
Necrosis Links Neurodegeneration and Neuroinflammation in Neurodegenerative Disease

Abstract
1. Introduction
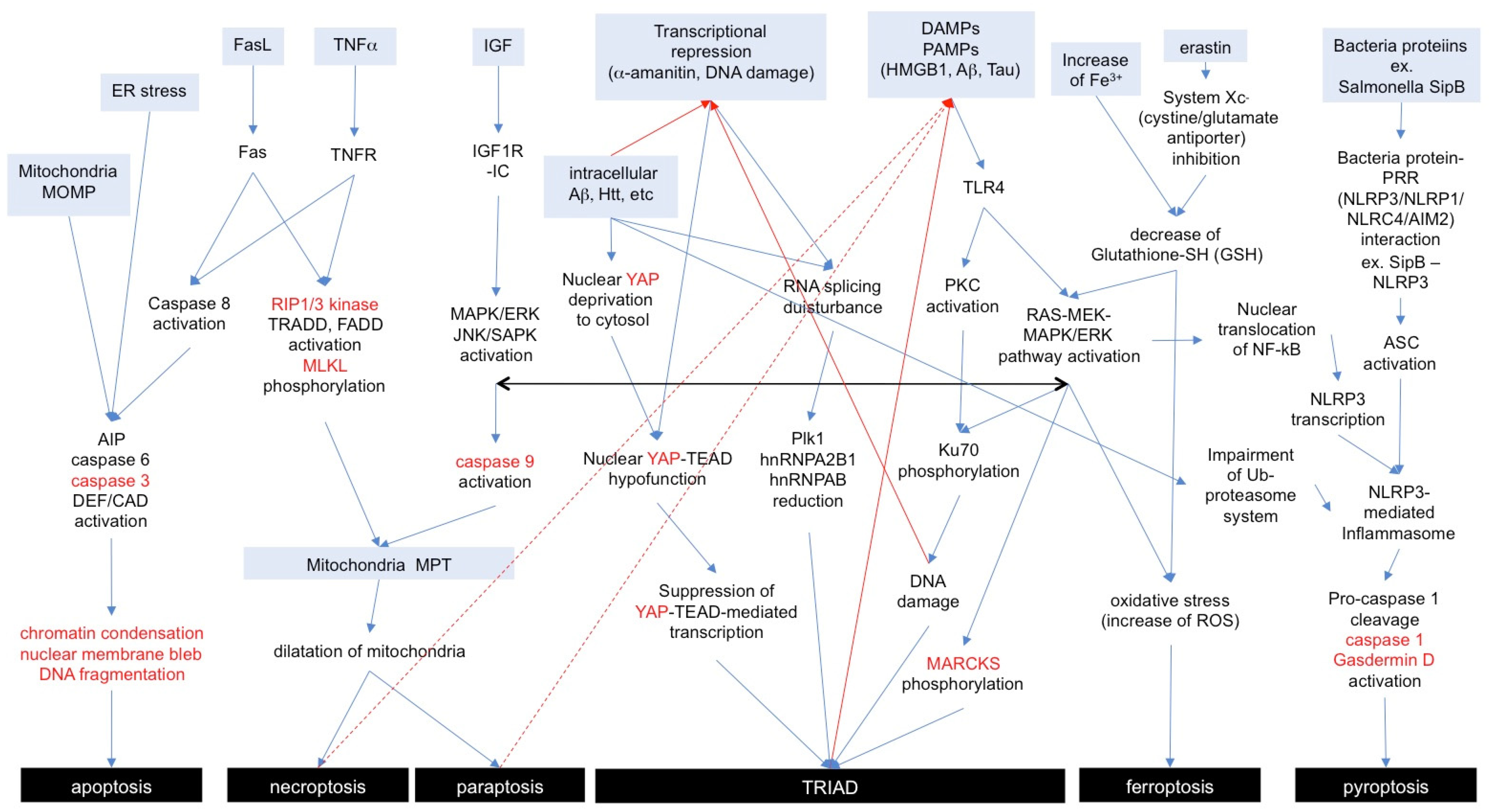
2. Variety of Necrosis Subtypes: Morphology, Biochemistry, and Signals
2.1. Necroptosis
2.2. Paraptosis
2.3. Pyroptosis
2.4. Ferroptosis
2.5. TRIAD
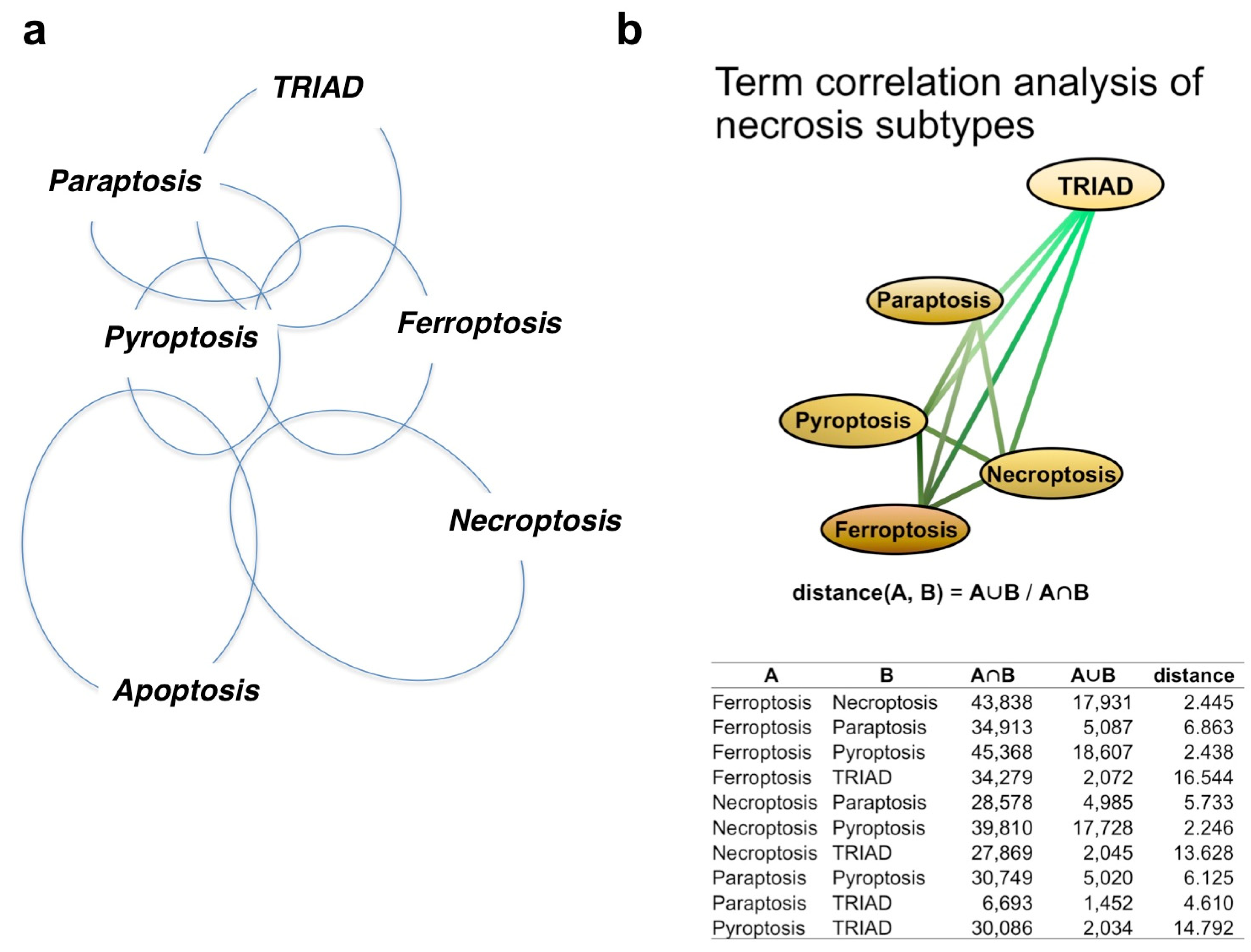
3. Discrepancies and Similarities of Necrosis Subtypes
3.1. Paraptosis vs. TRIAD
3.2. Ferroptosis vs. TRIAD
3.3. Hypothetical Relationships between Necrosis Subtypes and Apoptosis
4. Comparison of Necrosis Subtypes in Neurodegenerative Diseases
5. Molecules Linking Diseases to Neuronal Necrosis Subtypes
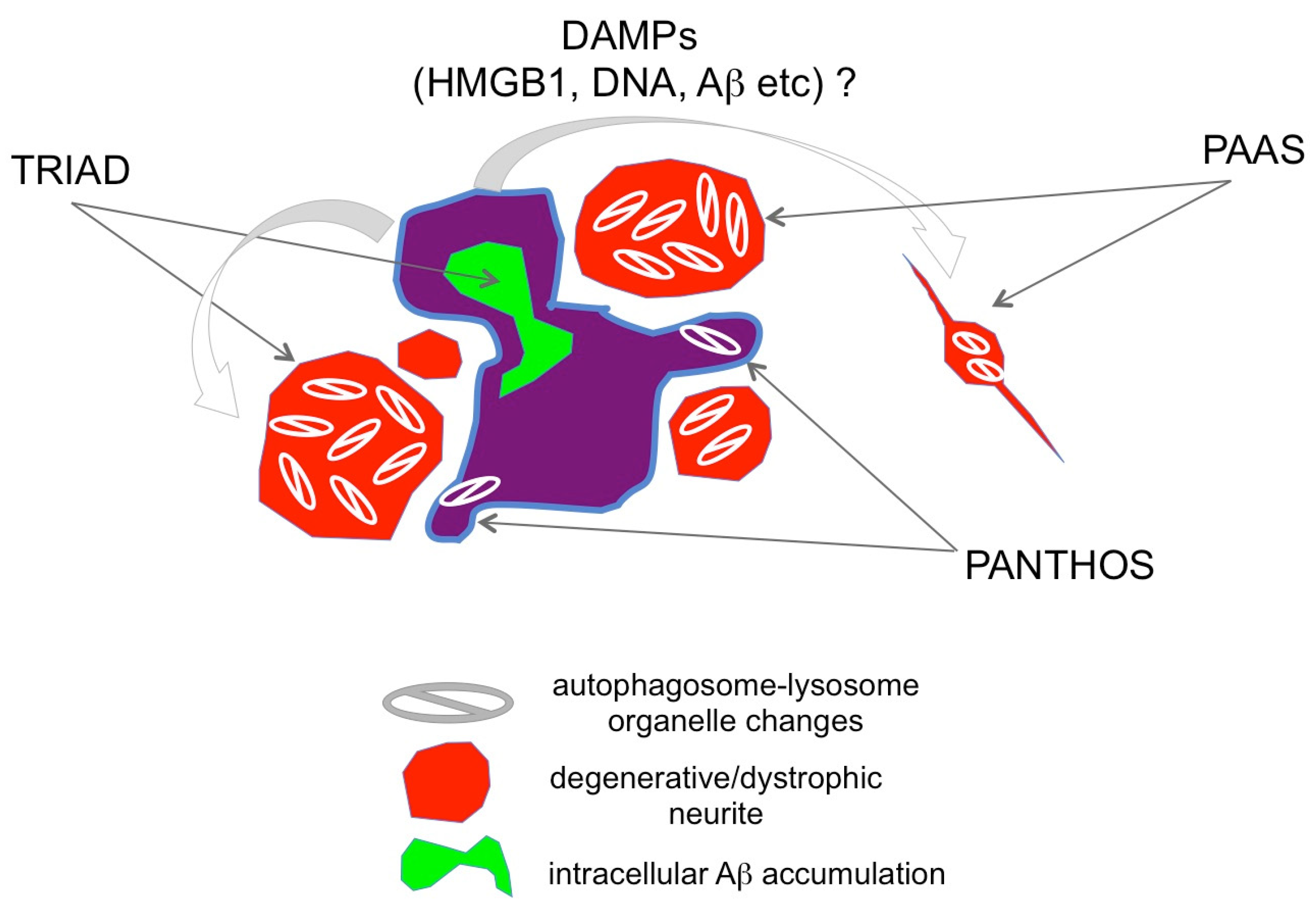
6. TRIAD, PANTHOS, and PAAS Could Be Multiple Sides of the Same Necrosis
7. TRIAD and Other Necrosis Subtypes in the Pathology of Parkinson’s Disease
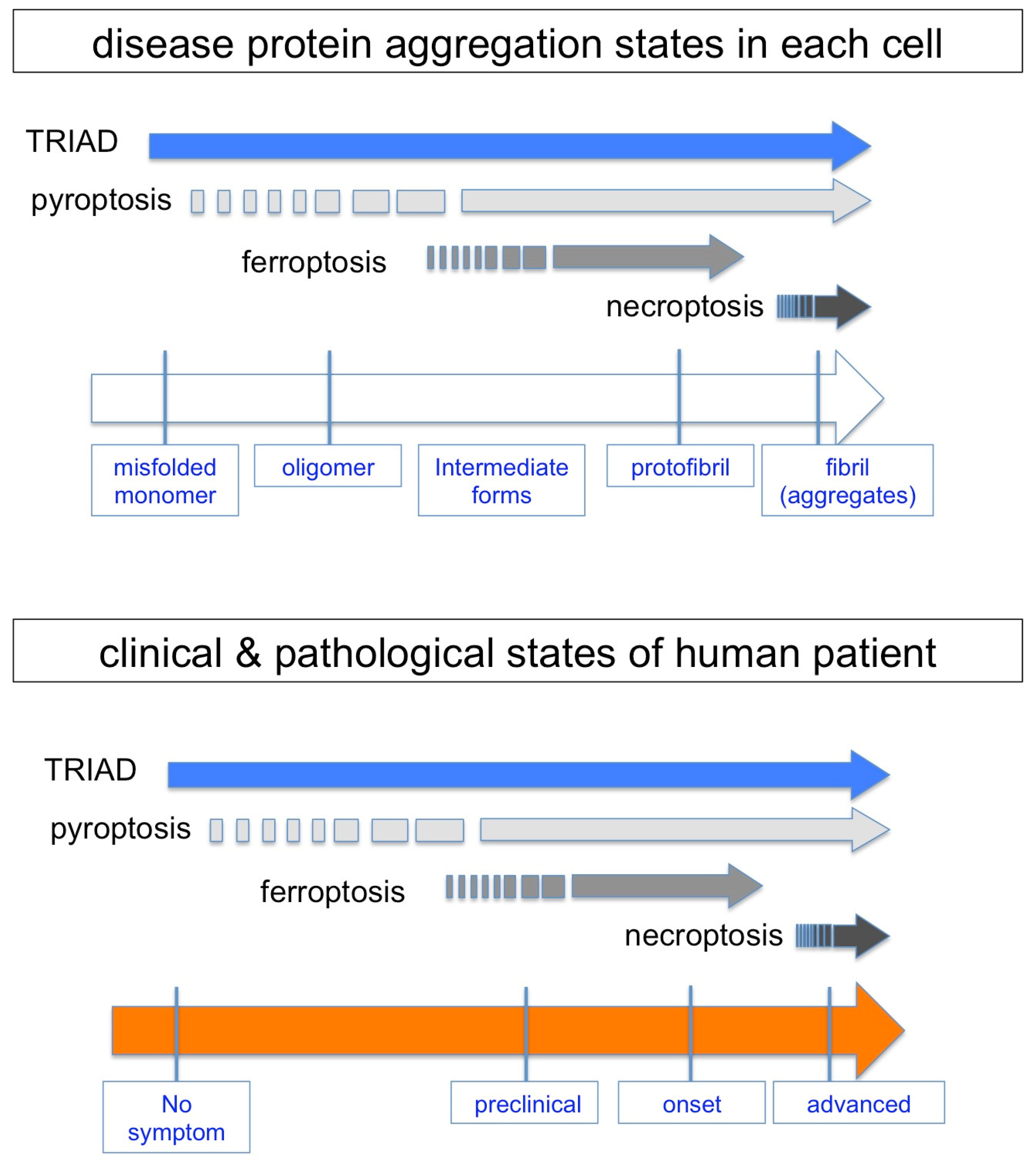
8. Protein Aggregation and Necrosis Subtypes
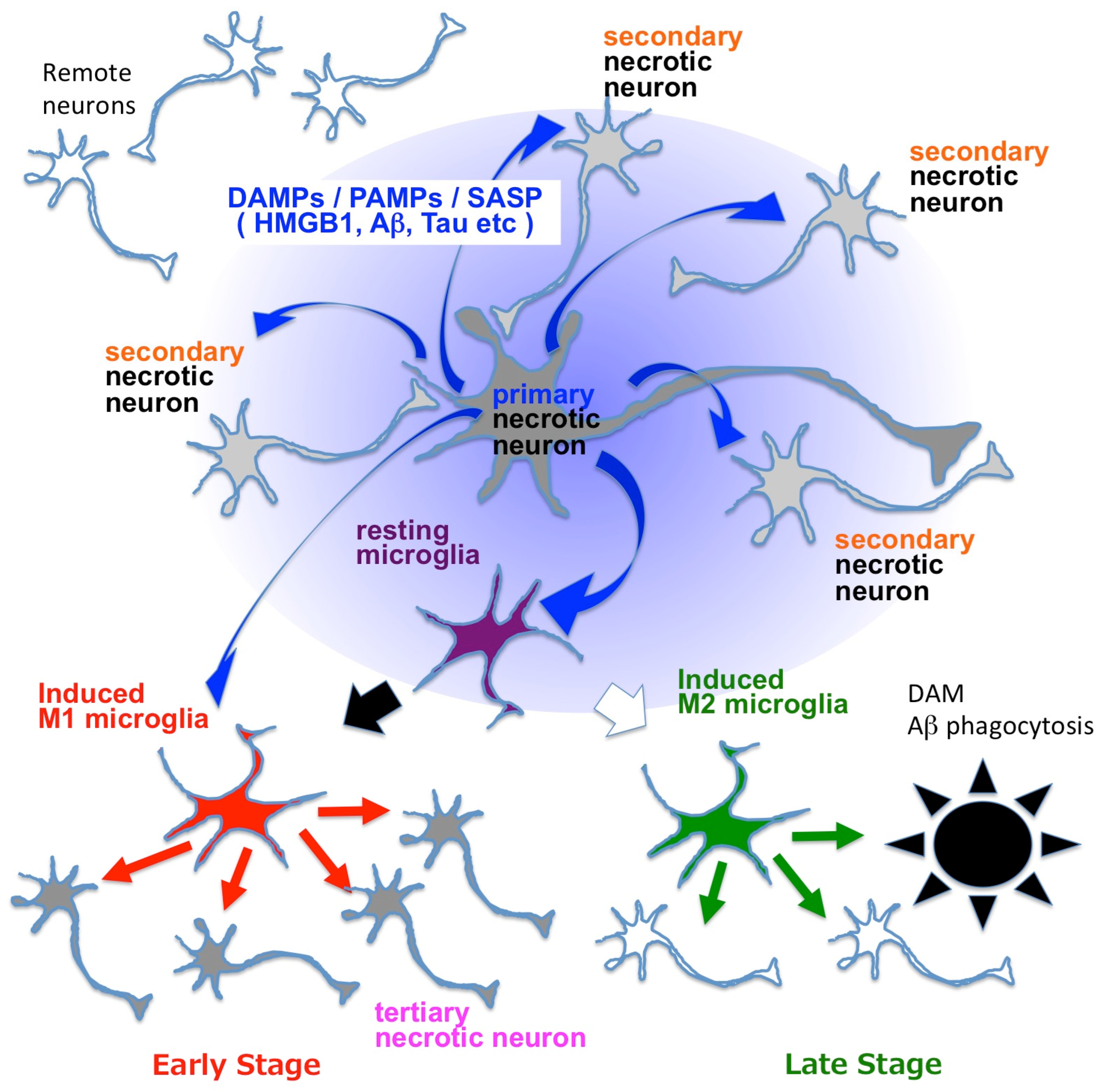
9. HMGB1 Released from Necrotic Neurons Induces Neuroinflammation


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disease Name | Investigated Cells or Animals | Morphological & Biochemical Phenotypes | Necrosis Subtype | Reference |
|---|---|---|---|---|
| Alzheimer’s disease | mouse model (5xFAD mouse, APP-KI mouse), human iPSC-derived neuron, human postmortem AD brain | ER enlargement, nuclear reduction and cytoplasmic translocation of YAP, dysfunction of TEAD-YAP transcription, MARCKS phosphorylation | TRIAD | [46,48,58] |
| culture cell, organotypic slice culture | iron accumulation, RAS-RAF-MEK pathway, ROS | ferroptosis | [50,51] | |
| Parkinson’s disease | cell culture, mouse model (a-Syn-BAC-Tg/GBA-hetero-KO mouse), human postmortem PD brain, human iPSC-derived neuron | MARCKS phosphorylation, activation of RAS-RAF-MEK pathway | TRIAD | [51,52,56] |
| cell culture, organotypic slice culture, mouse model (MPTP-treated mouse) human MRI, human postmortem PD brain | iron accumulation, neuromelanin accumulation, ROS | ferroptosis | [80,81,82] | |
| cell culture, mouse model (MPTP-treated mouse), human iPSC neuron | RIP kinase phosphorylation/activation, MLKL phosphorylation | necroptosis | [77,78] | |
| frontotemporal lober degeneration | mouse model (mutant PGRN-KI, mutant TDP43-KI, mutant VCP-KI, and mutant CHMP2B-KI) human iPSC-derived neuron, human postmortem FTLD brain | ER enlargement, nuclear reduction and cytoplasmic translocation of YAP, dysfunction of TEAD-YAP transcription, MARCKS phosphorylation | TRIAD | [47] |
| Huntington’s disease | cell culture, drosophila model, mouse model (R6/2 mouse, HdhQ111 knock-in mouse), human postmortem HD brain | ER enlargement, nuclear reduction and cytoplasmic translocation of YAP, dysfunction of TEAD-YAP transcription, MARCKS phosphorylation | TRIAD | [41,42,43,44] |
| amyotrophic lateral sclerosis | mouse model (G93ASOD1 transgenic mice) | YAPdeltaC decrease, p73 decrease | TRIAD | [57] |
| PKAN (Friedreich’s ataxia and Hallervorden-Spatz syndrome) | yeast cell, cell culture, mouse model, human postmortem brain | iron accumulation, ROS | ferroptosis | [53,54,55] |
| brain ischemia | mouse model of transient focal cerebral ischemia | MARCKS phosphorylation, nuclear reduction and cytoplasmic translocation of YAP | TRIAD | [46,58] |
| RIP kinase phosphorylation/activation, MLKL phosphorylation, autophagosome | necroptosis | [19,46,58] | ||
| caspase 1 activation | pyroptosis | [46,58] | ||
| caspase 9 activation | paraptosis | [46,58] | ||
| cancer | cell culture, xenograft model, zebrafish model, 3D cultures | caspase 9 activation | paraptosis | [26,27,28,29,30,31,32] |
| cell culture | activation of RAS-RAF-MEK pathway, increase of ROS | ferroptosis | [37,38,39,45] | |
