---|---|---|
| 1. Sulfasalazine | Rheumatoid arthritis and IBD | Breast [135], Colon [136], Brain [137] | Inhibitor of xCT, light subunit of system xC- → decreases levels of GSH, less active GPX4, more ROS and more lipid peroxidation |
| 2. Disulfiram | Antabuse | Brain [89], Liver [138], Naso-pharyngeal [139] | Increases levels of ROS, impairs mitochondrial homeostasis, increases iron levels, increases lipid peroxidation |
| 3. Sorafenib | Hepatocellular and renal cell carcinoma | Liver [91], Kidney [140], Lung [140], Colon [140], Pancreas [140], Skin [140] | System xC- inhibitor → decreases levels of GSH, leading to less active GPX4, more ROS and increases lipid peroxidation |
| 4. Auranofin | Rheumatoid arthritis | Cervix [141], Liver [142], Brain [99] | Inhibits TrxR, increases ROS, increases lipid peroxidation, activates hepcidin |
| 5. Metformin | Diabetes | Breast [143,144,145] | Inhibits autophagy, upregulates miR-324-3p and thereby inhibits GPX4, increases iron levels, and lipid ROS, decreases SLC7A11 stability via inhibition of UFMylation |
| 6. Artesunate | Anti-malaria agent | Brain [146], Lymphatic system [147,148], Liver [149] | Modulates the p38/ERK pathway, impairs STAT3 signalling, induces ER stress, activates the ATF4-CHOP-CHAC1 pathway, increases intracellular iron |
| 7. Statins | Cardio- vascular diseases | Breast [150], Non-tumorigenic tissue [151] | Inhibits HMG-CoA reductase → inhibits mevalonate pathway → inhibits biosynthesis of GPX4. Increases the levels of iron, increases the levels of ROS and lipid peroxidation, inhibits NRF2, SLC7A11, and GPX4 |
| 8. Vorinostat | Cutaneous T-cell lymphoma | Lung [115] | Increases the amount of lipid peroxidation/ROS, inhibits xCT |
| 9. Olaparib | BRCA mutated cancer | Ovarian [120,121] | Activates P53, inhibits SLC7A11, decreases levels of GSH, increases levels of lipid peroxidation, suppression of SCD1 |
| 10. Tyrosine kinase inhibitor | HER2+ Breast cancer | Breast [129,130], Brain [131], Lung [131] | Increases the levels of iron (Neratinib), increases ROS, increases lipid peroxidation increases transferrin expression, decreases ferroportin expression, degradation of heme-oxygenase (Lapatinib + Siramesine) |
5. Adverse Events of Ferroptosis Inducers as Radiosensitizers
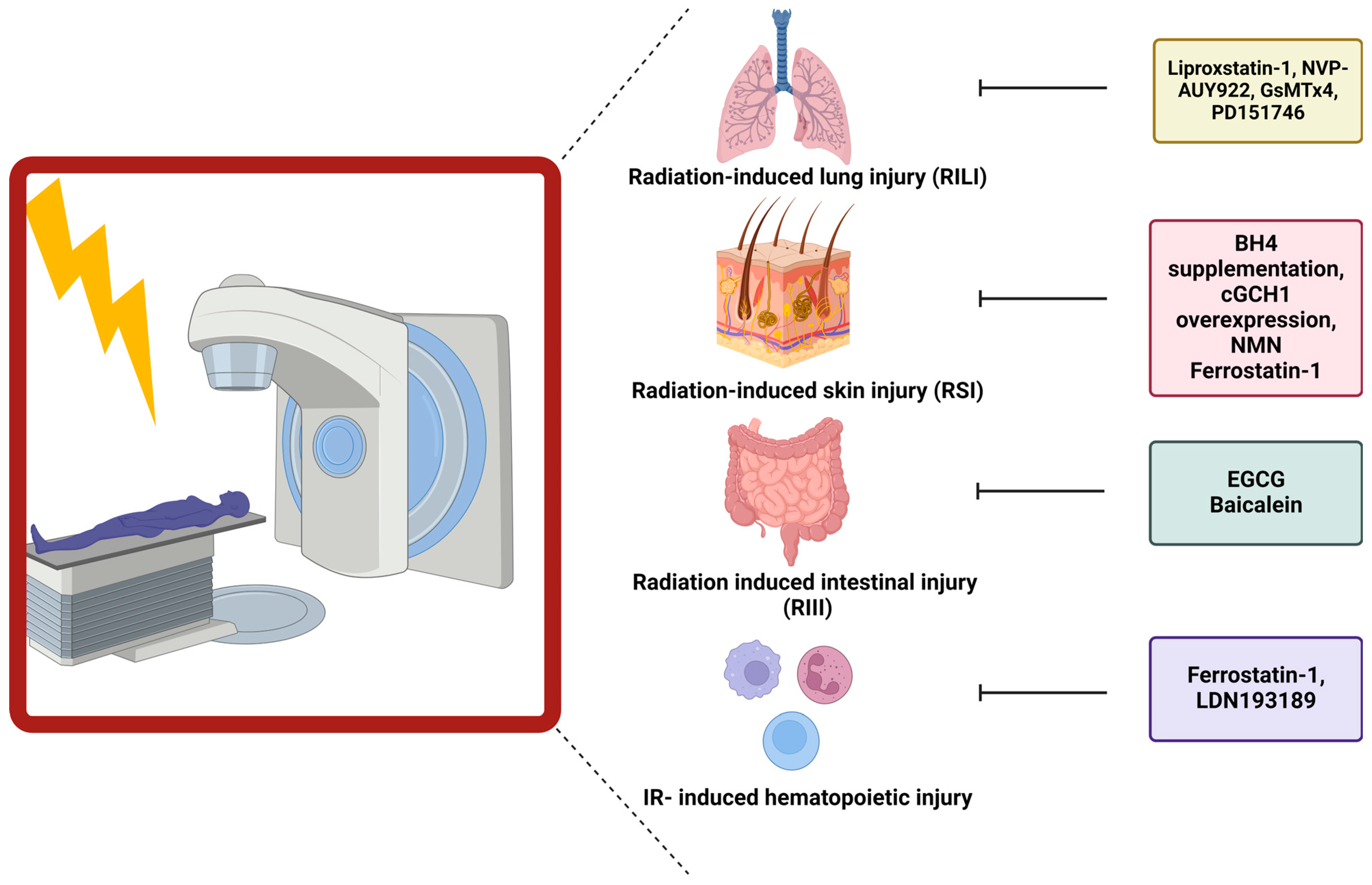
5.1. Interplay between Ferroptosis and Total Body Irradiation
5.2. Link between Ferroptosis and Radiation-Induced Skin Injury
5.3. Role of Ferroptosis in Radiation-Induced Lung Injury
| Tissue of Interest | Irradiation Source/Dose | Parameters Investigated |
|---|---|---|
| Lung | TBI γ-Ray (17 Gy) | Collagen deposition |
| Thorax irradiation X-ray (15 Gy) [164] | Szapiel score, Ashcroft score and hydroxyproline content | |
| Thorax irradiation X-ray (15 Gy) [165] | Lung injury score (Szapiel) | |
| Thorax irradiation X-ray (10 Gy) [166] | Collagen deposition and % apoptotic cells | |
| Gastro-intestinal system | TBI γ-ray (15 Gy) [160] | Villi height, % apoptosis and proliferation |
| TBI 60CO γ-source (9 Gy) [168] | Villus height, crypt depth, proliferation, crypts per circumference, apoptosis, DNA damage, and oxidative stress | |
| TBI (9.25 Gy) [169] | % Apoptosis and epithelial barrier disruption | |
| Skin | Electron beam (45 Gy) [162] | Number of skin appendages and epidermis depth |
| UVB lamp [163] | Epidermal and dermal thickness | |
| Hematopoietic system | TBI γ-Ray (4 Gy) [160] | Red blood cell count, white blood cell count, hemoglobin, platelets, percentage of lymphocytes, and percentage of neutrophil granulocytes |
| Whole body irradiation 137Cs source (10 Gy) [169] | Bone marrow hemorrhage, hepcidin content, iron content, white blood count, lymphocytes, red blood cell count, platelets, and hemoglobin | |
| Total body irradiation 137Cs source (8 Gy–10 Gy) [170] | Histology of the bone marrow, red blood cell count, white blood cell count, lymphocytes, monocytes, and platelets |
5.4. Ferroptosis Has Been Connected to Gastrointestinal Epithelial Injury
5.5. Ferroptosis Influences Hematopoietic Injury after Ionizing Radiation

6. Association between FLASH-RT and Ferroptosis
7. Conclusions and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Baumann, M.; Krause, M.; Overgaard, J.; Debus, J.; Bentzen, S.M.; Daartz, J.; Richter, C.; Zips, D.; Bortfeld, T. Radiation Oncology in the Era of Precision Medicine. Nat. Rev. Cancer 2016, 16, 234–249. [Google Scholar] [CrossRef]
- Abshire, D.; Lang, M.K. The Evolution of Radiation Therapy in Treating Cancer. Semin. Oncol. Nurs. 2018, 34, 151–157. [Google Scholar] [CrossRef]
- Nguyen, H.Q.; To, N.H.; Zadigue, P.; Kerbrat, S.; De La Taille, A.; Le Gouvello, S.; Belkacemi, Y. Ionizing Radiation-Induced Cellular Senescence Promotes Tissue Fibrosis after Radiotherapy. A Review. Crit. Rev. Oncol./Hematol. 2018, 129, 13–26. [Google Scholar] [CrossRef]
- Chen, H.; Han, Z.; Luo, Q.; Wang, Y.; Li, Q.; Zhou, L.; Zuo, H. Radiotherapy Modulates Tumor Cell Fate Decisions: A Review. Radiat. Oncol. 2022, 17, 196. [Google Scholar] [CrossRef]
- Jiao, Y.; Cao, F.; Liu, H. Radiation-Induced Cell Death and Its Mechanisms. Health Phys. 2022, 123, 376–386. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Yagoda, N.; von Rechenberg, M.; Zaganjor, E.; Bauer, A.J.; Yang, W.S.; Fridman, D.J.; Wolpaw, A.J.; Smukste, I.; Peltier, J.M.; Boniface, J.J.; et al. RAS–RAF–MEK-Dependent Oxidative Cell Death Involving Voltage-Dependent Anion Channels. Nature 2007, 447, 865–869. [Google Scholar] [CrossRef]
- Chen, X.; Comish, P.B.; Tang, D.; Kang, R. Characteristics and Biomarkers of Ferroptosis. Front. Cell Dev. Biol. 2021, 9, 637162. [Google Scholar] [CrossRef]
- Dolma, S.; Lessnick, S.L.; Hahn, W.C.; Stockwell, B.R. Identification of Genotype-Selective Antitumor Agents Using Synthetic Lethal Chemical Screening in Engineered Human Tumor Cells. Cancer Cell 2003, 3, 285–296. [Google Scholar] [CrossRef]
