
Formation of Pre-PCTA/DT Intermediates from 2-Chlorothiophenol on Silica Clusters: A Quantum Mechanical Study

Abstract
1. Introduction
2. Results
2.1. Silica Clusters
2.2. The Formation of 2-Chlorothiophenolate on Silica Clusters
2.3. Formation of Pre-PCDT Intermediates via the L–H Mechanism
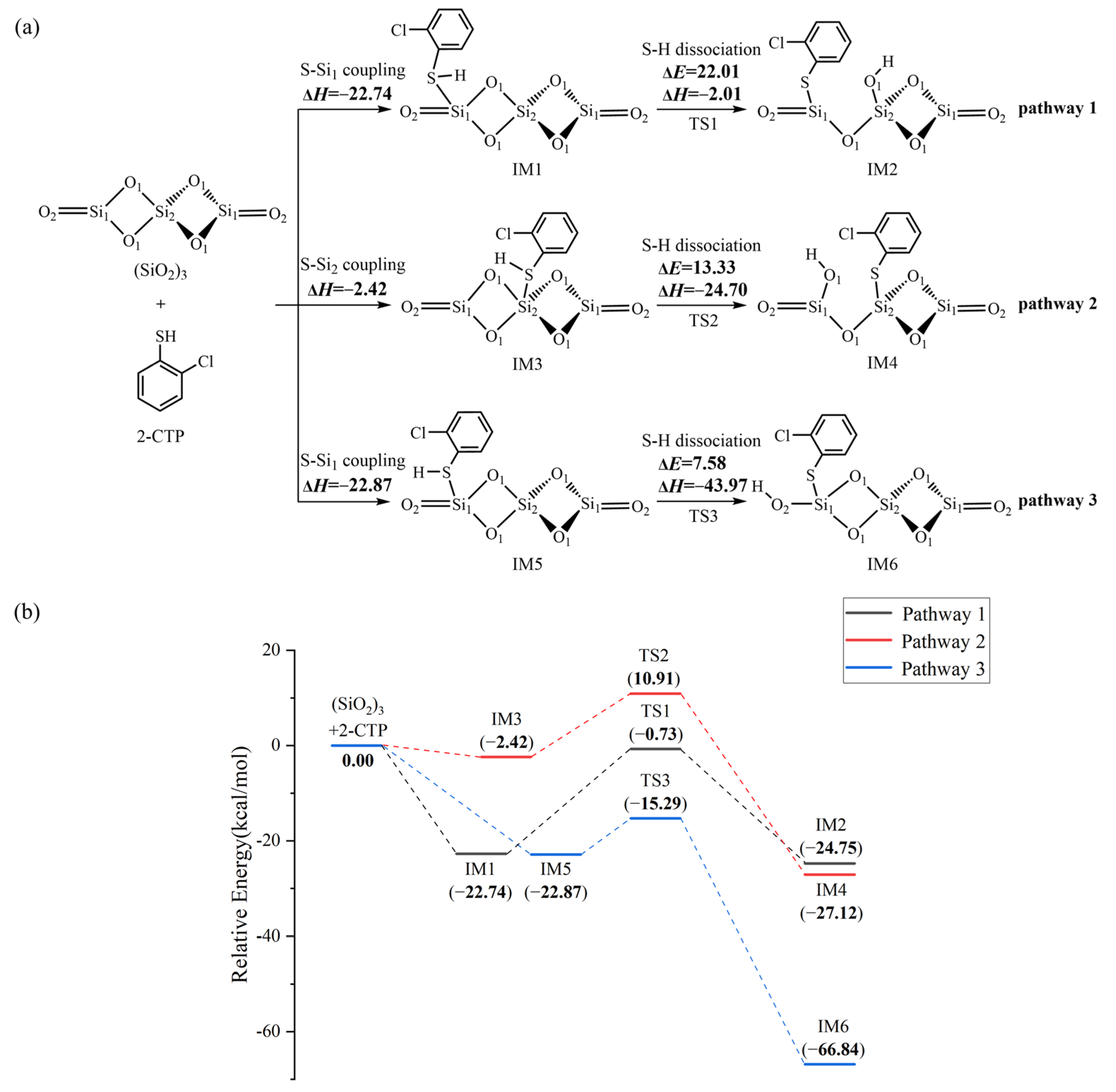
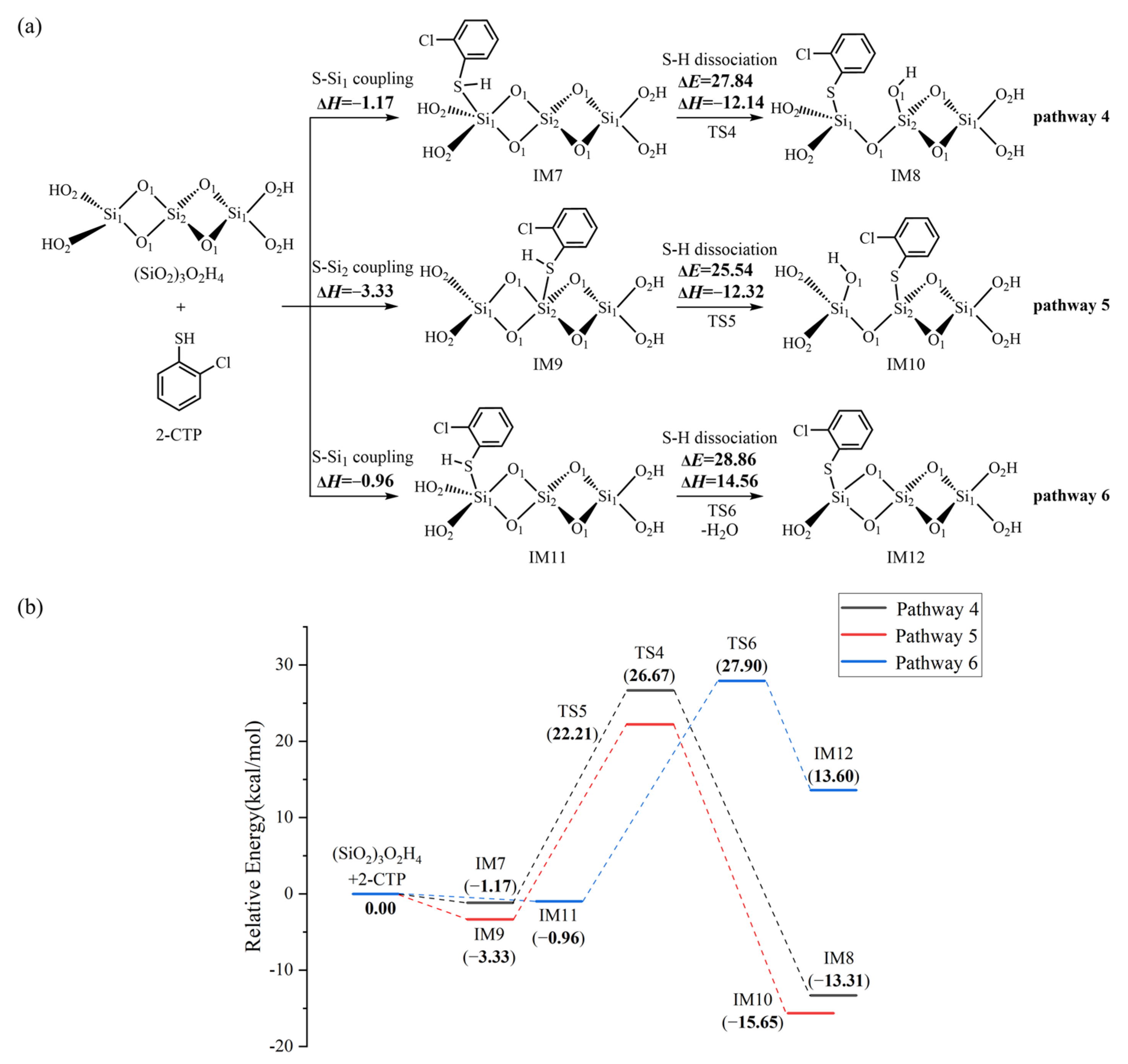
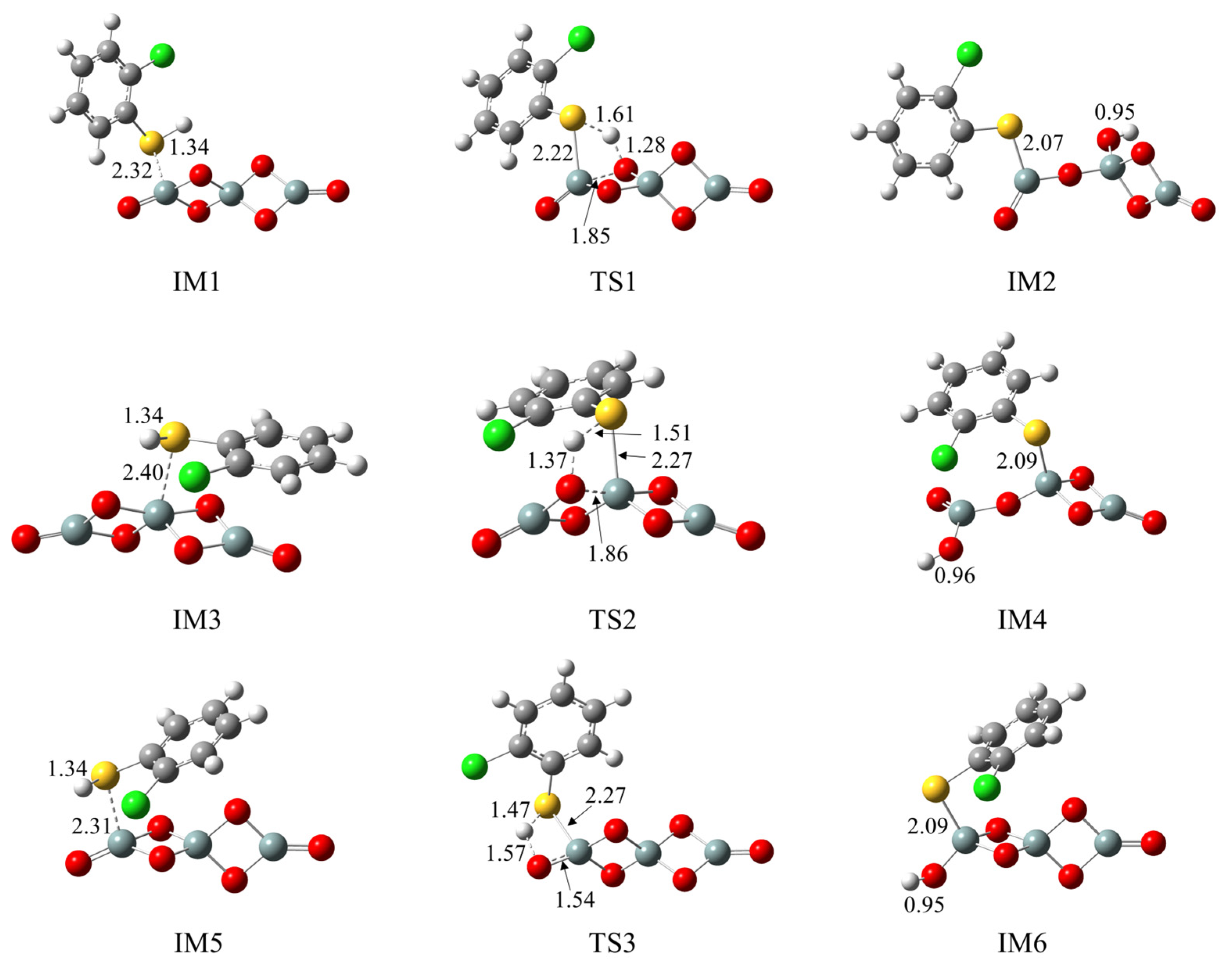
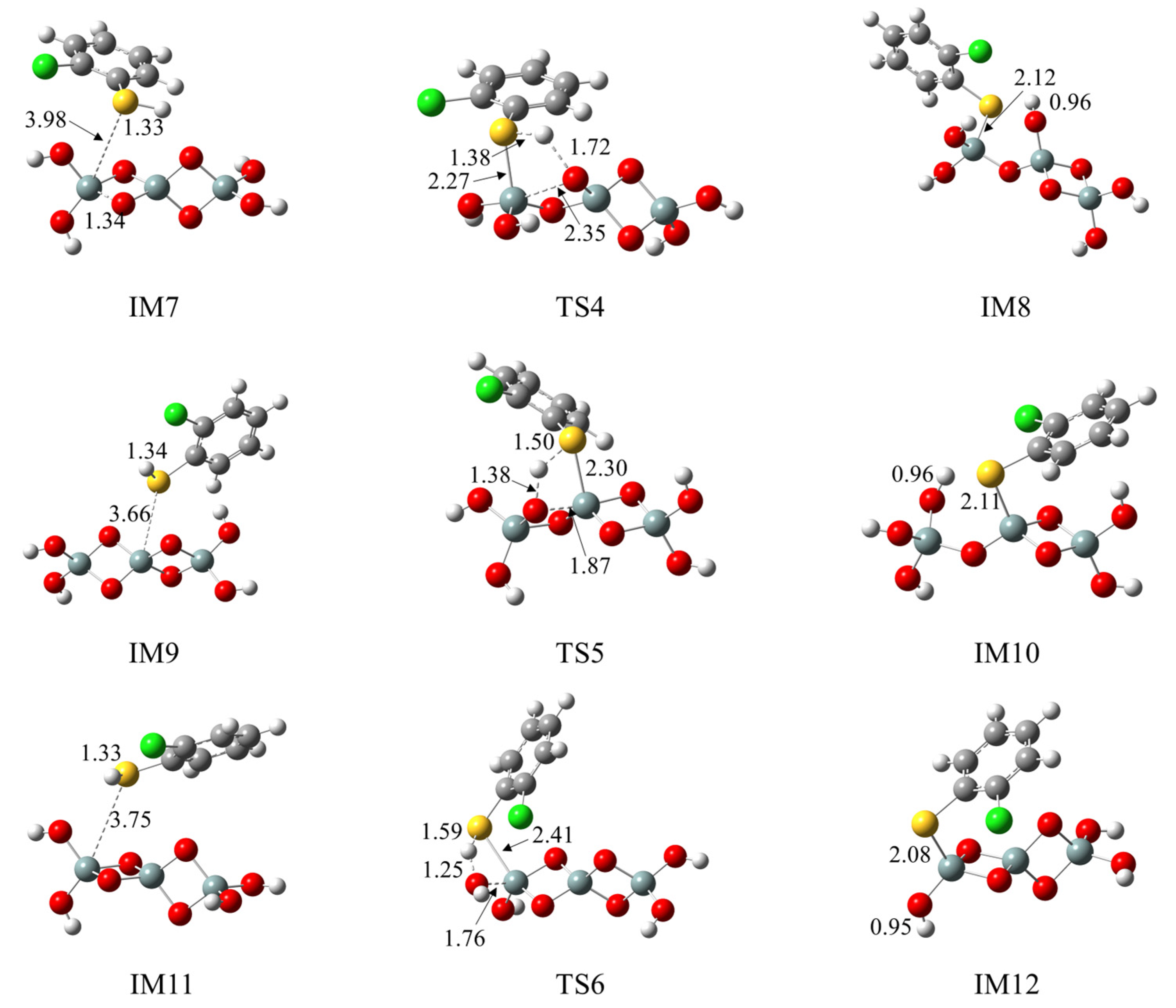
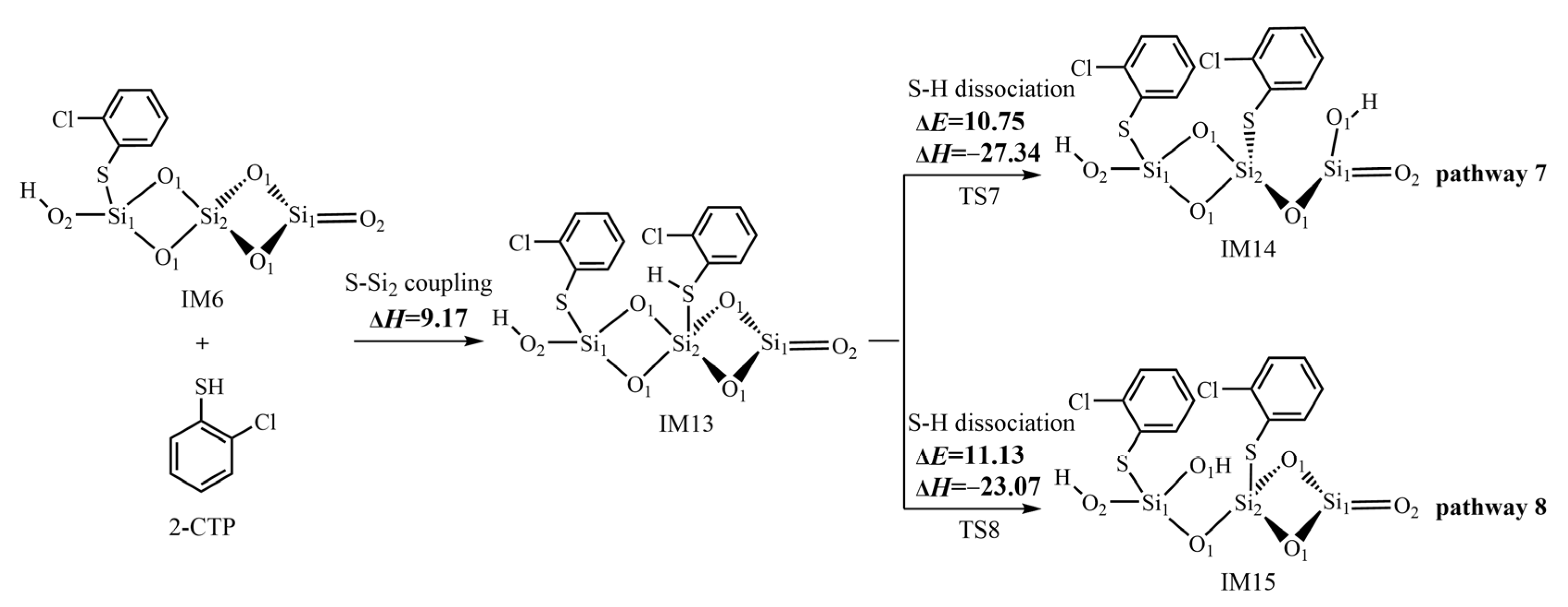
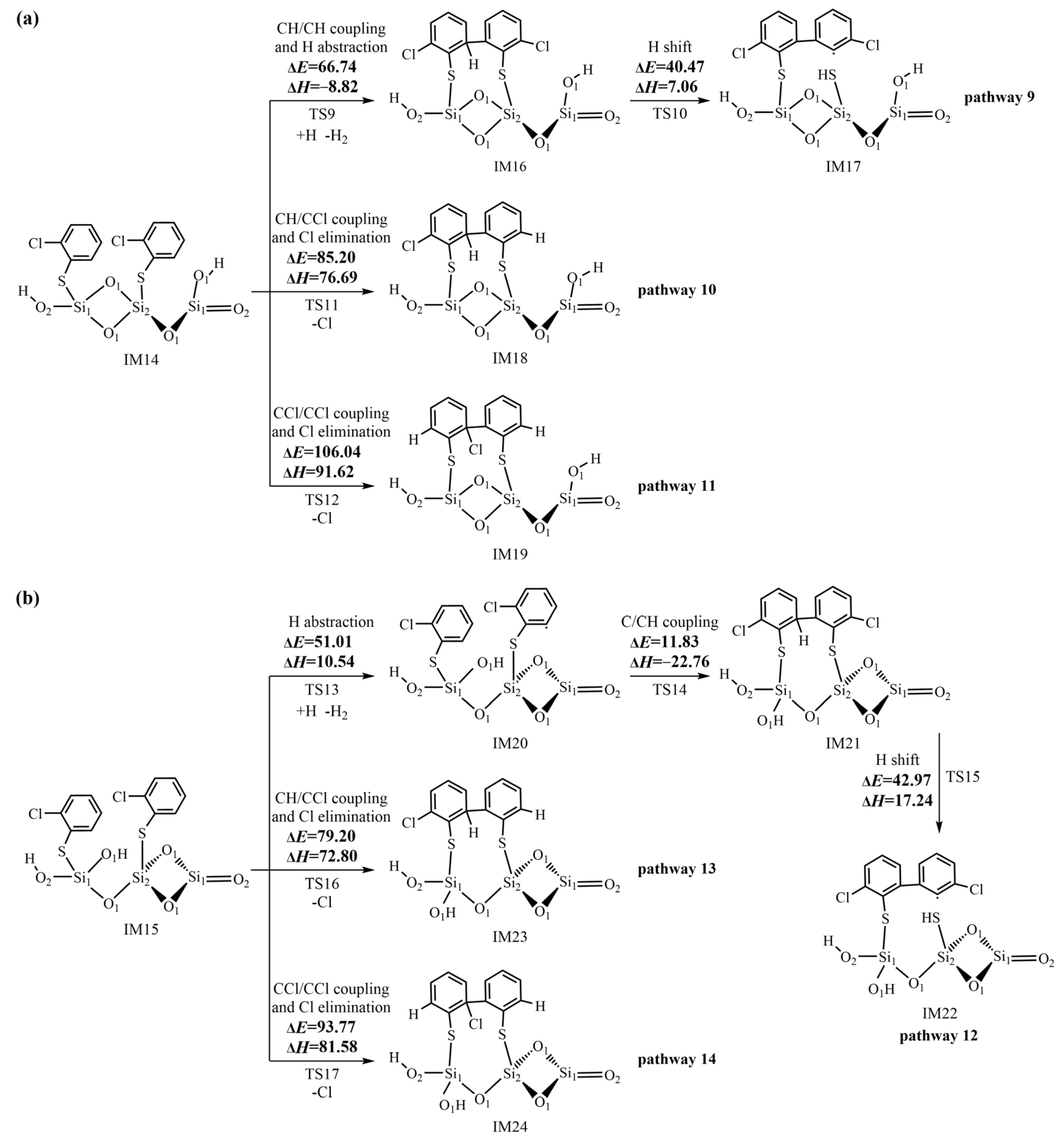
2.4. Formation of Pre-PCTA/DT Intermediates via the E–R Mechanism
2.5. Rate Constant Calculations
3. Discussion
3.1. The Formation of 2-Chlorothiophenolate on Silica Clusters
3.2. Formation of Pre-PCDT Intermediates via the L–H Mechanism
3.3. Formation of Pre-PCTA/DT Intermediates via the E–R Mechanism
3.4. Rate Constant Calculations
4. Materials and Methods
4.1. Density Functional Theory
4.2. Kinetic Calculations
4.3. Accuracy Verification
5. Conclusions
- (1)
- The adsorption and S-H dissociation of 2-CTP to form 2-chlorothiophenolate preferentially occur at the end of the dehydrated silica cluster, while those processes primarily take place in the middle of the hydroxylated silica cluster. Comparatively, the dehydrated silica cluster is more effective in catalyzing the conversion of 2-CTP to 2-chlorothiophenolate than the hydroxylated silica cluster.
- (2)
- 2-CTP, as a precursor adsorbed on the dehydrated silica cluster, can lead to the formation of pre-PCDTs via the L–H mechanism, while the E–R mechanism can result in the generation of pre-PCTAs. The formation of pre-PCTAs via the E–R mechanism is more prone to occur than the formation of pre-PCDTs through the L–H mechanism.
- (3)
- The silica is a relatively mild catalyst that can facilitate the conversion of 2-CTP to pre-PCTA/DTs. However, considering the high concentration of silica in fly ash, the catalytic effect of silica cannot be ignored.