| Salmonella infection | cell culture | Binding between SipB and Caspase-1, caspase-1 activation, PARP activation | pyroptosis | [33,34,35,36] |
10. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sormaz, M.; Murphy, C.; Wang, H.-T.; Hymers, M.; Karapanagiotidis, T.; Poerio, G.; Margulies, D.S.; Jefferies, E.; Smallwood, J. Default Mode Network Can Support the Level of Detail in Experience during Active Task States. Proc. Natl. Acad. Sci. USA 2018, 115, 9318–9323. [Google Scholar] [CrossRef] [PubMed]
- Menon, V.; Uddin, L.Q. Saliency, Switching, Attention and Control: A Network Model of Insula Function. Brain Struct. Funct. 2010, 214, 655–667. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M. NEURODEGENERATION. Alzheimer’s and Parkinson’s Diseases: The Prion Concept in Relation to Assembled Aβ, Tau, and α-Synuclein. Science 2015, 349, 1255555. [Google Scholar] [CrossRef] [PubMed]
- Fu, H.; Hardy, J.; Duff, K.E. Selective Vulnerability in Neurodegenerative Diseases. Nat. Neurosci. 2018, 21, 1350–1358. [Google Scholar] [CrossRef] [PubMed]
- Cotman, C.W.; Anderson, A.J. A Potential Role for Apoptosis in Neurodegeneration and Alzheimer’s Disease. Mol. Neurobiol. 1995, 10, 19–45. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.W.; Pettingell, W.H.; Jung, Y.K.; Kovacs, D.M.; Tanzi, R.E. Alternative Cleavage of Alzheimer-Associated Presenilins during Apoptosis by a Caspase-3 Family Protease. Science 1997, 277, 373–376. [Google Scholar] [CrossRef] [PubMed]
- Stadelmann, C.; Deckwerth, T.L.; Srinivasan, A.; Bancher, C.; Brück, W.; Jellinger, K.; Lassmann, H. Activation of Caspase-3 in Single Neurons and Autophagic Granules of Granulovacuolar Degeneration in Alzheimer’s Disease. Evidence for Apoptotic Cell Death. Am. J. Pathol. 1999, 155, 1459–1466. [Google Scholar] [CrossRef]
- Selznick, L.A.; Holtzman, D.M.; Han, B.H.; Gökden, M.; Srinivasan, A.N.; Johnson, E.M.J.; Roth, K.A. In Situ Immunodetection of Neuronal Caspase-3 Activation in Alzheimer Disease. J. Neuropathol. Exp. Neurol. 1999, 58, 1020–1026. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Albrecht, S.; Bourdeau, M.; Petzke, T.; Bergeron, C.; LeBlanc, A.C. Active Caspase-6 and Caspase-6-Cleaved Tau in Neuropil Threads, Neuritic Plaques, and Neurofibrillary Tangles of Alzheimer’s Disease. Am. J. Pathol. 2004, 165, 523–531. [Google Scholar] [CrossRef]
- Rohn, T.T.; Head, E.; Nesse, W.H.; Cotman, C.W.; Cribbs, D.H. Activation of Caspase-8 in the Alzheimer’s Disease Brain. Neurobiol. Dis. 2001, 8, 1006–1016. [Google Scholar] [CrossRef]
- Rohn, T.T.; Rissman, R.A.; Davis, M.C.; Kim, Y.E.; Cotman, C.W.; Head, E. Caspase-9 Activation and Caspase Cleavage of Tau in the Alzheimer’s Disease Brain. Neurobiol. Dis. 2002, 11, 341–354. [Google Scholar] [CrossRef] [PubMed]
- Perry, G.; Nunomura, A.; Lucassen, P.; Lassmann, H.; Smith, M.A. Apoptosis and Alzheimer’s Disease. Science 1998, 282, 1268–1269. [Google Scholar] [CrossRef] [PubMed]
- Gibson, R.M. Does Apoptosis Have a Role in Neurodegeneration? BMJ 2001, 322, 1539–1540. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Moujalled, D.; Strasser, A.; Liddell, J.R. Molecular Mechanisms of Cell Death in Neurological Diseases. Cell Death Differ. 2021, 28, 2029–2044. [Google Scholar] [CrossRef] [PubMed]
- Park, W.; Wei, S.; Kim, B.S.; Kim, B.; Bae, S.J.; Chae, Y.C.; Ryu, D.; Ha, K.T. Diversity and complexity of cell death: A historical review. Exp. Mol. Med. 2023, 55, 1573–1594. [Google Scholar] [CrossRef] [PubMed]
- Radosevich, J.A. Apoptosis and Beyond, 1st ed.; Wiley: Hoboken, NJ, USA, 2018. [Google Scholar]
- Vercammen, D.; Beyaert, R.; Denecker, G.; Goossens, V.; Van Loo, G.; Declercq, W.; Grooten, J.; Fiers, W.; Vandenabeele, P. Inhibition of Caspases Increases the Sensitivity of L929 Cells to Necrosis Mediated by Tumor Necrosis Factor. J. Exp. Med. 1998, 187, 1477–1485. [Google Scholar] [CrossRef] [PubMed]