- Yang, W.S.; Stockwell, B.R. Synthetic Lethal Screening Identifies Compounds Activating Iron-Dependent, Nonapoptotic Cell Death in Oncogenic-RAS-Harboring Cancer Cells. Chem. Biol. 2008, 15, 234–245. [Google Scholar] [CrossRef]
- Andreani, C.; Bartolacci, C.; Scaglioni, P.P. Ferroptosis: A Specific Vulnerability of RAS-Driven Cancers? Front. Oncol. 2022, 12, 923915. [Google Scholar] [CrossRef]
- Chen, X.; Kang, R.; Kroemer, G.; Tang, D. Broadening Horizons: The Role of Ferroptosis in Cancer. Nat. Rev. Clin. Oncol. 2021, 18, 280–296. [Google Scholar] [CrossRef]
- Lei, G.; Zhuang, L.; Gan, B. Targeting Ferroptosis as a Vulnerability in Cancer. Nat. Rev. Cancer 2022, 22, 381–396. [Google Scholar] [CrossRef]
- Hu, D.; Zhou, Z.; Wang, J.; Zhu, K. Screening of Ferroptosis-Related Genes with Prognostic Effect in Colorectal Cancer by Bioinformatic Analysis. Front. Mol. Biosci. 2022, 9, 979854. [Google Scholar] [CrossRef]
- Liu, C.; Liu, Y.; Yu, Y.; Zhao, Y.; Yu, A. Comprehensive Analysis of Ferroptosis-Related Genes and Prognosis of Cutaneous Melanoma. BMC Med. Genom. 2022, 15, 39. [Google Scholar] [CrossRef]
- Wang, N.; Gu, Y.; Li, L.; Chi, J.; Liu, X.; Xiong, Y.; Jiang, S.; Zhang, W.; Zhong, C. Identification of Novel Prognostic Risk Signature of Breast Cancer Based on Ferroptosis-Related Genes. Sci. Rep. 2022, 12, 13766. [Google Scholar] [CrossRef]
- Li, J.; Cao, F.; Yin, H.; Huang, Z.; Lin, Z.; Mao, N.; Sun, B.; Wang, G. Ferroptosis: Past, Present and Future. Cell Death Dis. 2020, 11, 88. [Google Scholar] [CrossRef]
- Jiang, X.; Stockwell, B.R.; Conrad, M. Ferroptosis: Mechanisms, Biology and Role in Disease. Nat. Rev. Mol. Cell Biol. 2021, 22, 266–282. [Google Scholar] [CrossRef]
- Stockwell, B.R.; Friedmann Angeli, J.P.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascón, S.; Hatzios, S.K.; Kagan, V.E.; et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef]
- Wiernicki, B.; Dubois, H.; Tyurina, Y.Y.; Hassannia, B.; Bayir, H.; Kagan, V.E.; Vandenabeele, P.; Wullaert, A.; Vanden Berghe, T. Excessive Phospholipid Peroxidation Distinguishes Ferroptosis from Other Cell Death Modes Including Pyroptosis. Cell Death Dis. 2020, 11, 922. [Google Scholar] [CrossRef]
- Zou, Y.; Li, H.; Graham, E.T.; Deik, A.A.; Eaton, J.K.; Wang, W.; Sandoval-Gomez, G.; Clish, C.B.; Doench, J.G.; Schreiber, S.L. Cytochrome P450 Oxidoreductase Contributes Tophospholipid Peroxidation in Ferroptosis. Nat. Chem. Biol. 2020, 16, 302–309. [Google Scholar] [CrossRef]
- Shah, R.; Shchepinov, M.S.; Pratt, D.A. Resolving the Role of Lipoxygenases in the Initiation and Execution of Ferroptosis. ACS Cent. Sci. 2018, 4, 387–396. [Google Scholar] [CrossRef]
- Yang, W.S.; Kim, K.J.; Gaschler, M.M.; Patel, M.; Shchepinov, M.S.; Stockwell, B.R. Peroxidation of Polyunsaturated Fatty Acids by Lipoxygenases Drives Ferroptosis. Proc. Natl. Acad. Sci. USA 2016, 113, E4966–E4975. [Google Scholar] [CrossRef]
- Wenzel, S.E.; Tyurina, Y.Y.; Zhao, J.; St. Croix, C.M.; Dar, H.H.; Mao, G.; Tyurin, V.A.; Anthonymuthu, T.S.; Kapralov, A.A.; Amoscato, A.A.; et al. PEBP1 Wardens Ferroptosis by Enabling Lipoxygenase Generation of Lipid Death Signals. Cell 2017, 171, 628–641.e26. [Google Scholar] [CrossRef]
- Wang, L.; Chen, X.; Yan, C. Ferroptosis: An Emerging Therapeutic Opportunity for Cancer. Genes Dis. 2020, 9, 334–346. [Google Scholar] [CrossRef]
- Kagan, V.E.; Mao, G.; Qu, F.; Angeli, J.P.F.; Doll, S.; Croix, C.S.; Dar, H.H.; Liu, B.; Tyurin, V.A.; Ritov, V.B.; et al. Oxidized Arachidonic and Adrenic PEs Navigate Cells to Ferroptosis. Nat. Chem. Biol. 2017, 13, 81–90. [Google Scholar] [CrossRef]
- Dixon, S.J.; Winter, G.E.; Musavi, L.S.; Lee, E.D.; Snijder, B.; Rebsamen, M.; Superti-Furga, G.; Stockwell, B.R. Human Haploid Cell Genetics Reveals Roles for Lipid Metabolism Genes in Nonapoptotic Cell Death. ACS Chem. Biol. 2015, 10, 1604–1609. [Google Scholar] [CrossRef]
- Doll, S.; Proneth, B.; Tyurina, Y.Y.; Panzilius, E.; Kobayashi, S.; Ingold, I.; Irmler, M.; Beckers, J.; Aichler, M.; Walch, A.; et al. ACSL4 Dictates Ferroptosis Sensitivity by Shaping Cellular Lipid Composition. Nat. Chem. Biol. 2017, 13, 91–98. [Google Scholar] [CrossRef]
- Yuan, H.; Li, X.; Zhang, X.; Kang, R.; Tang, D. Identification of ACSL4 as a Biomarker and Contributor of Ferroptosis. Biochem. Biophys. Res. Commun. 2016, 478, 1338–1343. [Google Scholar] [CrossRef]
- Bogdan, A.R.; Miyazawa, M.; Hashimoto, K.; Tsuji, Y. Regulators of Iron Homeostasis: New Players in Metabolism, Cell Death, and Disease. Trends Biochem. Sci. 2016, 41, 274–286. [Google Scholar] [CrossRef]
- Rochette, L.; Dogon, G.; Rigal, E.; Zeller, M.; Cottin, Y.; Vergely, C. Lipid Peroxidation and Iron Metabolism: Two Corner Stones in the Homeostasis Control of Ferroptosis. Int. J. Mol. Sci. 2022, 24, 449. [Google Scholar] [CrossRef]
- Morales, M.; Xue, X. Targeting Iron Metabolism in Cancer Therapy. Theranostics 2021, 11, 8412–8429. [Google Scholar] [CrossRef]
- Chen, X.; Yu, C.; Kang, R.; Tang, D. Iron Metabolism in Ferroptosis. Front. Cell Dev. Biol. 2020, 8, 590226. [Google Scholar] [CrossRef]
- Feng, H.; Schorpp, K.; Jin, J.; Yozwiak, C.E.; Hoffstrom, B.G.; Decker, A.M.; Rajbhandari, P.; Stokes, M.E.; Bender, H.G.; Csuka, J.M.; et al. Transferrin Receptor Is a Specific Ferroptosis Marker. Cell Rep. 2020, 30, 3411–3423.e7. [Google Scholar] [CrossRef]
- Zhang, Z.; Lu, M.; Chen, C.; Tong, X.; Li, Y.; Yang, K.; Lv, H.; Xu, J.; Qin, L. Holo-Lactoferrin: The Link between Ferroptosis and Radiotherapy in Triple-Negative Breast Cancer. Theranostics 2021, 11, 3167–3182. [Google Scholar] [CrossRef]
- Mumbauer, S.; Pascual, J.; Kolotuev, I.; Hamaratoglu, F. Ferritin Heavy Chain Protects the Developing Wing from Reactive Oxygen Species and Ferroptosis. PLoS Genet. 2019, 15, e1008396. [Google Scholar] [CrossRef]