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Benz, T.; Hagenmaier, H.; Lindig, C.; She, J. Occurrence of the sulphur analogue of octachlorodibenzo-p-dioxin in the environment and investigations on its potential source. Fresenius J. Anal. Chem. 1992, 344, 286–291. [Google Scholar] [CrossRef]
- Nakai, S.; Kishita, S.; Nomura, Y.; Hosomi, M. Polychlorinated dibenzothiophenes in Japanese environmental samples and their photodegradability and dioxin-like endocrine-disruption potential. Chemosphere 2007, 67, 1852–1857. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Qin, L.; Qu, R.; Feng, M.; Wei, Z.; Wang, L.; Wang, Z. Occurrence of polychlorinated diphenyl sulfides (PCDPSs) in surface sediments and surface water from the Nanjing section of the Yangtze River. Environ. Sci. Tech. 2014, 48, 11429–11436. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Dai, Y.; Zhang, R.; Shi, J.; Zhang, X.; Liu, B.; Feng, M. Occurrence, distribution and partitioning of polychlorinated dibenzothiophenes (PCDTs) in Chaohu Lake, Southeast China. Environ. Pollut. 2021, 277, 116751. [Google Scholar] [CrossRef] [PubMed]
- Sinkkonen, S.; Kolehmainen, E.; Paasivirta, J.; Koistinen, J.; Lahtipera, M.; Lammi, R. Identification and level estimation of chlorinated neutral aromatic sulfur compounds and their alkylated derivatives in pulp mill effluents and sediments. Chemosphere 1994, 28, 2049–2066. [Google Scholar] [CrossRef]
- Aurell, J.; Fick, J.; Haglund, P.; Marklund, S. Effects of sulfur on PCDD/F formation under stable and transient combustion conditions during MSW incineration. Chemosphere 2009, 76, 767–773. [Google Scholar] [CrossRef]
- Wiedmann, T.; Riehle, U.; Kurz, J.; Ballschmiter, K. HRGC-MS of polychlorinated phenanthrenes (PCPhen), dibenzothiophenes (PCDT), dibenzothianthrenes (PCTA), and phenoxathiins (PCPT). Fresenius J. Anal. Chem. 1997, 359, 176–188. [Google Scholar] [CrossRef]
