

PCNA Unloading Is Crucial for the Bypass of DNA Lesions Using Homologous Recombination


Abstract
1. Introduction
2. Results
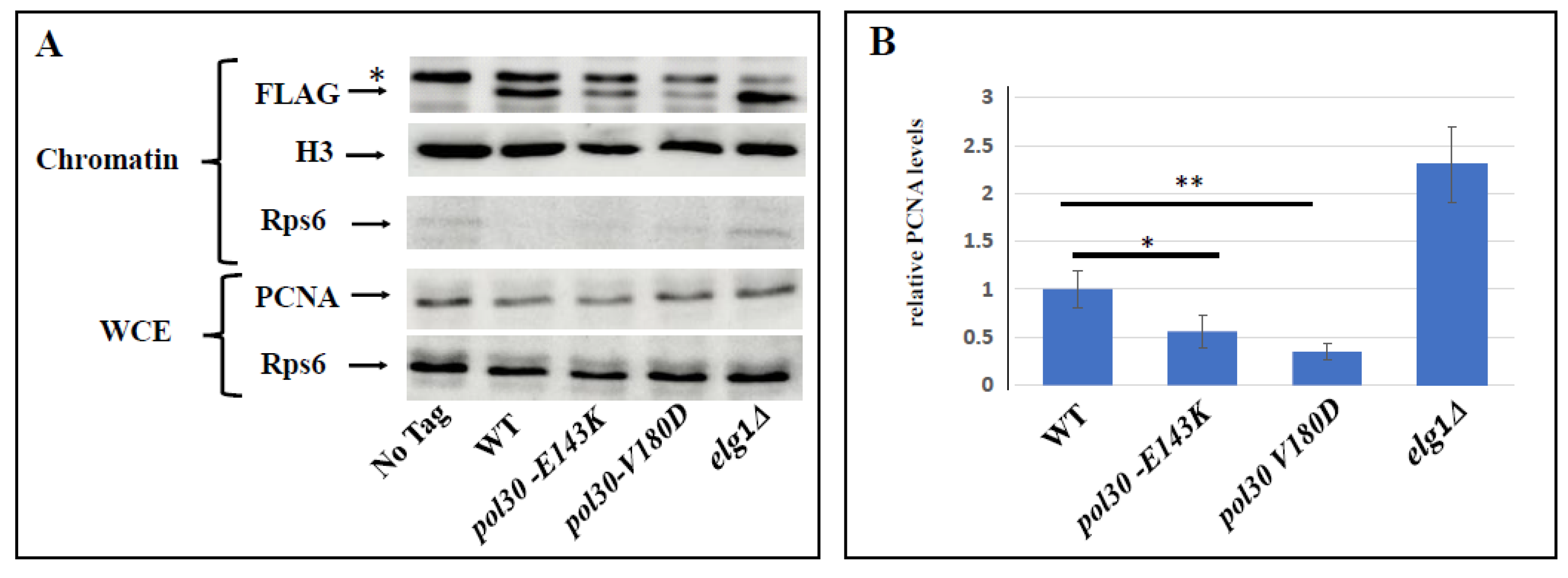
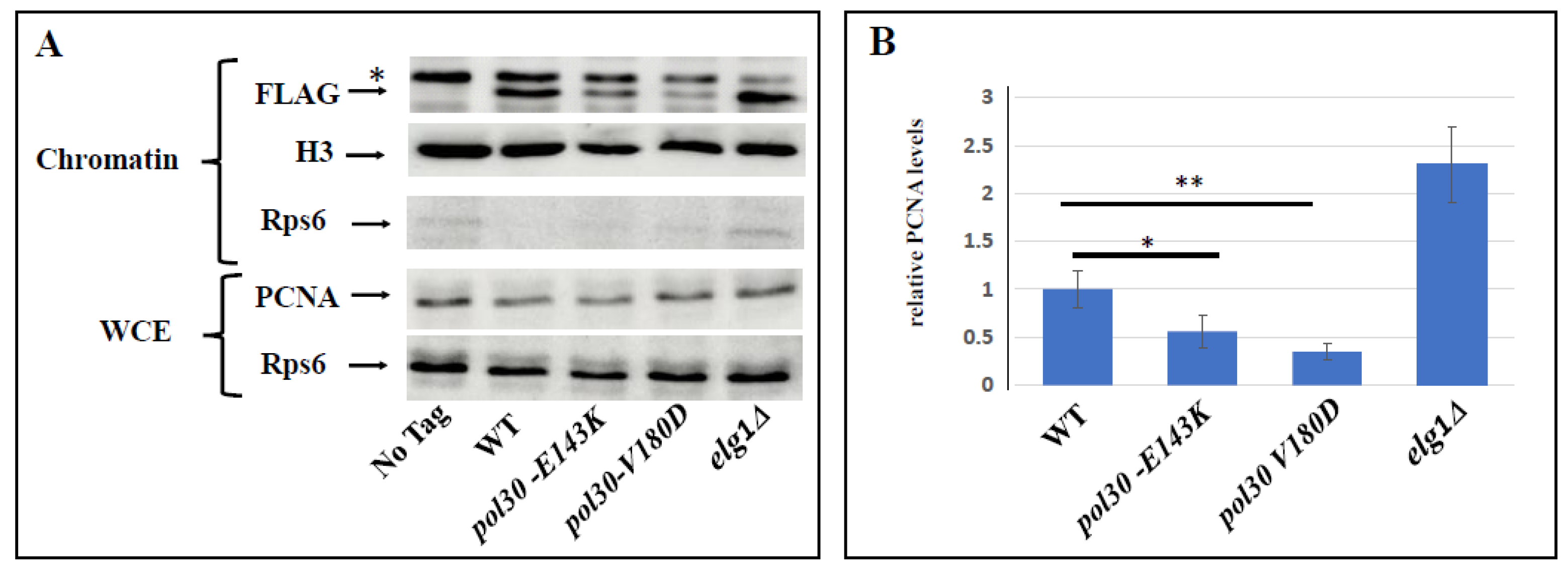
2.1. Spontaneous Unloading of PCNA Suppresses the Sensitivity Conferred by an Inactive DDT Mechanism
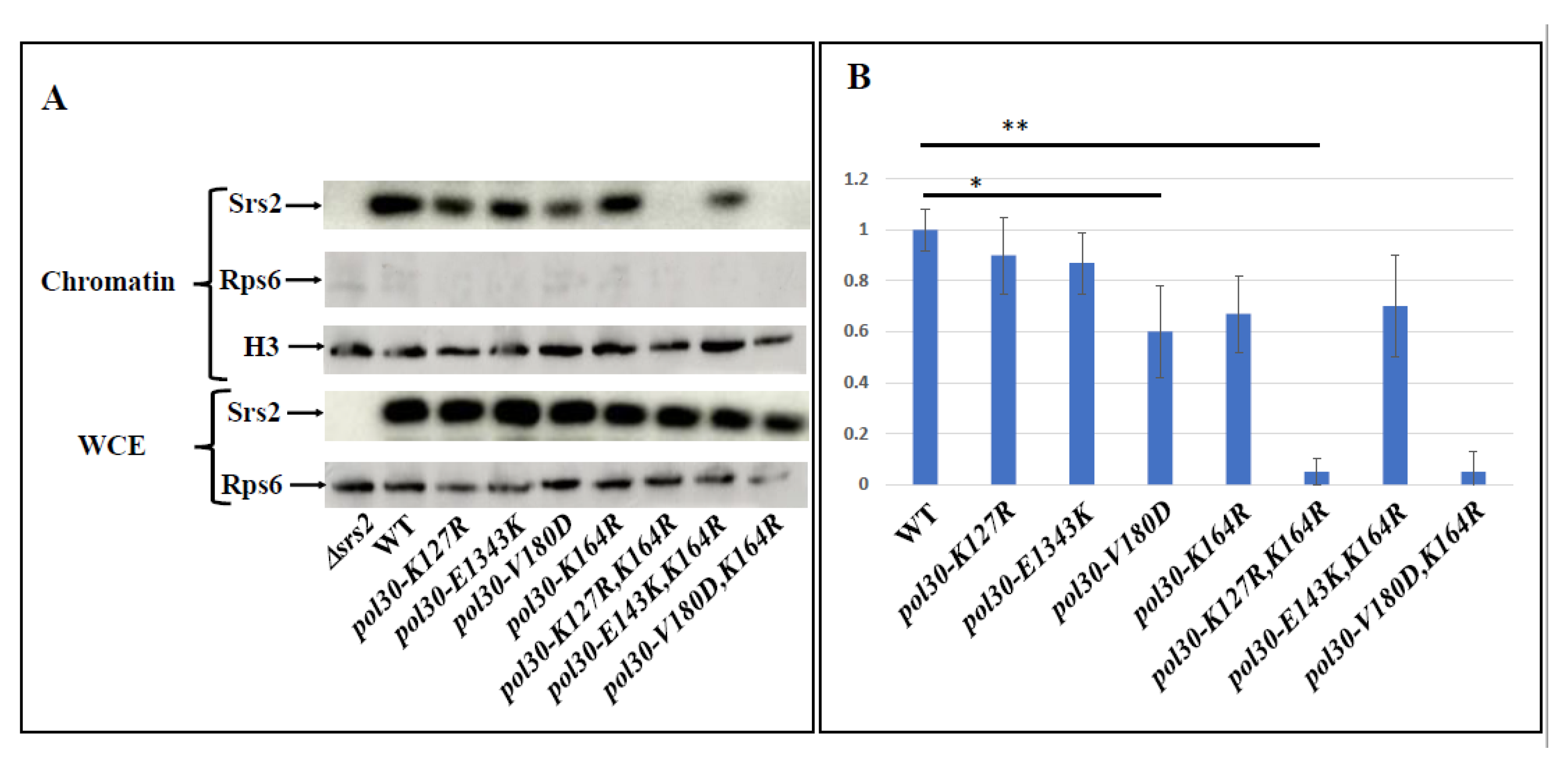
2.2. Unstable PCNA Results in Low Amount of Srs2 on the Chromatin
2.3. DPP Suppression of pol30-K164R Is Dependent on Homologous Recombination (HR)
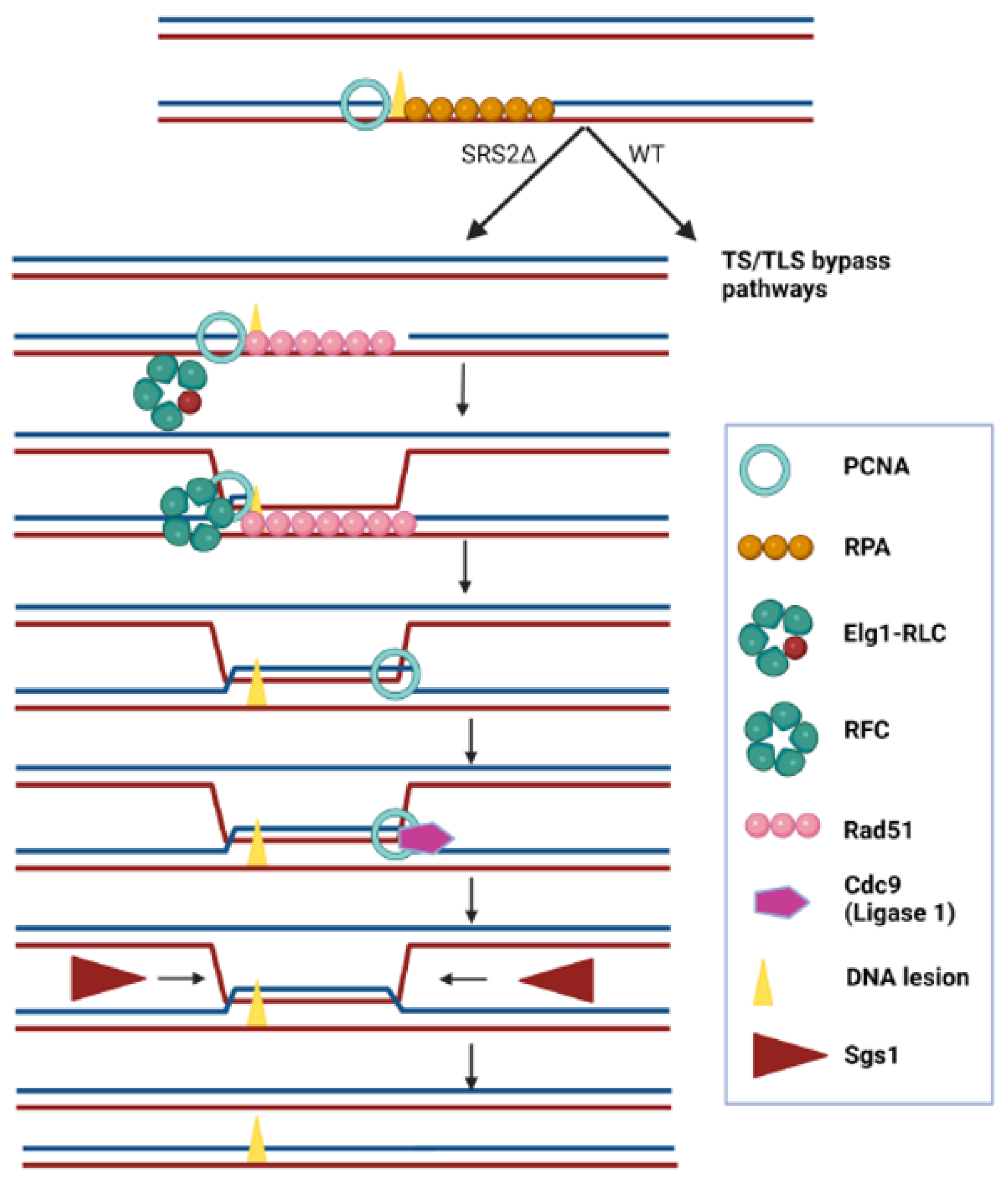