- Asperti, M.; Bellini, S.; Grillo, E.; Gryzik, M.; Cantamessa, L.; Ronca, R.; Maccarinelli, F.; Salvi, A.; De Petro, G.; Arosio, P.; et al. H-Ferritin Suppression and Pronounced Mitochondrial Respiration Make Hepatocellular Carcinoma Cells Sensitive to RSL3-Induced Ferroptosis. Free Radic. Biol. Med. 2021, 169, 294–303. [Google Scholar] [CrossRef]
- Brown, C.W.; Amante, J.J.; Chhoy, P.; Elaimy, A.L.; Liu, H.; Zhu, L.J.; Baer, C.E.; Dixon, S.J.; Mercurio, A.M. Prominin2 Drives Ferroptosis Resistance by Stimulating Iron Export. Dev. Cell 2019, 51, 575–586.e4. [Google Scholar] [CrossRef] [PubMed]
- Hou, W.; Xie, Y.; Song, X.; Sun, X.; Lotze, M.T.; Zeh, H.J.; Kang, R.; Tang, D. Autophagy Promotes Ferroptosis by Degradation of Ferritin. Autophagy 2016, 12, 1425–1428. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Jiang, H.; Van De Gucht, M.; De Ridder, M. Hypoxic Radioresistance: Can ROS Be the Key to Overcome It? Cancers 2019, 11, 112. [Google Scholar] [CrossRef]
- Jiang, H.; Wang, H.; De Ridder, M. Targeting Antioxidant Enzymes as a Radiosensitizing Strategy. Cancer Lett. 2018, 438, 154–164. [Google Scholar] [CrossRef]
- Lee, H.; Zandkarimi, F.; Zhang, Y.; Meena, J.K.; Kim, J.; Zhuang, L.; Tyagi, S.; Ma, L.; Westbrook, T.F.; Steinberg, G.R.; et al. Energy-Stress-Mediated AMPK Activation Inhibits Ferroptosis. Nat. Cell Biol. 2020, 22, 225–234. [Google Scholar] [CrossRef]
- Li, C.; Dong, X.; Du, W.; Shi, X.; Chen, K.; Zhang, W.; Gao, M. LKB1-AMPK Axis Negatively Regulates Ferroptosis by Inhibiting Fatty Acid Synthesis. Signal Transduct. Target. Ther. 2020, 5, 187. [Google Scholar] [CrossRef]
- Gan, B. Mitochondrial Regulation of Ferroptosis. J. Cell Biol. 2021, 220, e202105043. [Google Scholar] [CrossRef]
- Friedmann Angeli, J.P.; Schneider, M.; Proneth, B.; Tyurina, Y.Y.; Tyurin, V.A.; Hammond, V.J.; Herbach, N.; Aichler, M.; Walch, A.; Eggenhofer, E.; et al. Inactivation of the Ferroptosis Regulator Gpx4 Triggers Acute Renal Failure in Mice. Nat. Cell Biol. 2014, 16, 1180–1191. [Google Scholar] [CrossRef]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of Ferroptotic Cancer Cell Death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef]
- Ingold, I.; Berndt, C.; Schmitt, S.; Doll, S.; Poschmann, G.; Buday, K.; Roveri, A.; Peng, X.; Porto Freitas, F.; Seibt, T.; et al. Selenium Utilization by GPX4 Is Required to Prevent Hydroperoxide-Induced Ferroptosis. Cell 2018, 172, 409–422.e21. [Google Scholar] [CrossRef]
- Koppula, P.; Zhuang, L.; Gan, B. Cystine Transporter SLC7A11/xCT in Cancer: Ferroptosis, Nutrient Dependency, and Cancer Therapy. Protein Cell 2021, 12, 599–620. [Google Scholar] [CrossRef]
- Bonifácio, V.D.B.; Pereira, S.A.; Serpa, J.; Vicente, J.B. Cysteine Metabolic Circuitries: Druggable Targets in Cancer. Br. J. Cancer 2021, 124, 862–879. [Google Scholar] [CrossRef]
- Soula, M.; Weber, R.A.; Zilka, O.; Alwaseem, H.; La, K.; Yen, F.; Molina, H.; Garcia-Bermudez, J.; Pratt, D.A.; Birsoy, K. Metabolic Determinants of Cancer Cell Sensitivity to Canonical Ferroptosis Inducers. Nat. Chem. Biol. 2020, 16, 1351–1360. [Google Scholar] [CrossRef] [PubMed]
- Kraft, V.A.N.; Bezjian, C.T.; Pfeiffer, S.; Ringelstetter, L.; Müller, C.; Zandkarimi, F.; Merl-Pham, J.; Bao, X.; Anastasov, N.; Kössl, J.; et al. GTP Cyclohydrolase 1/Tetrahydrobiopterin Counteract Ferroptosis through Lipid Remodeling. ACS Cent. Sci. 2020, 6, 41–53. [Google Scholar] [CrossRef]
- Bersuker, K.; Hendricks, J.M.; Li, Z.; Magtanong, L.; Ford, B.; Tang, P.H.; Roberts, M.A.; Tong, B.; Maimone, T.J.; Zoncu, R.; et al. The CoQ Oxidoreductase FSP1 Acts Parallel to GPX4 to Inhibit Ferroptosis. Nature 2019, 575, 688–692. [Google Scholar] [CrossRef]
- Doll, S.; Freitas, F.P.; Shah, R.; Aldrovandi, M.; da Silva, M.C.; Ingold, I.; Goya Grocin, A.; Xavier da Silva, T.N.; Panzilius, E.; Scheel, C.H.; et al. FSP1 Is a Glutathione-Independent Ferroptosis Suppressor. Nature 2019, 575, 693–698. [Google Scholar] [CrossRef]
- Mao, C.; Liu, X.; Zhang, Y.; Lei, G.; Yan, Y.; Lee, H.; Koppula, P.; Wu, S.; Zhuang, L.; Fang, B.; et al. DHODH-Mediated Ferroptosis Defence Is a Targetable Vulnerability in Cancer. Nature 2021, 593, 586–590. [Google Scholar] [CrossRef]
- Lang, X.; Green, M.D.; Wang, W.; Yu, J.; Choi, J.E.; Jiang, L.; Liao, P.; Zhou, J.; Zhang, Q.; Dow, A.; et al. Radiotherapy and Immunotherapy Promote Tumoral Lipid Oxidation and Ferroptosis via Synergistic Repression of SLC7A11. Cancer Discov. 2019, 9, 1673–1685. [Google Scholar] [CrossRef]
- Ye, L.F.; Chaudhary, K.R.; Zandkarimi, F.; Harken, A.D.; Kinslow, C.J.; Upadhyayula, P.S.; Dovas, A.; Higgins, D.M.; Tan, H.; Zhang, Y.; et al. Radiation-Induced Lipid Peroxidation Triggers Ferroptosis and Synergizes with Ferroptosis Inducers. ACS Chem. Biol. 2020, 15, 469–484. [Google Scholar] [CrossRef]
- Lei, G.; Zhang, Y.; Koppula, P.; Liu, X.; Zhang, J.; Lin, S.H.; Ajani, J.A.; Xiao, Q.; Liao, Z.; Wang, H.; et al. The Role of Ferroptosis in Ionizing Radiation-Induced Cell Death and Tumor Suppression. Cell Res. 2020, 30, 146–162. [Google Scholar] [CrossRef]
- Kerkhove, L.; Geirnaert, F.; Rifi, A.L.; Law, K.L.; Gutiérrez, A.; Oudaert, I.; Corbet, C.; Gevaert, T.; Dufait, I.; De Ridder, M. Repurposing Sulfasalazine as a Radiosensitizer in Hypoxic Human Colorectal Cancer. Cancers 2023, 15, 2363. [Google Scholar] [CrossRef]
- Xie, Y.; Zhu, S.; Song, X.; Sun, X.; Fan, Y.; Liu, J.; Zhong, M.; Yuan, H.; Zhang, L.; Billiar, T.R.; et al. The Tumor Suppressor P53 Limits Ferroptosis by Blocking DPP4 Activity. Cell Rep. 2017, 20, 1692–1704. [Google Scholar] [CrossRef]
- Jiang, L.; Kon, N.; Li, T.; Wang, S.-J.; Su, T.; Hibshoosh, H.; Baer, R.; Gu, W. Ferroptosis as a P53-Mediated Activity during Tumour Suppression. Nature 2015, 520, 57–62. [Google Scholar] [CrossRef]
- Overgaard, J. Hypoxic Radiosensitization: Adored and Ignored. J. Clin. Oncol. 2007, 25, 4066–4074. [Google Scholar] [CrossRef]
- Hall, E.J.; Giaccia, A.J. Radiobiology for the Radiologist, 7th ed.; Wolters Kluwer: Beijing, China, 2010. [Google Scholar]