- Sato, S.; Matsumura, A.; Urushigawa, Y.; Metwally, M.; Al-Muzaini, S. Structural analysis of weathered oil from Kuwait’s environment. Environ. Int. 1998, 24, 77–87. [Google Scholar] [CrossRef]
- Pruell, R.; Rubinstein, N.; Taplin, B.; LiVolsi, J.; Bowen, R. Accumulation of polychlorinated organic contaminants from sediment by three benthic marine species. Arch. Environ. Contam. Toxicol. 1993, 24, 290–297. [Google Scholar] [CrossRef]
- Dar, T.; Altarawneh, M.; Dlugogorski, B.Z. Quantum chemical study on formation of PCDT/TA from 2-chlorothiophenol precursor. Environ. Sci. Tech. 2013, 47, 11040–11047. [Google Scholar] [CrossRef] [PubMed]
- Qu, X.; Wang, H.; Zhang, Q.; Shi, X.; Xu, F.; Wang, W. Mechanistic and kinetic studies on the homogeneous gas-phase formation of PCDD/Fs from 2, 4, 5-trichlorophenol. Environ. Sci. Tech. 2009, 43, 4068–4075. [Google Scholar] [CrossRef]
- Xu, F.; Shi, X.; Li, Y.; Zhang, Q. Mechanistic and kinetic studies on the homogeneous gas-phase formation of PCTA/DTs from 2, 4-dichlorothiophenol and 2, 4, 6-trichlorothiophenol. Int. J. Mol. Sci. 2015, 16, 20449–20467. [Google Scholar] [CrossRef]
- Li, H.; Liu, W.; Tang, C.; Lei, R.; Zhu, W. Emission profiles and formation pathways of 2, 3, 7, 8-substituted and non-2, 3, 7, 8-substituted polychlorinated dibenzo-p-dioxins and dibenzofurans in secondary copper smelters. Sci. Total Environ. 2019, 649, 473–481. [Google Scholar] [CrossRef] [PubMed]
- Sinkkonen, S.; Vattulainen, A.; Aittola, J.P.; Paasivirta, J.; Tarhanen, J.; Lahtiperä, M. Metal reclamation produces sulphur analogues of toxic dioxins and furans. Chemosphere 1994, 28, 1279–1288. [Google Scholar] [CrossRef]
- Bei, J.; Xu, X.; Zhan, M.; Li, X.; Jiao, W.; Khachatryan, L.; Wu, A. Revealing the mechanism of dioxin formation from municipal solid waste gasification in a reducing atmosphere. Environ. Sci. Tech. 2022, 56, 14539–14549. [Google Scholar] [CrossRef]
- Stanmore, B. The formation of dioxins in combustion systems. Combust. Flame 2004, 136, 398–427. [Google Scholar] [CrossRef]