- Kawahara, A.; Ohsawa, Y.; Matsumura, H.; Uchiyama, Y.; Nagata, S. Caspase-Independent Cell Killing by Fas-Associated Protein with Death Domain. J. Cell Biol. 1998, 143, 1353–1360. [Google Scholar] [CrossRef] [PubMed]
- Degterev, A.; Huang, Z.; Boyce, M.; Li, Y.; Jagtap, P.; Mizushima, N.; Cuny, G.D.; Mitchison, T.J.; Moskowitz, M.A.; Yuan, J. Chemical Inhibitor of Nonapoptotic Cell Death with Therapeutic Potential for Ischemic Brain Injury. Nat. Chem. Biol. 2005, 1, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Holler, N.; Zaru, R.; Micheau, O.; Thome, M.; Attinger, A.; Valitutti, S.; Bodmer, J.L.; Schneider, P.; Seed, B.; Tschopp, J. Fas Triggers an Alternative, Caspase-8-Independent Cell Death Pathway Using the Kinase RIP as Effector Molecule. Nat. Immunol. 2000, 1, 489–495. [Google Scholar] [CrossRef]
- Hitomi, J.; Christofferson, D.E.; Ng, A.; Yao, J.; Degterev, A.; Xavier, R.J.; Yuan, J. Identification of a Molecular Signaling Network That Regulates a Cellular Necrotic Cell Death Pathway. Cell 2008, 135, 1311–1323. [Google Scholar] [CrossRef]
- Kroemer, G.; Levine, B. Autophagic Cell Death: The Story of a Misnomer. Nat. Rev. Mol. Cell Biol. 2008, 9, 1004–1010. [Google Scholar] [CrossRef] [PubMed]
- Denton, D.; Kumar, S. Autophagy-Dependent Cell Death. Cell Death Differ. 2019, 26, 605–616. [Google Scholar] [CrossRef] [PubMed]
- Sperandio, S.; De Belle, I.; Bredesen, D.E. An Alternative, Nonapoptotic Form of Programmed Cell Death. Proc. Natl. Acad. Sci. USA 2000, 97, 14376–14381. [Google Scholar] [CrossRef] [PubMed]
- Sperandio, S.; Poksay, K.; de Belle, I.; Lafuente, M.J.; Liu, B.; Nasir, J.; Bredesen, D.E. Paraptosis: Mediation by MAP Kinases and Inhibition by AIP-1/Alix. Cell Death Differ. 2004, 11, 1066–1075. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.; Kim, I.Y.; Saha, S.; Choi, K.S. Paraptosis in the Anti-Cancer Arsenal of Natural Products. Pharmacol. Ther. 2016, 162, 120–133. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.Y.; Shim, M.J.; Lee, D.M.; Lee, A.R.; Kim, M.A.; Yoon, M.J.; Kwon, M.R.; Lee, H.I.; Seo, M.J.; Choi, Y.W.; et al. Loperamide Overcomes the Resistance of Colon Cancer Cells to Bortezomib by Inducing CHOP-Mediated Paraptosis-like Cell Death. Biochem. Pharmacol. 2019, 162, 41–54. [Google Scholar] [CrossRef] [PubMed]
- Nedungadi, D.; Binoy, A.; Vinod, V.; Vanuopadath, M.; Nair, S.S.; Nair, B.G.; Mishra, N. Ginger Extract Activates Caspase Independent Paraptosis in Cancer Cells via ER Stress, Mitochondrial Dysfunction, AIF Translocation and DNA Damage. Nutr. Cancer 2021, 73, 147–159. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.R.; Seo, M.J.; Kim, J.; Lee, D.M.; Kim, I.Y.; Yoon, M.J.; Hoon, H.; Choi, K.S. Lercanidipine Synergistically Enhances Bortezomib Cytotoxicity in Cancer Cells via Enhanced Endoplasmic Reticulum Stress and Mitochondrial Ca2+ Overload. Int. J. Mol. Sci. 2019, 20, 6112. [Google Scholar] [CrossRef] [PubMed]
- Lizák, B.; Birk, J.; Zana, M.; Kosztyi, G.; Kratschmar, D.V.; Odermatt, A.; Zimmermann, R.; Geiszt, M.; Appenzeller-Herzog, C.; Bánhegyi, G. Ca2+ Mobilization-Dependent Reduction of the Endoplasmic Reticulum Lumen Is Due to Influx of Cytosolic Glutathione. BMC Biol. 2020, 18, 19. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Lee, D.M.; Seo, M.J.; Lee, H.J.; Choi, K.S. Intracellular Ca2+ Imbalance Critically Contributes to Paraptosis. Front. Cell Dev. Biol. 2020, 8, 607844. [Google Scholar] [CrossRef]
- Hanson, S.; Dharan, A.; PV, J.; Pal, S.; Nair, B.G.; Kar, R.; Mishra, N. Paraptosis: A Unique Cell Death Mode for Targeting Cancer. Front. Pharmacol. 2023, 14, 1159409. [Google Scholar] [CrossRef] [PubMed]