- Su, J.; Zhao, Q.; Zheng, Z.; Wang, H.; Bian, C.; Meng, L.; Xin, Y.; Jiang, X. Prospective Application of Ferroptosis in Hypoxic Cells for Tumor Radiotherapy. Antioxidants 2022, 11, 921. [Google Scholar] [CrossRef] [PubMed]
- Wicks, E.E.; Semenza, G.L. Hypoxia-Inducible Factors: Cancer Progression and Clinical Translation. J. Clin. Investig. 2022, 132, e159839. [Google Scholar] [CrossRef] [PubMed]
- Fan, Z.; Yang, G.; Zhang, W.; Liu, Q.; Liu, G.; Liu, P.; Xu, L.; Wang, J.; Yan, Z.; Han, H.; et al. Hypoxia Blocks Ferroptosis of Hepatocellular Carcinoma via Suppression of METTL14 Triggered YTHDF2-dependent Silencing of SLC7A11. J. Cell. Mol. Med. 2021, 25, 10197–10212. [Google Scholar] [CrossRef] [PubMed]
- Xiong, J.; Nie, M.; Fu, C.; Chai, X.; Zhang, Y.; He, L.; Sun, S. Hypoxia Enhances HIF1α Transcription Activity by Upregulating KDM4A and Mediating H3K9me3, Thus Inducing Ferroptosis Resistance in Cervical Cancer Cells. Stem Cells Int. 2022, 2022, 1608806. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Tang, H.; Zheng, J.; Yang, K. The PER1/HIF-1alpha Negative Feedback Loop Promotes Ferroptosis and Inhibits Tumor Progression in Oral Squamous Cell Carcinoma. Transl. Oncol. 2022, 18, 101360. [Google Scholar] [CrossRef] [PubMed]
- Fuhrmann, D.C.; Mondorf, A.; Beifuß, J.; Jung, M.; Brüne, B. Hypoxia Inhibits Ferritinophagy, Increases Mitochondrial Ferritin, and Protects from Ferroptosis. Redox Biol. 2020, 36, 101670. [Google Scholar] [CrossRef] [PubMed]
- Ni, S.; Yuan, Y.; Qian, Z.; Zhong, Z.; Lv, T.; Kuang, Y.; Yu, B. Hypoxia Inhibits RANKL-Induced Ferritinophagy and Protects Osteoclasts from Ferroptosis. Free Radic. Biol. Med. 2021, 169, 271–282. [Google Scholar] [CrossRef]
- Wen, K.; Yan, Y.; Shi, J.; Hu, L.; Wang, W.; Liao, H.; Li, H.; Zhu, Y.; Mao, K.; Xiao, Z. Construction and Validation of a Combined Ferroptosis and Hypoxia Prognostic Signature for Hepatocellular Carcinoma. Front. Mol. Biosci. 2021, 8, 809672. [Google Scholar] [CrossRef]
- Yang, M.; Chen, P.; Liu, J.; Zhu, S.; Kroemer, G.; Klionsky, D.J.; Lotze, M.T.; Zeh, H.J.; Kang, R.; Tang, D. Clockophagy Is a Novel Selective Autophagy Process Favoring Ferroptosis. Sci. Adv. 2019, 5, eaaw2238. [Google Scholar] [CrossRef]
- Zou, Y.; Palte, M.J.; Deik, A.A.; Li, H.; Eaton, J.K.; Wang, W.; Tseng, Y.-Y.; Deasy, R.; Kost-Alimova, M.; Dančík, V.; et al. A GPX4-Dependent Cancer Cell State Underlies the Clear-Cell Morphology and Confers Sensitivity to Ferroptosis. Nat. Commun. 2019, 10, 1617. [Google Scholar] [CrossRef] [PubMed]
- Mastrogiannaki, M.; Matak, P.; Keith, B.; Simon, M.C.; Vaulont, S.; Peyssonnaux, C. HIF-2α, but Not HIF-1α, Promotes Iron Absorption in Mice. J. Clin. Investig. 2009, 119, 1159–1166. [Google Scholar] [CrossRef]
- Singhal, R.; Mitta, S.R.; Das, N.K.; Kerk, S.A.; Sajjakulnukit, P.; Solanki, S.; Andren, A.; Kumar, R.; Olive, K.P.; Banerjee, R.; et al. HIF-2α Activation Potentiates Oxidative Cell Death in Colorectal Cancers by Increasing Cellular Iron. J. Clin. Investig. 2021, 131, e143691. [Google Scholar] [CrossRef] [PubMed]
- Shen, D.; Luo, J.; Chen, L.; Ma, W.; Mao, X.; Zhang, Y.; Zheng, J.; Wang, Y.; Wan, J.; Wang, S.; et al. PARPi Treatment Enhances Radiotherapy-Induced Ferroptosis and Antitumor Immune Responses via the cGAS Signaling Pathway in Colorectal Cancer. Cancer Lett. 2022, 550, 215919. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Cai, H.; Xu, C.; Zhai, H.; Lux, F.; Xie, Y.; Feng, L.; Du, L.; Liu, Y.; Sun, X.; et al. AGuIX Nanoparticles Enhance Ionizing Radiation-Induced Ferroptosis on Tumor Cells by Targeting the NRF2-GPX4 Signaling Pathway. J. Nanobiotechnol. 2022, 20, 449. [Google Scholar] [CrossRef]
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, J.; McNamee, C.; et al. Drug Repurposing: Progress, Challenges and Recommendations. Nat. Rev. Drug Discov. 2019, 18, 41–58. [Google Scholar] [CrossRef] [PubMed]
- Singhal, S.; Mehta, J.; Desikan, R.; Ayers, D.; Roberson, P.; Eddlemon, P.; Munshi, N.; Anaissie, E.; Wilson, C.; Dhodapkar, M.; et al. Antitumor Activity of Thalidomide in Refractory Multiple Myeloma. N. Engl. J. Med. 1999, 341, 1565–1571. [Google Scholar] [CrossRef]
- Sun, S.; Shen, J.; Jiang, J.; Wang, F.; Min, J. Targeting Ferroptosis Opens New Avenues for the Development of Novel Therapeutics. Signal Transduct. Target. Ther. 2023, 8, 372. [Google Scholar] [CrossRef]
- Sleire, L.; Skeie, B.S.; Netland, I.A.; Førde, H.E.; Dodoo, E.; Selheim, F.; Leiss, L.; Heggdal, J.I.; Pedersen, P.-H.; Wang, J.; et al. Drug Repurposing: Sulfasalazine Sensitizes Gliomas to Gamma Knife Radiosurgery by Blocking Cystine Uptake through System Xc−, Leading to Glutathione Depletion. Oncogene 2015, 34, 5951–5959. [Google Scholar] [CrossRef]
- Nagane, M.; Kanai, E.; Shibata, Y.; Shimizu, T.; Yoshioka, C.; Maruo, T.; Yamashita, T. Sulfasalazine, an Inhibitor of the Cystine-Glutamate Antiporter, Reduces DNA Damage Repair and Enhances Radiosensitivity in Murine B16F10 Melanoma. PLoS ONE 2018, 13, e0195151. [Google Scholar] [CrossRef]
- Takeuchi, S.; Wada, K.; Nagatani, K.; Otani, N.; Osada, H.; Nawashiro, H. Sulfasalazine and Temozolomide with Radiation Therapy for Newly Diagnosed Glioblastoma. Neurol. India 2014, 62, 42. [Google Scholar] [CrossRef]
- Nie, D.; Chen, C.; Li, Y.; Zeng, C. Disulfiram, an Aldehyde Dehydrogenase Inhibitor, Works as a Potent Drug against Sepsis and Cancer via NETosis, Pyroptosis, Apoptosis, Ferroptosis, and Cuproptosis. Blood Sci. 2022, 4, 152–154. [Google Scholar] [CrossRef]
- Xu, Y.; Lu, L.; Luo, J.; Wang, L.; Zhang, Q.; Cao, J.; Jiao, Y. Disulfiram Alone Functions as a Radiosensitizer for Pancreatic Cancer Both In Vitro and In Vivo. Front. Oncol. 2021, 11, 683695. [Google Scholar] [CrossRef]
- Lee, Y.E.; Choi, S.A.; Kwack, P.A.; Kim, H.J.; Kim, I.H.; Wang, K.-C.; Phi, J.H.; Lee, J.Y.; Chong, S.; Park, S.-H.; et al. Repositioning Disulfiram as a Radiosensitizer against Atypical Teratoid/Rhabdoid Tumor. Neuro-Oncol. 2017, 19, 1079–1087. [Google Scholar] [CrossRef]