- Wang, M.; Liu, G.; Yang, L.; Zheng, M. Framework of the Integrated Approach to Formation Mechanisms of Typical Combustion Byproducts-Polyhalogenated Dibenzo-p-dioxins/Dibenzofurans (PXDD/Fs). Environ. Sci. Tech. 2023, 57, 2217–2234. [Google Scholar] [CrossRef]
- Nganai, S.; Lomnicki, S. Surface catalysed PCDD/F formation from precursors-high PCDF yield does not indicate de novo mechanism! Int. J. Environ. Pollut. 2017, 61, 208–222. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Altarawneh, M.; Dlugogorski, B.Z.; Kennedy, E.M.; Mackie, J.C. Catalytic effect of CuO and other transition metal oxides in formation of dioxins: Theoretical investigation of reaction between 2, 4, 5-trichlorophenol and CuO. Environ. Sci. Tech. 2007, 41, 5708–5715. [Google Scholar] [CrossRef]
- Yang, L.; Liu, G.; Zheng, M.; Zhao, Y.; Jin, R.; Wu, X.; Xu, Y. Molecular mechanism of dioxin formation from chlorophenol based on electron paramagnetic resonance spectroscopy. Environ. Sci. Tech. 2017, 51, 4999–5007. [Google Scholar] [CrossRef]
- Lomnicki, S.; Truong, H.; Vejerano, E.; Dellinger, B. Copper oxide-based model of persistent free radical formation on combustion-derived particulate matter. Environ. Sci. Tech. 2008, 42, 4982–4988. [Google Scholar] [CrossRef]
- Khachatryan, L.; Lomnicki, S.; Dellinger, B. An expanded reaction kinetic model of the CuO surface-mediated formation of PCDD/F from pyrolysis of 2-chlorophenol. Chemosphere 2007, 68, 1741–1750. [Google Scholar] [CrossRef]
- Nganai, S.; Dellinger, B.; Lomnicki, S. PCDD/PCDF ratio in the precursor formation model over CuO surface. Environ. Sci. Tech. 2014, 48, 13864–13870. [Google Scholar] [CrossRef]
- Nganai, S.; Lomnicki, S.M.; Dellinger, B. Formation of PCDD/Fs from the copper oxide-mediated pyrolysis and oxidation of 1, 2-dichlorobenzene. Environ. Sci. Tech. 2011, 45, 1034–1040. [Google Scholar] [CrossRef]