- Hilbi, H.; Moss, J.E.; Hersh, D.; Chen, Y.; Arondel, J.; Banerjee, S.; Flavell, R.A.; Yuan, J.; Sansonetti, P.J.; Zychlinsky, A. Shigella-Induced Apoptosis Is Dependent on Caspase-1 Which Binds to IpaB. J. Biol. Chem. 1998, 273, 32895–32900. [Google Scholar] [CrossRef] [PubMed]
- Hersh, D.; Monack, D.M.; Smith, M.R.; Ghori, N.; Falkow, S.; Zychlinsky, A. The Salmonella Invasin SipB Induces Macrophage Apoptosis by Binding to Caspase-1. Proc. Natl. Acad. Sci. USA 1999, 96, 2396–2401. [Google Scholar] [CrossRef] [PubMed]
- Brennan, M.A.; Cookson, B.T. Salmonella Induces Macrophage Death by Caspase-1-Dependent Necrosis. Mol. Microbiol. 2000, 38, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Cookson, B.T.; Brennan, M.A. Pro-Inflammatory Programmed Cell Death. Trends Microbiol. 2001, 9, 113–114. [Google Scholar] [CrossRef] [PubMed]
- Dolma, S.; Lessnick, S.L.; Hahn, W.C.; Stockwell, B.R. Identification of Genotype-Selective Antitumor Agents Using Synthetic Lethal Chemical Screening in Engineered Human Tumor Cells. Cancer Cell 2003, 3, 285–296. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.S.; Stockwell, B.R. Synthetic Lethal Screening Identifies Compounds Activating Iron-Dependent, Nonapoptotic Cell Death in Oncogenic-RAS-Harboring Cancer Cells. Chem. Biol. 2008, 15, 234–245. [Google Scholar] [CrossRef]
- Yagoda, N.; von Rechenberg, M.; Zaganjor, E.; Bauer, A.J.; Yang, W.S.; Fridman, D.J.; Wolpaw, A.J.; Smukste, I.; Peltier, J.M.; Boniface, J.J.; et al. RAS-RAF-MEK-Dependent Oxidative Cell Death Involving Voltage-Dependent Anion Channels. Nature 2007, 447, 864–868. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Hoshino, M.; Qi, M.L.; Yoshimura, N.; Miyashita, T.; Tagawa, K.; Wada, Y.; Enokido, Y.; Marubuchi, S.; Harjes, P.; Arai, N.; et al. Transcriptional Repression Induces a Slowly Progressive Atypical Neuronal Death Associated with Changes of YAP Isoforms and P73. J. Cell Biol. 2006, 172, 589–604. [Google Scholar] [CrossRef] [PubMed]
- Mao, Y.; Tamura, T.; Yuki, Y.; Abe, D.; Tamada, Y.; Imoto, S.; Tanaka, H.; Homma, H.; Tagawa, K.; Miyano, S.; et al. The HnRNP-Htt Axis Regulates Necrotic Cell Death Induced by Transcriptional Repression through Impaired RNA Splicing. Cell Death Dis. 2016, 7, e2207. [Google Scholar] [CrossRef]
- Yamanishi, E.; Hasegawa, K.; Fujita, K.; Ichinose, S.; Yagishita, S.; Murata, M.; Tagawa, K.; Akashi, T.; Eishi, Y.; Okazawa, H. A Novel Form of Necrosis, TRIAD, Occurs in Human Huntington’s Disease. Acta Neuropathol. Commun. 2017, 5, 19. [Google Scholar] [CrossRef] [PubMed]
- Mao, Y.; Chen, X.; Xu, M.; Fujita, K.; Motoki, K.; Sasabe, T.; Homma, H.; Murata, M.; Tagawa, K.; Tamura, T.; et al. Targeting TEAD/YAP-Transcription-Dependent Necrosis, TRIAD, ameliorates Huntington’s Disease Pathology. Hum. Mol. Genet. 2016, 25, 4749–4770. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Gundelach, J.H.; Bram, R.J. Cycloheximide Promotes Paraptosis Induced by Inhibition of Cyclophilins in Glioblastoma Multiforme. Cell Death Dis. 2017, 8, e2807. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, H.; Homma, H.; Fujita, K.; Kondo, K.; Yamada, S.; Jin, X.; Waragai, M.; Ohtomo, G.; Iwata, A.; Tagawa, K.; et al. YAP-Dependent Necrosis Occurs in Early Stages of Alzheimer’s Disease and Regulates Mouse Model Pathology. Nat. Commun. 2020, 11, 507. [Google Scholar] [CrossRef] [PubMed]
- Homma, H.; Tanaka, H.; Jin, M.; Jin, X.; Huang, Y.; Yoshioka, Y.; Bertens, C.J.; Tsumaki, K.; Kondo, K.; Shiwaku, H.; et al. DNA Damage in Embryonic Neural Stem Cell Determines FTLDs’ Fate via Early-Stage Neuronal Necrosis. Life Sci. Alliance 2021, 4, e202101022. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, H.; Kondo, K.; Fujita, K.; Homma, H.; Tagawa, K.; Jin, X.; Jin, M.; Yoshioka, Y.; Takayama, S.; Masuda, H.; et al. HMGB1 Signaling Phosphorylates Ku70 and Impairs DNA Damage Repair in Alzheimer’s Disease Pathology. Commun. Biol. 2021, 4, 1175. [Google Scholar] [CrossRef] [PubMed]