- Wang, R.; Shen, J.; Yan, H.; Gao, X.; Dong, T.; Wang, P.; Zhou, J. The Evolving Role of Disulfiram in Radiobiology and the Treatment of Breast Cancer. OncoTargets Ther. 2020, 13, 10441–10446. [Google Scholar] [CrossRef]
- Rae, C.; Tesson, M.; Babich, J.W.; Boyd, M.; Sorensen, A.; Mairs, R.J. The Role of Copper in Disulfiram-Induced Toxicity and Radiosensitization of Cancer Cells. J. Nucl. Med. 2013, 54, 953–960. [Google Scholar] [CrossRef] [PubMed]
- Qiu, C.; Zhang, X.; Huang, B.; Wang, S.; Zhou, W.; Li, C.; Li, X.; Wang, J.; Yang, N. Disulfiram, a Ferroptosis Inducer, Triggers Lysosomal Membrane Permeabilization by Up-Regulating ROS in Glioblastoma. OncoTargets Ther. 2020, 13, 10631–10640. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, S.M.; Adnane, L.; Newell, P.; Villanueva, A.; Llovet, J.M.; Lynch, M. Preclinical Overview of Sorafenib, a Multikinase Inhibitor That Targets Both Raf and VEGF and PDGF Receptor Tyrosine Kinase Signaling. Mol. Cancer Ther. 2008, 7, 3129–3140. [Google Scholar] [CrossRef] [PubMed]
- Louandre, C.; Ezzoukhry, Z.; Godin, C.; Barbare, J.-C.; Mazière, J.-C.; Chauffert, B.; Galmiche, A. Iron-Dependent Cell Death of Hepatocellular Carcinoma Cells Exposed to Sorafenib: Iron-Dependent Cytotoxicity of Sorafenib. Int. J. Cancer 2013, 133, 1732–1742. [Google Scholar] [CrossRef]
- Dixon, S.J.; Patel, D.N.; Welsch, M.; Skouta, R.; Lee, E.D.; Hayano, M.; Thomas, A.G.; Gleason, C.E.; Tatonetti, N.P.; Slusher, B.S.; et al. Pharmacological Inhibition of Cystine–Glutamate Exchange Induces Endoplasmic Reticulum Stress and Ferroptosis. eLife 2014, 3, e02523. [Google Scholar] [CrossRef]
- Jeong, Y.K.; Kim, M.-S.; Lee, J.Y.; Kim, E.H.; Kim, W.; Ha, H.; Jeong, J.-H. Sorafenib Acts Synergistically in Combination with Radiotherapy without Causing Intestinal Damage in Colorectal Cancer. Tumori 2013, 99, 176–182. [Google Scholar] [CrossRef]
- Dai, X.-F.; Ding, J.; Zhang, R.-G.; Ren, J.-H.; Ma, C.-M.C.; Wu, G. Radiosensitivity Enhancement of Human Hepatocellular Carcinoma Cell Line SMMC-7721 by Sorafenib through the MEK/ERK Signal Pathway. Int. J. Radiat. Biol. 2013, 89, 724–731. [Google Scholar] [CrossRef]
- Laban, S.; Steinmeister, L.; Gleißner, L.; Grob, T.J.; Grénman, R.; Petersen, C.; Gal, A.; Knecht, R.; Dikomey, E.; Kriegs, M. Sorafenib Sensitizes Head and Neck Squamous Cell Carcinoma Cells to Ionizing Radiation. Radiother. Oncol. 2013, 109, 286–292. [Google Scholar] [CrossRef]
- Kim, E.H.; Kim, M.-S.; Jung, W.-G. The Mechanisms Responsible for the Radiosensitizing Effects of Sorafenib on Colon Cancer Cells. Oncol. Rep. 2014, 32, 2421–2428. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Bouzakoura, S.; de Mey, S.; Jiang, H.; Law, K.; Dufait, I.; Corbet, C.; Verovski, V.; Gevaert, T.; Feron, O.; et al. Auranofin Radiosensitizes Tumor Cells through Targeting Thioredoxin Reductase and Resulting Overproduction of Reactive Oxygen Species. Oncotarget 2017, 8, 35728–35742. [Google Scholar] [CrossRef]
- Rodman, S.N.; Spence, J.M.; Ronnfeldt, T.J.; Zhu, Y.; Solst, S.R.; O’Neill, R.A.; Allen, B.G.; Guan, X.; Spitz, D.R.; Fath, M.A. Enhancement of Radiation Response in Breast Cancer Stem Cells by Inhibition of Thioredoxin- and Glutathione-Dependent Metabolism. Radiat. Res. 2016, 186, 385. [Google Scholar] [CrossRef]
- Van Loenhout, J.; Freire Boullosa, L.; Quatannens, D.; De Waele, J.; Merlin, C.; Lambrechts, H.; Lau, H.W.; Hermans, C.; Lin, A.; Lardon, F.; et al. Auranofin and Cold Atmospheric Plasma Synergize to Trigger Distinct Cell Death Mechanisms and Immunogenic Responses in Glioblastoma. Cells 2021, 10, 2936. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, J.M.; Jandrey, E.H.F.; Koyama, F.C.; Leite, K.R.M.; Camargo, A.A.; Costa, É.T.; Perez, R.O.; Asprino, P.F. Metformin as an Alternative Radiosensitizing Agent to 5-Fluorouracil During Neoadjuvant Treatment for Rectal Cancer. Dis. Colon. Rectum 2020, 63, 918–926. [Google Scholar] [CrossRef]
- Song, C.W.; Lee, H.; Dings, R.P.M.; Williams, B.; Powers, J.; Santos, T.D.; Choi, B.-H.; Park, H.J. Metformin Kills and Radiosensitizes Cancer Cells and Preferentially Kills Cancer Stem Cells. Sci. Rep. 2012, 2, 362. [Google Scholar] [CrossRef]
- 102 De Mey, S.; Jiang, H.; Corbet, C.; Wang, H.; Dufait, I.; Law, K.; Bastien, E.; Verovski, V.; Gevaert, T.; Feron, O.; et al. Antidiabetic Biguanides radiosensitize hypoxic colorectal cancer cells through a decrease in oxygen consumption. Front Pharmacol. 2018, 9, 1073. [Google Scholar] [CrossRef]
- Tojo, M.; Miyato, H.; Koinuma, K.; Horie, H.; Tsukui, H.; Kimura, Y.; Kaneko, Y.; Ohzawa, H.; Yamaguchi, H.; Yoshimura, K.; et al. Metformin Combined with Local Irradiation Provokes Abscopal Effects in a Murine Rectal Cancer Model. Sci. Rep. 2022, 12, 7290. [Google Scholar] [CrossRef]
- Fei, Z.; Gu, W.; Xie, R.; Su, H.; Jiang, Y. Artesunate Enhances Radiosensitivity of Esophageal Cancer Cells by Inhibiting the Repair of DNA Damage. J. Pharmacol. Sci. 2018, 138, 131–137. [Google Scholar] [CrossRef]
- Reichert, S.; Reinboldt, V.; Hehlgans, S.; Efferth, T.; Rödel, C.; Rödel, F. A Radiosensitizing Effect of Artesunate in Glioblastoma Cells Is Associated with a Diminished Expression of the Inhibitor of Apoptosis Protein Survivin. Radiother. Oncol. 2012, 103, 394–401. [Google Scholar] [CrossRef]
- Luo, J.; Zhu, W.; Tang, Y.; Cao, H.; Zhou, Y.; Ji, R.; Zhou, X.; Lu, Z.; Yang, H.; Zhang, S.; et al. Artemisinin Derivative Artesunate Induces Radiosensitivity in Cervical Cancer Cells in Vitro and in Vivo. Radiat. Oncol. 2014, 9, 84. [Google Scholar] [CrossRef]
- Jiang, W.; Hu, J.-W.; He, X.-R.; Jin, W.-L.; He, X.-Y. Statins: A Repurposed Drug to Fight Cancer. J. Exp. Clin. Cancer Res. 2021, 40, 241. [Google Scholar] [CrossRef]