- Lomnicki, S.; Dellinger, B. A detailed mechanism of the surface-mediated formation of PCDD/F from the oxidation of 2-chlorophenol on a CuO/silica surface. J. Phys. Chem. A 2003, 107, 4387–4395. [Google Scholar] [CrossRef]
- Liu, G.; Jiang, X.; Wang, M.; Dong, S.; Zheng, M. Comparison of PCDD/F levels and profiles in fly ash samples from multiple industrial thermal sources. Chemosphere 2015, 133, 68–74. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Meng, A.; Long, Y.; Li, Q.; Zhang, Y. A review of dioxin-related substances during municipal solid waste incineration. Waste Manag. 2015, 36, 106–118. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Zheng, M.; Zhao, Y.; Yang, H.; Yang, L.; Jin, R.; Xu, Y.; Xiao, K.; Liu, W.; Liu, G. Thermochemical formation of polychlorinated dibenzo-p-dioxins and dibenzofurans on the fly ash matrix from metal smelting sources. Chemosphere 2018, 191, 825–831. [Google Scholar] [CrossRef]
- Fujimori, T.; Tanino, Y.; Takaoka, M. Coexistence of Cu, Fe, Pb, and Zn oxides and chlorides as a determinant of chlorinated aromatics generation in municipal solid waste incinerator fly ash. Environ. Sci. Tech. 2014, 48, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Li, W.; Xiong, Z.; Xia, D.; Yang, C.; Wang, W.; Sun, Y. Synergistic effect of iron and copper oxides on the formation of persistent chlorinated aromatics in iron ore sintering based on in situ XPS analysis. J. Hazard. Mater. 2019, 366, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Liu, G.; Jiang, X.; Zheng, M.; Yang, L.; Zhao, Y.; Jin, R. Thermochemical formation of polybrominated dibenzo-p-dioxins and dibenzofurans mediated by secondary copper smelter fly ash, and implications for emission reduction. Environ. Sci. Tech. 2016, 50, 7470–7479. [Google Scholar] [CrossRef]
- Liu, S.; Liu, G.; Yang, L.; Liu, X.; Wang, M.; Qin, L.; Zheng, M. Metal-catalyzed formation of organic pollutants intermediated by organic free radicals. Environ. Sci. Tech. 2022, 56, 14550–14561. [Google Scholar] [CrossRef] [PubMed]