- Sperandio, S.; Poksay, K.S.; Schilling, B.; Crippen, D.; Gibson, B.W.; Bredesen, D.E. Identification of New Modulators and Protein Alterations in Non-Apoptotic Programmed Cell Death. J. Cell. Biochem. 2010, 111, 1401–1412. [Google Scholar] [CrossRef] [PubMed]
- Khattar, N.; Triebswetter, C.; Kiely, M.; Ferrucci, L.; Resnick, S.M.; Spencer, R.G.; Bouhrara, M. Investigation of the Association between Cerebral Iron Content and Myelin Content in Normative Aging Using Quantitative Magnetic Resonance Neuroimaging. Neuroimage 2021, 239, 118267. [Google Scholar] [CrossRef]
- Sfera, A.; Bullock, K.; Price, A.; Inderias, L.; Osorio, C. Ferrosenescence: The Iron Age of Neurodegeneration? Mech. Ageing Dev. 2018, 174, 63–75. [Google Scholar] [CrossRef]
- Zecca, L.; Youdim, M.B.H.; Riederer, P.; Connor, J.R.; Crichton, R.R. Iron, Brain Ageing and Neurodegenerative Disorders. Nat. Rev. Neurosci. 2004, 5, 863–873. [Google Scholar] [CrossRef]
- Gordon, N. Friedreich’s Ataxia and Iron Metabolism. Brain Dev. 2000, 22, 465–468. [Google Scholar] [CrossRef]
- Mühlenhoff, U.; Richhardt, N.; Ristow, M.; Kispal, G.; Lill, R. The Yeast Frataxin Homolog Yfh1p Plays a Specific Role in the Maturation of Cellular Fe/S Proteins. Hum. Mol. Genet. 2002, 11, 2025–2036. [Google Scholar] [CrossRef] [PubMed]
- Ke, Y.; Ming Qian, Z. Iron Misregulation in the Brain: A Primary Cause of Neurodegenerative Disorders. Lancet Neurol. 2003, 2, 246–253. [Google Scholar] [CrossRef] [PubMed]
- Fujita, K.; Homma, H.; Kondo, K.; Ikuno, M.; Yamakado, H.; Tagawa, K.; Murayama, S.; Takahashi, R.; Okazawa, H. Ser46-Phosphorylated MARCKS Is a Marker of Neurite Degeneration at the Pre-Aggregation Stage in PD/DLB Pathology. eNeuro 2018, 5. [Google Scholar] [CrossRef]
- Morimoto, N.; Nagai, M.; Miyazaki, K.; Kurata, T.; Takehisa, Y.; Ikeda, Y.; Kamiya, T.; Okazawa, H.; Abe, K. Progressive Decrease in the Level of YAPdeltaCs, Prosurvival Isoforms of YAP, in the Spinal Cord of Transgenic Mouse Carrying a Mutant SOD1 Gene. J. Neurosci. Res. 2009, 87, 928–936. [Google Scholar] [CrossRef]
- Fujita, K.; Motoki, K.; Tagawa, K.; Chen, X.; Hama, H.; Nakajima, K.; Homma, H.; Tamura, T.; Watanabe, H.; Katsuno, M.; et al. HMGB1, a Pathogenic Molecule That Induces Neurite Degeneration via TLR4-MARCKS, Is a Potential Therapeutic Target for Alzheimer’s Disease. Sci. Rep. 2016, 6, 31895. [Google Scholar] [CrossRef]
- Agresti, A.; Bianchi, M.E. HMGB proteins and gene expression. Curr. Opin. Genet. Dev. 2003, 13, 170–178. [Google Scholar] [CrossRef] [PubMed]
- Travers, A.E. Priming the nucleosome: A role for HMGB proteins? EMBO Rep. 2003, 4, 131–136. [Google Scholar] [CrossRef]
- Lotze, M.T.; Tracey, K.J. High-mobility group box 1 protein (HMGB1): Nuclear weapon in the immune arsenal. Nat. Rev. Immunol. 2005, 5, 331–342. [Google Scholar] [CrossRef]
- Scaffidi, P.; Misteli, T.; Bianchi, M.E. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature 2002, 418, 191–195. [Google Scholar] [CrossRef]
- Guo, H.; Callaway, J.B.; Ting, J.P.-Y. Inflammasomes: Mechanism of Action, Role in Disease, and Therapeutics. Nat. Med. 2015, 21, 677–687. [Google Scholar] [CrossRef] [PubMed]
- Broz, P.; Dixit, V.M. Inflammasomes: Mechanism of Assembly, Regulation and Signalling. Nat. Rev. Immunol. 2016, 16, 407–420. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Wang, X.; Huang, L.; Qian, H.; Liu, W. Mechanism of Pyroptosis in Neurodegenerative Diseases and Its Therapeutic Potential by Traditional Chinese Medicine. Front. Pharmacol. 2023, 14, 1122104. [Google Scholar] [CrossRef] [PubMed]