- Lacerda, L.; Reddy, J.P.; Liu, D.; Larson, R.; Li, L.; Masuda, H.; Brewer, T.; Debeb, B.G.; Xu, W.; Hortobágyi, G.N.; et al. Simvastatin Radiosensitizes Differentiated and Stem-Like Breast Cancer Cell Lines and Is Associated With Improved Local Control in Inflammatory Breast Cancer Patients Treated With Postmastectomy Radiation. Stem Cells Transl. Med. 2014, 3, 849–856. [Google Scholar] [CrossRef]
- Gutt, R.; Tonlaar, N.; Kunnavakkam, R.; Karrison, T.; Weichselbaum, R.R.; Liauw, S.L. Statin Use and Risk of Prostate Cancer Recurrence in Men Treated With Radiation Therapy. J. Clin. Oncol. 2010, 28, 2653–2659. [Google Scholar] [CrossRef] [PubMed]
- Mace, A.G.; Gantt, G.A.; Skacel, M.; Pai, R.; Hammel, J.P.; Kalady, M.F. Statin Therapy Is Associated With Improved Pathologic Response to Neoadjuvant Chemoradiation in Rectal Cancer. Dis. Colon. Rectum 2013, 56, 1217–1227. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-A.; Shih, H.-W.; Lin, Y.-C.; Hsu, H.-Y.; Wu, T.-F.; Tsai, C.-H.; Wu, C.-L.; Wu, H.-Y.; Hsieh, J.-T.; Tang, C.-H.; et al. Simvastatin Sensitizes Radioresistant Prostate Cancer Cells by Compromising DNA Double-Strand Break Repair. Front. Pharmacol. 2018, 9, 600. [Google Scholar] [CrossRef] [PubMed]
- Efimova, E.V.; Ricco, N.; Labay, E.; Mauceri, H.J.; Flor, A.C.; Ramamurthy, A.; Sutton, H.G.; Weichselbaum, R.R.; Kron, S.J. HMG-CoA Reductase Inhibition Delays DNA Repair and Promotes Senescence After Tumor Irradiation. Mol. Cancer Ther. 2018, 17, 407–418. [Google Scholar] [CrossRef] [PubMed]
- d’Hose, D.; Mignion, L.; Hamelin, L.; Sonveaux, P.; Jordan, B.F.; Gallez, B. Statins Alleviate Tumor Hypoxia in Prostate Cancer Models by Decreasing Oxygen Consumption: An Opportunity for Radiosensitization? Biomolecules 2022, 12, 1418. [Google Scholar] [CrossRef]
- Li, G.; Tian, Y.; Zhu, W.-G. The Roles of Histone Deacetylases and Their Inhibitors in Cancer Therapy. Front. Cell Dev. Biol. 2020, 8, 576946. [Google Scholar] [CrossRef]
- Zhang, T.; Sun, B.; Zhong, C.; Xu, K.; Wang, Z.; Hofman, P.; Nagano, T.; Legras, A.; Breadner, D.; Ricciuti, B.; et al. Targeting Histone Deacetylase Enhances the Therapeutic Effect of Erastin-Induced Ferroptosis in EGFR-Activating Mutant Lung Adenocarcinoma. Transl. Lung Cancer Res. 2021, 10, 1857–1872. [Google Scholar] [CrossRef] [PubMed]
- Saelen, M.G.; Ree, A.H.; Kristian, A.; Fleten, K.G.; Furre, T.; Hektoen, H.H.; Flatmark, K. Radiosensitization by the Histone Deacetylase Inhibitor Vorinostat under Hypoxia and with Capecitabine in Experimental Colorectal Carcinoma. Radiat. Oncol. 2012, 7, 165. [Google Scholar] [CrossRef]
- Zhu, Z.; Wu, M.; Sun, J.; Huangfu, Z.; Yin, L.; Yong, W.; Sun, J.; Wang, G.; Meng, F.; Zhong, Z. Redox-Sensitive Iodinated Polymersomes Carrying Histone Deacetylase Inhibitor as a Dual-Functional Nano-Radiosensitizer for Enhanced Radiotherapy of Breast Cancer. Drug Deliv. 2021, 28, 2301–2309. [Google Scholar] [CrossRef] [PubMed]
- Gerelchuluun, A.; Maeda, J.; Manabe, E.; Brents, C.; Sakae, T.; Fujimori, A.; Chen, D.; Tsuboi, K.; Kato, T. Histone Deacetylase Inhibitor Induced Radiation Sensitization Effects on Human Cancer Cells after Photon and Hadron Radiation Exposure. Int. J. Mech. Sci. 2018, 19, 496. [Google Scholar] [CrossRef]
- Arora, S.; Balasubramaniam, S.; Zhang, H.; Berman, T.; Narayan, P.; Suzman, D.; Bloomquist, E.; Tang, S.; Gong, Y.; Sridhara, R.; et al. FDA Approval Summary: Olaparib Monotherapy or in Combination with Bevacizumab for the Maintenance Treatment of Patients with Advanced Ovarian Cancer. Oncologist 2021, 26, e164–e172. [Google Scholar] [CrossRef]
- Hong, T.; Lei, G.; Chen, X.; Li, H.; Zhang, X.; Wu, N.; Zhao, Y.; Zhang, Y.; Wang, J. PARP Inhibition Promotes Ferroptosis via Repressing SLC7A11 and Synergizes with Ferroptosis Inducers in BRCA-Proficient Ovarian Cancer. Redox Biol. 2021, 42, 101928. [Google Scholar] [CrossRef]
- Tang, S.; Shen, Y.; Wei, X.; Shen, Z.; Lu, W.; Xu, J. Olaparib Synergizes with Arsenic Trioxide by Promoting Apoptosis and Ferroptosis in Platinum-Resistant Ovarian Cancer. Cell Death Dis. 2022, 13, 826. [Google Scholar] [CrossRef]
- Nile, D.L.; Rae, C.; Hyndman, I.J.; Gaze, M.N.; Mairs, R.J. An Evaluation in Vitro of PARP-1 Inhibitors, Rucaparib and Olaparib, as Radiosensitisers for the Treatment of Neuroblastoma. BMC Cancer 2016, 16, 621. [Google Scholar] [CrossRef]
- Kageyama, S.; Junyan, D.; Hojo, H.; Motegi, A.; Nakamura, M.; Tsuchihara, K.; Akimoto, T. PARP Inhibitor Olaparib Sensitizes Esophageal Carcinoma Cells to Fractionated Proton Irradiation. J. Radiat. Res. 2020, 61, 177–186. [Google Scholar] [CrossRef]
- Tian, S.; Lou, L.; Tian, M.; Lu, G.; Tian, J.; Chen, X. MAPK4 Deletion Enhances Radiation Effects and Triggers Synergistic Lethality with Simultaneous PARP1 Inhibition in Cervical Cancer. J. Exp. Clin. Cancer Res. 2020, 39, 143. [Google Scholar] [CrossRef]
- Qin, C.; Ji, Z.; Zhai, E.; Xu, K.; Zhang, Y.; Li, Q.; Jing, H.; Wang, X.; Song, X. PARP Inhibitor Olaparib Enhances the Efficacy of Radiotherapy on XRCC2-Deficient Colorectal Cancer Cells. Cell Death Dis. 2022, 13, 505. [Google Scholar] [CrossRef] [PubMed]
- Barcellini, A.; Loap, P.; Murata, K.; Villa, R.; Kirova, Y.; Okonogi, N.; Orlandi, E. PARP Inhibitors in Combination with Radiotherapy: To Do or Not to Do? Cancers 2021, 13, 5380. [Google Scholar] [CrossRef]
- Thomson, R.J.; Moshirfar, M.; Ronquillo, Y. Tyrosine Kinase Inhibitors. In StatPearls (Internet); StatPearlsPublisher: St. Petersburg, FL, USA, 2023. [Google Scholar]
- Gutierrez, C.; Schiff, R. HER2: Biology, Detection, and Clinical Implications. Arch. Pathol. Lab. Med. 2011, 135, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Nagpal, A.; Redvers, R.P.; Ling, X.; Ayton, S.; Fuentes, M.; Tavancheh, E.; Diala, I.; Lalani, A.; Loi, S.; David, S.; et al. Neoadjuvant Neratinib Promotes Ferroptosis and Inhibits Brain Metastasis in a Novel Syngeneic Model of Spontaneous HER2+ve Breast Cancer Metastasis. Breast Cancer Res. 2019, 21, 94. [Google Scholar] [CrossRef]