- Vejerano, E.; Lomnicki, S.M.; Dellinger, B. Formation and stabilization of combustion-generated, environmentally persistent radicals on Ni (II) O supported on a silica surface. Environ. Sci. Tech. 2012, 46, 9406–9411. [Google Scholar] [CrossRef] [PubMed]
- Nganai, S.; Lomnicki, S.; Dellinger, B. Ferric oxide mediated formation of PCDD/Fs from 2-monochlorophenol. Environ. Sci. Tech. 2009, 43, 368–373. [Google Scholar] [CrossRef] [PubMed]
- Mosallanejad, S.; Dlugogorski, B.Z.; Kennedy, E.M.; Stockenhuber, M.; Lomnicki, S.M.; Assaf, N.W.; Altarawneh, M. Formation of PCDD/Fs in oxidation of 2-chlorophenol on neat silica surface. Environ. Sci. Tech. 2016, 50, 1412–1418. [Google Scholar] [CrossRef] [PubMed]
- Alderman, S.L.; Dellinger, B. FTIR investigation of 2-chlorophenol chemisorption on a silica surface from 200 to 500 C. J. Phys. Chem. A 2005, 109, 7725–7731. [Google Scholar] [CrossRef] [PubMed]
- Pan, W.; Zhong, W.; Zhang, D.; Liu, C. Theoretical study of the reactions of 2-chlorophenol over the dehydrated and hydroxylated silica clusters. J. Phys. Chem. A 2012, 116, 430–436. [Google Scholar] [CrossRef] [PubMed]
- Longo, R.C.; Ueda, H.; Cho, K.; Ranjan, A.; Ventzek, P.L. Mechanisms for graphene growth on SiO2 using plasma-enhanced chemical vapor deposition: A density functional theory study. ACS Appl. Mater. Interfaces 2022, 14, 9492–9503. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Zhang, R. A structural model of one-dimensional thin silica nanowires. Chem. Phys. Lett. 2004, 394, 437–440. [Google Scholar] [CrossRef]
- Hosono, H.; Ikuta, Y.; Kinoshita, T.; Kajihara, K.; Hirano, M. Physical disorder and optical properties in the vacuum ultraviolet region of amorphous SiO2. Phys. Rev. Lett. 2001, 87, 175501–175505. [Google Scholar] [CrossRef]