- von Herrmann, K.M.; Salas, L.A.; Martinez, E.M.; Young, A.L.; Howard, J.M.; Feldman, M.S.; Christensen, B.C.; Wilkins, O.M.; Lee, S.L.; Hickey, W.F.; et al. NLRP3 Expression in Mesencephalic Neurons and Characterization of a Rare NLRP3 Polymorphism Associated with Decreased Risk of Parkinson’s Disease. NPJ Park. Dis. 2018, 4, 24. [Google Scholar] [CrossRef] [PubMed]
- Panicker, N.; Kam, T.-I.; Wang, H.; Neifert, S.; Chou, S.-C.; Kumar, M.; Brahmachari, S.; Jhaldiyal, A.; Hinkle, J.T.; Akkentli, F.; et al. Neuronal NLRP3 Is a Parkin Substrate That Drives Neurodegeneration in Parkinson’s Disease. Neuron 2022, 110, 2422–2437.e9. [Google Scholar] [CrossRef] [PubMed]
- Molnár, K.; Nógrádi, B.; Kristóf, R.; Mészáros, Á.; Pajer, K.; Siklós, L.; Nógrádi, A.; Wilhelm, I.; Krizbai, I.A. Motoneuronal Inflammasome Activation Triggers Excessive Neuroinflammation and Impedes Regeneration after Sciatic Nerve Injury. J. Neuroinflammation 2022, 19, 68. [Google Scholar] [CrossRef] [PubMed]
- Anerillas, C.; Mazan-Mamczarz, K.; Herman, A.B.; Munk, R.; Lam, K.-W.G.; Calvo-Rubio, M.; Garrido, A.; Tsitsipatis, D.; Martindale, J.L.; Altés, G.; et al. The YAP-TEAD Complex Promotes Senescent Cell Survival by Lowering Endoplasmic Reticulum Stress. Nat. Aging 2023, 3, 1237–1250. [Google Scholar] [CrossRef] [PubMed]
- West, A.; Brodsky, I.; Rahner, C.; Woo, D.K.; Erdjument-Bromage, H.; Tempst, P.; Walsh, M.C.; Choi, Y.; Shadel, G.S.; Ghosh, S. TLR signalling augments macrophage bactericidal activity through mitochondrial ROS. Nature 2011, 472, 476–480. [Google Scholar] [CrossRef]
- Yuan, P.; Zhang, M.; Tong, L.; Morse, T.M.; McDougal, R.A.; Ding, H.; Chan, D.; Cai, Y.; Grutzendler, J. PLD3 Affects Axonal Spheroids and Network Defects in Alzheimer’s Disease. Nature 2022, 612, 328–337. [Google Scholar] [CrossRef]
- Lee, J.-H.; Yang, D.-S.; Goulbourne, C.N.; Im, E.; Stavrides, P.; Pensalfini, A.; Chan, H.; Bouchet-Marquis, C.; Bleiwas, C.; Berg, M.J.; et al. Faulty Autolysosome Acidification in Alzheimer’s Disease Mouse Models Induces Autophagic Build-up of Aβ in Neurons, Yielding Senile Plaques. Nat. Neurosci. 2022, 25, 688–701. [Google Scholar] [CrossRef] [PubMed]
- Graham, D.I.; Lantos, P.L. Chapter 4: Aging and Dementia. In Greenfield’s Neuropathol (2 Volume Set), 7th ed.; CRC Press: Boca Raton, FL, USA, 2002; Volume 2, p. 210. [Google Scholar]
- Jellinger, K.A. Cell death mechanisms in Parkinson’s disease. J. Neural Transm. 2000, 107, 1–29. [Google Scholar] [CrossRef] [PubMed]
- Yamakado, H.; Moriwaki, Y.; Yamasaki, N.; Miyakawa, T.; Kurisu, J.; Uemura, K.; Inoue, H.; Takahashi, M.; Takahashi, R. α-Synuclein BAC transgenic mice as a model for Parkinson’s disease manifested decreased anxiety-like behavior and hyperlocomotion. Neurosci. Res. 2012, 73, 173–177. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M.; Spillantini, M.G.; Del Tredici, K.; Braak, H. 100 years of Lewy pathology. Nat. Rev. Neurol. 2013, 9, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Iannielli, A.; Bido, S.; Folladori, L.; Segnali, A.; Cancellieri, C.; Maresca, A.; Massimino, L.; Rubio, A.; Morabito, G.; Caporali, L.; et al. Pharmacological Inhibition of Necroptosis Protects from Dopaminergic Neuronal Cell Death in Parkinson’s Disease Models. Cell Rep. 2018, 22, 2066–2079. [Google Scholar] [CrossRef] [PubMed]
- Alegre-Cortés, E.; Muriel-González, A.; Canales-Cortés, S.; Uribe-Carretero, E.; Martínez-Chacón, G.; Aiastui, A.; López de Munain, A.; Niso-Santano, M.; Gonzalez-Polo, R.A.; Fuentes, J.M.; et al. Toxicity of Necrostatin-1 in Parkinson’s Disease Models. Antioxidants 2020, 9, 524. [Google Scholar] [CrossRef]
- Wang, Z.L.; Yuan, L.; Li, W.; Li, J.Y. Ferroptosis in Parkinson’s disease: Glia-neuron crosstalk. Trends Mol. Med. 2022, 28, 258–269. [Google Scholar] [CrossRef] [PubMed]