- Ma, S.; Henson, E.S.; Chen, Y.; Gibson, S.B. Ferroptosis Is Induced Following Siramesine and Lapatinib Treatment of Breast Cancer Cells. Cell Death Dis. 2016, 7, e2307. [Google Scholar] [CrossRef] [PubMed]
- Villalpando-Rodriguez, G.E.; Blankstein, A.R.; Konzelman, C.; Gibson, S.B. Lysosomal Destabilizing Drug Siramesine and the Dual Tyrosine Kinase Inhibitor Lapatinib Induce a Synergistic Ferroptosis through Reduced Heme Oxygenase-1 (HO-1) Levels. Oxidative Med. Cell. Longev. 2019, 2019, 9561281. [Google Scholar] [CrossRef]
- Sambade, M.J.; Kimple, R.J.; Camp, J.T.; Peters, E.; Livasy, C.A.; Sartor, C.I.; Shields, J.M. Lapatinib in Combination With Radiation Diminishes Tumor Regrowth in HER2+ and Basal-Like/EGFR+ Breast Tumor Xenografts. Int. J. Radiat. Oncol. Biol. Phys. 2010, 77, 575–581. [Google Scholar] [CrossRef][Green Version]
- Yu, T.; Cho, B.J.; Choi, E.J.; Park, J.M.; Kim, D.H.; Kim, I.A. Radiosensitizing Effect of Lapatinib in Human Epidermal Growth Factor Receptor 2-Positive Breast Cancer Cells. Oncotarget 2016, 7, 79089–79100. [Google Scholar] [CrossRef] [PubMed]
- Schneider, S.; Thurnher, D.; Kadletz, L.; Seemann, R.; Brunner, M.; Kotowski, U.; Schmid, R.; Lill, C.; Heiduschka, G. Effects of Neratinib and Combination with Irradiation and Chemotherapy in Head and Neck Cancer Cells. Oral Dis. 2016, 22, 797–804. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Yang, C.; Jian, L.; Guo, S.; Chen, R.; Li, K.; Qu, F.; Tao, K.; Fu, Y.; Luo, F.; et al. Sulfasalazine-induced Ferroptosis in Breast Cancer Cells Is Reduced by the Inhibitory Effect of Estrogen Receptor on the Transferrin Receptor. Oncol. Rep. 2019, 42, 826–838. [Google Scholar] [CrossRef] [PubMed]
- Ma, M.; Chen, G.; Wang, P.; Lu, W.; Zhu, C.; Song, M.; Yang, J.; Wen, S.; Xu, R.; Hu, Y.; et al. Xc− Inhibitor Sulfasalazine Sensitizes Colorectal Cancer to Cisplatin by a GSH-Dependent Mechanism. Cancer Lett. 2015, 368, 88–96. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Guo, C.; Gao, T.; Ma, D.; Su, X.; Pang, Q.; Zhang, R. Hypoxia Enhances Glioma Resistance to Sulfasalazine-Induced Ferroptosis by Upregulating SLC7A11 via PI3K/AKT/HIF-1α Axis. Oxidative Med. Cell. Longev. 2022, 2022, 7862430. [Google Scholar] [CrossRef] [PubMed]
- Ren, X.; Li, Y.; Zhou, Y.; Hu, W.; Yang, C.; Jing, Q.; Zhou, C.; Wang, X.; Hu, J.; Wang, L.; et al. Overcoming the Compensatory Elevation of NRF2 Renders Hepatocellular Carcinoma Cells More Vulnerable to Disulfiram/Copper-Induced Ferroptosis. Redox Biol. 2021, 46, 102122. [Google Scholar] [CrossRef]
- Li, Y.; Chen, F.; Chen, J.; Chan, S.; He, Y.; Liu, W.; Zhang, G. Disulfiram/Copper Induces Antitumor Activity against Both Nasopharyngeal Cancer Cells and Cancer-Associated Fibroblasts through ROS/MAPK and Ferroptosis Pathways. Cancers 2020, 12, 138. [Google Scholar] [CrossRef]
- Lachaier, E.; Louandre, C.; Godin, C.; Saidak, Z.; Baert, M.; Diouf, M.; Chauffert, B.; Galmiche, A. Sorafenib Induces Ferroptosis in Human Cancer Cell Lines Originating from Different Solid Tumors. Anticancer. Res. 2014, 34, 6417–6422. [Google Scholar]
- Yang, Z.; Huang, S.; Liu, Y.; Chang, X.; Liang, Y.; Li, X.; Xu, Z.; Wang, S.; Lu, Y.; Liu, Y.; et al. Biotin-Targeted Au(I) Radiosensitizer for Cancer Synergistic Therapy by Intervening with Redox Homeostasis and Inducing Ferroptosis. J. Med. Chem. 2022, 65, 8401–8415. [Google Scholar] [CrossRef]
- Yang, L.; Wang, H.; Yang, X.; Wu, Q.; An, P.; Jin, X.; Liu, W.; Huang, X.; Li, Y.; Yan, S.; et al. Auranofin Mitigates Systemic Iron Overload and Induces Ferroptosis via Distinct Mechanisms. Signal Transduct. Target. Ther. 2020, 5, 138. [Google Scholar] [CrossRef]
- Hou, Y.; Cai, S.; Yu, S.; Lin, H. Metformin Induces Ferroptosis by Targeting miR-324-3p/GPX4 Axis in Breast Cancer. Acta Biochim. Biophys. Sin. 2021, 53, 333–341. [Google Scholar] [CrossRef]
- Yang, J.; Zhou, Y.; Xie, S.; Wang, J.; Li, Z.; Chen, L.; Mao, M.; Chen, C.; Huang, A.; Chen, Y.; et al. Metformin Induces Ferroptosis by Inhibiting UFMylation of SLC7A11 in Breast Cancer. J. Exp. Clin. Cancer Res. 2021, 40, 206. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Qin, C.; Zhou, Y.; Chen, Y.; Mao, M.; Yang, J. Metformin May Induce Ferroptosis by Inhibiting Autophagy via lncRNA H19 in Breast Cancer. FEBS Open Bio. 2022, 12, 146–153. [Google Scholar] [CrossRef]
- Song, Q.; Peng, S.; Che, F.; Zhu, X. Artesunate Induces Ferroptosis via Modulation of P38 and ERK Signaling Pathway in Glioblastoma Cells. J. Pharmacol. Sci. 2022, 148, 300–306. [Google Scholar] [CrossRef]
- Wang, N.; Zeng, G.-Z.; Yin, J.-L.; Bian, Z.-X. Artesunate Activates the ATF4-CHOP-CHAC1 Pathway and Affects Ferroptosis in Burkitt’s Lymphoma. Biochem. Biophys. Res. Commun. 2019, 519, 533–539. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wang, F.; Wu, P.; Gong, S.; Gao, J.; Tao, H.; Shen, Q.; Wang, S.; Zhou, Z.; Jia, Y. Artesunate Induces Apoptosis, Autophagy and Ferroptosis in Diffuse Large B Cell Lymphoma Cells by Impairing STAT3 Signaling. Cell. Signal. 2021, 88, 110167. [Google Scholar] [CrossRef]
- Li, Z.; Dai, H.; Huang, X.; Feng, J.; Deng, J.; Wang, Z.; Yang, X.; Liu, Y.; Wu, Y.; Chen, P.; et al. Artesunate Synergizes with Sorafenib to Induce Ferroptosis in Hepatocellular Carcinoma. Acta Pharmacol. Sin. 2021, 42, 301–310. [Google Scholar] [CrossRef]
- Yao, X.; Xie, R.; Cao, Y.; Tang, J.; Men, Y.; Peng, H.; Yang, W. Simvastatin Induced Ferroptosis for Triple-Negative Breast Cancer Therapy. J. Nanobiotechnol. 2021, 19, 311. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Qu, H.; Chen, Y.; Luo, X.; Chen, C.; Xiao, B.; Ding, X.; Zhao, P.; Lu, Y.; Chen, A.F.; et al. Atorvastatin Induces Mitochondria-Dependent Ferroptosis via the Modulation of Nrf2-xCT/GPx4 Axis. Front. Cell Dev. Biol. 2022, 10, 806081. [Google Scholar] [CrossRef]
- Gosselin, T.K.; Mautner, B.; Wilkinson, K. Amifostine as a Radioprotectant. Clin. J. Oncol. Nurs. 2002, 6, 175–176. [Google Scholar] [CrossRef]