- Xu, F.; Shi, X.; Zhang, Q.; Wang, W. Formation of chlorotriophenoxy radicals from complete series reactions of chlorotriophenols with H and OH radicals. Int. J. Mol. Sci. 2015, 16, 18714–18731. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision A.02; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Zhao, Y.; Truhlar, D.G. Hybrid meta density functional theory methods for thermochemistry, thermochemical kinetics, and noncovalent interactions: The MPW1B95 and MPWB1K models and comparative assessments for hydrogen bonding and van der Waals interactions. J. Phys. Chem. A 2004, 108, 6908–6918. [Google Scholar] [CrossRef]
- Fukui, K. The path of chemical reactions-the IRC approach. Acc. Chem. Res. 1981, 14, 363–368. [Google Scholar] [CrossRef]
- Canneaux, S.; Bohr, F.; Henon, E. KiSThelP: A program to predict thermodynamic properties and rate constants from quantum chemistry results. J. Comput. Chem. 2014, 35, 82–93. [Google Scholar] [CrossRef] [PubMed]
- Larson, N.W.; Nicolaisen, F.M. Far-infrared gas spectra of phenol, 4-fluorophenol, thiophenol and some deuterated species: Barrier to internal rotation. J. Mol. Struct. 1974, 22, 29–43. [Google Scholar] [CrossRef]
- Nyquist, R.A.; Evans, J.C. The vibrational spectra of p-chlorobenzenethiol. Spectrochim. Acta 1961, 17, 795–801. [Google Scholar] [CrossRef]
- Xu, H.; Han, Z.; Zhang, D.J.; Liu, C.B. Theoretical elucidation of the dual role of [HMIm]BF4 ionic liquid as catalyst and extractant in the oxidative desulfurization of dibenzothiophene. J. Mol. Catal. A Chem. 2015, 398, 297–303. [Google Scholar] [CrossRef]
- Lee, S.Y. Density functional theory calculation of molecular structure and vibrational spectra of dibenzothiophene in the ground and the lowest triplet state. J. Phys. Chem. A 2001, 105, 8093–8097. [Google Scholar] [CrossRef]
- Johnson, R.D., III. NIST Computational Chemistry Comparison and Benchmark Database, NIST Standard Reference Database; Department of Commerce, USA, Release 16a, August. 2013. Available online: http://cccbdb.nist.gov/ (accessed on 28 August 2015).