- Biondetti, E.; Santin, M.D.; Valabrègue, R.; Mangone, G.; Gaurav, R.; Pyatigorskaya, N.; Hutchison, M.; Yahia-Cherif, L.; Villain, N.; Habert, M.O.; et al. The spatiotemporal changes in dopamine, neuromelanin and iron characterizing Parkinson’s disease. Brain 2021, 144, 3114–3125. [Google Scholar] [CrossRef] [PubMed]
- Do Van, B.; Gouel, F.; Jonneaux, A.; Timmerman, K.; Gelé, P.; Pétrault, M.; Bastide, M.; Laloux, C.; Moreau, C.; Bordet, R.; et al. Ferroptosis, a newly characterized form of cell death in Parkinson’s disease that is regulated by PKC. Neurobiol. Dis. 2016, 94, 169–178. [Google Scholar] [CrossRef]
- Ito, K.; Eguchi, Y.; Imagawa, Y.; Akai, S.; Mochizuki, H.; Tsujimoto, Y. MPP+ induces necrostatin-1- and ferrostatin-1-sensitive necrotic death of neuronal SH-SY5Y cells. Cell Death Discov. 2017, 3, 17013. [Google Scholar] [CrossRef]
- Shoji, M.; Iwakami, N.; Takeuchi, S.; Waragai, M.; Suzuki, M.; Kanazawa, I.; Lippa, C.F.; Ono, S.; Okazawa, H. JNK activation is associated with intracellular beta-amyloid accumulation. Brain Res. Mol. Brain Res. 2000, 85, 221–233. [Google Scholar] [CrossRef]
- LaFerla, F.M.; Green, K.N.; Oddo, S. Intracellular amyloid-β in Alzheimer’s disease. Nat. Rev. Neurosci. 2007, 8, 499–509. [Google Scholar] [CrossRef] [PubMed]
- Okazawa, H. Ultra-Early Phase pathologies of Alzheimer’s disease and other neurodegenerative diseases. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2017, 93, 361–377. [Google Scholar] [CrossRef] [PubMed]
- Okazawa, H. Intracellular amyloid hypothesis for ultra-early phase pathology of Alzheimer’s disease. Neuropathology 2021, 41, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Goehler, H.; Lalowski, M.; Stelzl, U.; Waelter, S.; Stroedicke, M.; Worm, U.; Droege, A.; Lindenberg, K.S.; Knoblich, M.; Haenig, C.; et al. A protein interaction network links GIT1, an enhancer of huntingtin aggregation, to Huntington’s disease. Mol. Cell 2004, 15, 853–865. [Google Scholar] [CrossRef] [PubMed]
- Stelzl, U.; Worm, U.; Lalowski, M.; Haenig, C.; Brembeck, F.H.; Goehler, H.; Stroedicke, M.; Zenkner, M.; Schoenherr, A.; Koeppen, S.; et al. A human protein-protein interaction network: A resource for annotating the proteome. Cell 2005, 122, 957–968. [Google Scholar] [CrossRef]
- Lim, J.; Hao, T.; Shaw, C.; Patel, A.J.; Szabó, G.; Rual, J.F.; Fisk, C.J.; Li, N.; Smolyar, A.; Hill, D.E.; et al. A protein-protein interaction network for human inherited ataxias and disorders of Purkinje cell degeneration. Cell 2006, 125, 801–814. [Google Scholar] [CrossRef] [PubMed]
- Enokido, Y.; Tamura, T.; Ito, H.; Arumughan, A.; Komuro, A.; Shiwaku, H.; Sone, M.; Foulle, R.; Sawada, H.; Ishiguro, H.; et al. Mutant huntingtin impairs Ku70-mediated DNA repair. J. Cell Biol. 2010, 189, 425–443. [Google Scholar] [CrossRef] [PubMed]
- Moalem, S.; Percy, M.E.; Andrews, D.F.; Kruck, T.P.; Wong, S.; Dalton, A.J.; Mehta, P.; Fedor, B.; Warren, A.C. Are hereditary hemochromatosis mutations involved in Alzheimer disease? Am. J. Med. Genet. 2000, 93, 58–66. [Google Scholar] [CrossRef]
- Berlin, D.; Chong, G.; Chertkow, H.; Bergman, H.; Phillips, N.A.; Schipper, H.M. Evaluation of HFE (hemochromatosis) mutations as genetic modifiers in sporadic AD and MCI. Neurobiol. Aging 2004, 25, 465–474. [Google Scholar] [CrossRef]
- Caccamo, A.; Branca, C.; Piras, I.S.; Ferreira, E.; Huentelman, M.J.; Liang, W.S.; Readhead, B.; Dudley, J.T.; Spangenberg, E.E.; Green, K.N.; et al. Necroptosis activation in Alzheimer’s disease. Nat. Neurosci. 2017, 20, 1236–1246. [Google Scholar] [CrossRef]
- Yu, P.; Zhang, X.; Liu, N.; Tang, L.; Peng, C.; Chen, X. Pyroptosis: Mechanisms and diseases. Signal Transduct. Target. Ther. 2021, 6, 128. [Google Scholar] [CrossRef]
- Hanamsagar, R.; Hanke, M.L.; Kielian, T. Toll-like Receptor (TLR) and Inflammasome Actions in the Central Nervous System. Trends Immunol. 2012, 33, 333–342. [Google Scholar] [CrossRef]