- Montay-Gruel, P.; Meziani, L.; Yakkala, C.; Vozenin, M.-C. Expanding the Therapeutic Index of Radiation Therapy by Normal Tissue Protection. Br. J. Radiol. 2018, 92, 20180008. [Google Scholar] [CrossRef]
- Barnett, G.C.; West, C.M.L.; Dunning, A.M.; Elliott, R.M.; Coles, C.E.; Pharoah, P.D.P.; Burnet, N.G. Normal Tissue Reactions to Radiotherapy: Towards Tailoring Treatment Dose by Genotype. Nat. Rev. Cancer 2009, 9, 134–142. [Google Scholar] [CrossRef]
- Straub, J.M.; New, J.; Hamilton, C.D.; Lominska, C.; Shnayder, Y.; Thomas, S.M. Radiation-Induced Fibrosis: Mechanisms and Implications for Therapy. J. Cancer Res. Clin. Oncol. 2015, 141, 1985–1994. [Google Scholar] [CrossRef]
- Linkermann, A.; Stockwell, B.R.; Krautwald, S.; Anders, H.-J. Regulated Cell Death and Inflammation: An Auto-Amplification Loop Causes Organ Failure. Nat. Rev. Immunol. 2014, 14, 759–767. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Tan, Y.; Wang, R.; Li, X. Role of Ferroptosis in Fibrotic Diseases. J. Inflamm. Res. 2022, 15, 3689–3708. [Google Scholar] [CrossRef]
- Xu, C.; Liu, Z.; Xiao, J. Ferroptosis: A Double-Edged Sword in Gastrointestinal Disease. Int. J. Mol. Sci. 2021, 22, 12403. [Google Scholar] [CrossRef]
- Du, X.; Dong, R.; Wu, Y.; Ni, B. Physiological Effects of Ferroptosis on Organ Fibrosis. Oxidative Med. Cell. Longev. 2022, 2022, 5295434. [Google Scholar] [CrossRef]
- Zhang, J.; Li, K.; Zhang, Q.; Zhu, Z.; Huang, G.; Tian, H. Polycysteine as a New Type of Radio-Protector Ameliorated Tissue Injury through Inhibiting Ferroptosis in Mice. Cell Death Dis. 2021, 12, 195. [Google Scholar] [CrossRef] [PubMed]
- Thermozier, S.; Hou, W.; Zhang, X.; Shields, D.; Fisher, R.; Bayir, H.; Kagan, V.; Yu, J.; Liu, B.; Bahar, I.; et al. Anti-Ferroptosis Drug Enhances Total-Body Irradiation Mitigation by Drugs That Block Apoptosis and Necroptosis. Radiat. Res. 2020, 193, 435. [Google Scholar] [CrossRef] [PubMed]
- Xue, J.; Yu, C.; Sheng, W.; Zhu, W.; Luo, J.; Zhang, Q.; Yang, H.; Cao, H.; Wang, W.; Zhou, J.; et al. The Nrf2/GCH1/BH4 Axis Ameliorates Radiation-Induced Skin Injury by Modulating the ROS Cascade. J. Investig. Dermatol. 2017, 137, 2059–2068. [Google Scholar] [CrossRef]
- Feng, Z.; Qin, Y.; Huo, F.; Jian, Z.; Li, X.; Geng, J.; Li, Y.; Wu, J. NMN Recruits GSH to Enhance GPX4-Mediated Ferroptosis Defense in UV Irradiation Induced Skin Injury. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2022, 1868, 166287. [Google Scholar] [CrossRef]
- Li, X.; Duan, L.; Yuan, S.; Zhuang, X.; Qiao, T.; He, J. Ferroptosis Inhibitor Alleviates Radiation-Induced Lung Fibrosis (RILF) via down-Regulation of TGF-Β1. J. Inflamm. 2019, 16, 11. [Google Scholar] [CrossRef]
- Li, X.; Zhuang, X.; Qiao, T. Role of Ferroptosis in the Process of Acute Radiation-Induced Lung Injury in Mice. Biochem. Biophys. Res. Commun. 2019, 519, 240–245. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Wu, D.; Deng, S.; Li, J.; Zhang, F.; Zou, Y.; Zhang, T.; Xu, Y. NVP-AUY922 Alleviates Radiation-Induced Lung Injury via Inhibition of Autophagy-Dependent Ferroptosis. Cell Death Discov. 2022, 8, 86. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.-W.; Zhang, H.; Huang, J.-Q.; Wang, S.-N.; Lu, Y.; Cheng, B.; Dong, S.-H.; Wang, Y.-Y.; Li, F.-S.; Li, Y.-W. PIEZO1 Ion Channel Mediates Ionizing Radiation-Induced Pulmonary Endothelial Cell Ferroptosis via Ca2+/Calpain/VE-Cadherin Signaling. Front. Mol. Biosci. 2021, 8, 725274. [Google Scholar] [CrossRef]
- Xie, L.-W.; Cai, S.; Zhao, T.-S.; Li, M.; Tian, Y. Green Tea Derivative (−)-Epigallocatechin-3-Gallate (EGCG) Confers Protection against Ionizing Radiation-Induced Intestinal Epithelial Cell Death Both in Vitro and in Vivo. Free Radic. Biol. Med. 2020, 161, 175–186. [Google Scholar] [CrossRef]
- Dar, H.H.; Epperly, M.W.; Tyurin, V.A.; Amoscato, A.A.; Anthonymuthu, T.S.; Souryavong, A.B.; Kapralov, A.A.; Shurin, G.V.; Samovich, S.N.; St. Croix, C.M.; et al. P. aeruginosa Augments Irradiation Injury via 15-Lipoxygenase–Catalyzed Generation of 15-HpETE-PE and Induction of Theft-Ferroptosis. JCI Insight 2022, 7, e156013. [Google Scholar] [CrossRef]
- Zhang, X.; Xing, X.; Liu, H.; Feng, J.; Tian, M.; Chang, S.; Liu, P.; Zhang, H. Ionizing Radiation Induces Ferroptosis in Granulocyte-Macrophage Hematopoietic Progenitor Cells of Murine Bone Marrow. Int. J. Radiat. Biol. 2020, 96, 584–595. [Google Scholar] [CrossRef]
- Zhang, X.; Tian, M.; Li, X.; Zheng, C.; Wang, A.; Feng, J.; Hu, X.; Chang, S.; Zhang, H. Hematopoietic Protection and Mechanisms of Ferrostatin-1 on Hematopoietic Acute Radiation Syndrome of Mice. Int. J. Radiat. Biol. 2021, 97, 464–473. [Google Scholar] [CrossRef]
- Vilaplana-Lopera, N.; Abu-Halawa, A.; Walker, E.; Kim, J.; Moon, E.J. Ferroptosis, a Key to Unravel the Enigma of the FLASH Effect? Br. J. Radiol. 2022, 95, 20220825. [Google Scholar] [CrossRef]
- Kale, R.K.; Sitasawad, S.L. Radiation Induced Lipid Peroxidation in Liposomes. Int. J. Radiat. Appl. Instrumentation. Part C. Radiat. Phys. Chem. 1990, 36, 361–364. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kerkhove, L.; Geirnaert, F.; Dufait, I.; De Ridder, M. Ferroptosis: Frenemy of Radiotherapy. Int. J. Mol. Sci. 2024, 25, 3641. https://doi.org/10.3390/ijms25073641
Kerkhove L, Geirnaert F, Dufait I, De Ridder M. Ferroptosis: Frenemy of Radiotherapy. International Journal of Molecular Sciences. 2024; 25(7):3641. https://doi.org/10.3390/ijms25073641
Chicago/Turabian StyleKerkhove, Lisa, Febe Geirnaert, Inès Dufait, and Mark De Ridder. 2024. "Ferroptosis: Frenemy of Radiotherapy" International Journal of Molecular Sciences 25, no. 7: 3641. https://doi.org/10.3390/ijms25073641
APA StyleKerkhove, L., Geirnaert, F., Dufait, I., & De Ridder, M. (2024). Ferroptosis: Frenemy of Radiotherapy. International Journal of Molecular Sciences, 25(7), 3641. https://doi.org/10.3390/ijms25073641