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reactions | Arrhenius Formulas |
|---|---|
| IM1 → IM2 via TS1 | k(T) = (8.63 × 1012) exp (−11,138/T) |
| IM3 → IM4 via TS2 | k(T) = (8.24 × 1011) exp (−6651/T) |
| IM5 → IM6 via TS3 | k(T) = (4.55 × 1013) exp (−3936/T) |
| IM7 → IM8 via TS4 | k(T) = (8.84 × 1011) exp (−13,886/T) |
| IM9 → IM10 via TS5 | k(T) = (3.33 × 1010) exp (−12,643/T) |
| IM11 → IM12 via TS6 | k(T) = (1.63 × 1010) exp (−13,915/T) |
| IM13 → IM14 via TS7 | k(T) = (1.46 × 1013) exp (−5576/T) |
| IM13 → IM15 via TS8 | k(T) = (1.53 × 1012) exp (−5482/T) |
| IM14 → IM18 + Cl via TS11 | k(T) = (1.61 × 1012) exp (−43,242/T) |
| IM14 → IM19+ Cl via TS12 | k(T) = (3.10 × 1012) exp (−53,879/T) |
| IM15 → IM20 + H2 via TS13 | k(T) = (5.44 × 10−14) exp (−25,324/T) |
| IM15 → IM23 + Cl via TS16 | k(T) = (1.16 × 1012) exp (−40,134/T) |
| IM15 → IM24 + Cl via TS17 | k(T) = (3.19 × 1011) exp (−47,477/T) |
| IM6 + 2-CTP → IM25 + Cl via TS18 | k(T) = (1.03 × 10−14) exp (−37,954/T) |
| IM6 + 2-CTP → IM26 + Cl via TS19 | k(T) = (5.02 × 10−15) exp (−41,857/T) |
| IM6 + 2-CTP → IM27 + H2 via TS20 | k(T) = (1.88 × 10−15) exp (−52,003/T) |
| IM6 + 2-CTP → IM28 + Cl via TS21 | k(T) = (2.75 × 10−14) exp (−29,308/T) |
| IM29 + 2-CP → IM30 + Cl via TS22 | k(T) = (7.78 × 10−16) exp (−33,859/T) |
| IM29 + 2-CP → IM31 + Cl via TS23 | k(T) = (7.83 × 10−16) exp (−38,866/T) |
| IM29 + 2-CP → IM32 + H2 via TS24 | k(T) = (1.61 × 10−16) exp (−56,864/T) |
| IM29 + 2-CP → IM33 + Cl via TS25 | k(T) = (2.05 × 10−15) exp (−33,514/T) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, F.; Wang, X.; Li, Y.; Hu, Y.; Zhou, Y.; Hadizadeh, M.H. Formation of Pre-PCTA/DT Intermediates from 2-Chlorothiophenol on Silica Clusters: A Quantum Mechanical Study. Int. J. Mol. Sci. 2024, 25, 3485. https://doi.org/10.3390/ijms25063485
Xu F, Wang X, Li Y, Hu Y, Zhou Y, Hadizadeh MH. Formation of Pre-PCTA/DT Intermediates from 2-Chlorothiophenol on Silica Clusters: A Quantum Mechanical Study. International Journal of Molecular Sciences. 2024; 25(6):3485. https://doi.org/10.3390/ijms25063485
Chicago/Turabian StyleXu, Fei, Xiaotong Wang, Ying Li, Yongxia Hu, Ying Zhou, and Mohammad Hassan Hadizadeh. 2024. "Formation of Pre-PCTA/DT Intermediates from 2-Chlorothiophenol on Silica Clusters: A Quantum Mechanical Study" International Journal of Molecular Sciences 25, no. 6: 3485. https://doi.org/10.3390/ijms25063485
APA StyleXu, F., Wang, X., Li, Y., Hu, Y., Zhou, Y., & Hadizadeh, M. H. (2024). Formation of Pre-PCTA/DT Intermediates from 2-Chlorothiophenol on Silica Clusters: A Quantum Mechanical Study. International Journal of Molecular Sciences, 25(6), 3485. https://doi.org/10.3390/ijms25063485