- Hickman, S.; Izzy, S.; Sen, P.; Morsett, L.; El Khoury, J. Microglia in Neurodegeneration. Nat. Neurosci. 2018, 21, 1359–1369. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.; Jiang, J.; Tan, Y.; Chen, S. Microglia in Neurodegenerative Diseases: Mechanism and Potential Therapeutic Targets. Signal Transduct. Target. Ther. 2023, 8, 359. [Google Scholar] [CrossRef] [PubMed]
- Keren-Shaul, H.; Spinrad, A.; Weiner, A.; Matcovitch-Natan, O.; Dvir-Szternfeld, R.; Ulland, T.K.; David, E.; Baruch, K.; Lara-Astaiso, D.; Toth, B.; et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 2017, 169, 1276–1290.e17. [Google Scholar] [CrossRef]
- Jin, M.; Shiwaku, H.; Tanaka, H.; Obita, T.; Ohuchi, S.; Yoshioka, Y.; Jin, X.; Kondo, K.; Fujita, K.; Homma, H.; et al. Tau Activates Microglia via the PQBP1-CGAS-STING Pathway to Promote Brain Inflammation. Nat. Commun. 2021, 12, 6565. [Google Scholar] [CrossRef] [PubMed]
- Eyolfson, E.; Khan, A.; Mychasiuk, R.; Lohman, A.W. Microglia dynamics in adolescent traumatic brain injury. J. Neuroinflammation 2020, 17, 326. [Google Scholar] [CrossRef]
- Jin, M.; Jin, X.; Homma, H.; Fujita, K.; Tanaka, H.; Murayama, S.; Akatsu, H.; Tagawa, K.; Okazawa, H. Prediction and verification of the AD-FTLD common pathomechanism based on dynamic molecular network analysis. Commun. Biol. 2021, 4, 961. [Google Scholar] [CrossRef]





| Origin of Discovery | Original Cell Type | Biochemical Features | Morphological Features | Exclusion Criteria | |
|---|---|---|---|---|---|
| Necroptosis | FasL- and TNFa-induced cell death in apoptosis-deficient conditions | L929 cells with zVAD.fmk Jurkat-derived cell line that is deficient in caspase-8 (JB-6) |
|
|
|
| Paraptosis | Cell death induced by forced expression of intracellular domain of insulin-like growth factor 1 receptor (IGF1R-IC) | 293T cells 293, MCF-7, Cos-7, and primary mouse embryonic fibroblasts |
|
|
|
| Pyroptosis | Cell death induced by Salmonella invasin SipB in macrophages | Macrophage |
|
|
|
| Ferroptosis | Chemical screening to find anti-cancer candidate drugs that are effective on Ras-mutated cancer cells | RAS mutated cancer cells |
|
|
|
| TRIAD | Neuronal cell death induced by RNA polymerase II-specific inhibitor, alpha-amanitin | Neuron |
|
|
|
| pS46-MARCKS | Nuclear YAP Reduction | pMLKL & pRIP | Cleaved Caspase 1 | Cleaved Caspase 9 | |
|---|---|---|---|---|---|
| TRIAD | ◯ | ◯ | ✖ | ||
| necroptosis | ◯ | ||||
| pyroptosis | ◯ | ||||
| paraptosis | ◯ |
| pS46-MARCKS | Nuclear YAP Reduction | pMLKL & pRIP | Cleaved Caspase 1 | Cleaved Caspase 9 | |
|---|---|---|---|---|---|
| TRIAD | ◯ | ◯ | ✖ | ✖ | ✖ |
| necroptosis | ◯ | ✖ | ◯ | ||
| pyroptosis | ◯ | △ | ◯ | ||
| paraptosis | ◯ | ✖ | ◯ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Homma, H.; Tanaka, H.; Fujita, K.; Okazawa, H. Necrosis Links Neurodegeneration and Neuroinflammation in Neurodegenerative Disease. Int. J. Mol. Sci. 2024, 25, 3636. https://doi.org/10.3390/ijms25073636
Homma H, Tanaka H, Fujita K, Okazawa H. Necrosis Links Neurodegeneration and Neuroinflammation in Neurodegenerative Disease. International Journal of Molecular Sciences. 2024; 25(7):3636. https://doi.org/10.3390/ijms25073636
Chicago/Turabian StyleHomma, Hidenori, Hikari Tanaka, Kyota Fujita, and Hitoshi Okazawa. 2024. "Necrosis Links Neurodegeneration and Neuroinflammation in Neurodegenerative Disease" International Journal of Molecular Sciences 25, no. 7: 3636. https://doi.org/10.3390/ijms25073636
APA StyleHomma, H., Tanaka, H., Fujita, K., & Okazawa, H. (2024). Necrosis Links Neurodegeneration and Neuroinflammation in Neurodegenerative Disease. International Journal of Molecular Sciences, 25(7), 3636. https://doi.org/10.3390/ijms25073636