2.4. Suppression of the Synthetic Sickness of elg1∆srs2∆ Double Mutants by Spontaneous Unloading of PCNA
3. Discussion
4. Materials and Methods
4.1. Chromatin Fractionation Assay
4.2. DNA Damage Sensitivity Assays
4.3. Western Blot Analysis
4.4. Tetrad Dissection
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Boiteux, S.; Jinks-Robertson, S. DNA Repair Mechanisms and the Bypass of DNA Damage in Saccharomyces Cerevisiae. Genetics 2013, 193, 1025–1064. [Google Scholar] [CrossRef]
- Moldovan, G.-L.; Pfander, B.; Jentsch, S. PCNA, the Maestro of the Replication Fork. Cell 2007, 129, 665–679. [Google Scholar] [CrossRef]
- Stelter, P.; Ulrich, H.D. Control of Spontaneous and Damage-Induced Mutagenesis by SUMO and Ubiquitin Conjugation. Nature 2003, 425, 188–191. [Google Scholar] [CrossRef]
- Choe, K.N.; Moldovan, G.L. Forging Ahead through Darkness: PCNA, Still the Principal Conductor at the Replication Fork. Mol. Cell 2017, 65, 380–392. [Google Scholar] [CrossRef] [PubMed]
- Madru, C.; Henneke, G.; Raia, P.; Hugonneau-Beaufet, I.; Pehau-Arnaudet, G.; England, P.; Lindahl, E.; Delarue, M.; Carroni, M.; Sauguet, L. Structural Basis for the Increased Processivity of D-Family DNA Polymerases in Complex with PCNA. Nat. Commun. 2020, 11, 1591. [Google Scholar] [CrossRef]
- Matunis, M.J. On the Road to Repair: PCNA Encounters SUMO and Ubiquitin Modifications. Mol. Cell 2002, 10, 441–442. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Qin, Z.; Zhang, X.; Xiao, W. Roles of Sequential Ubiquitination of PCNA in DNA-Damage Tolerance. FEBS Lett. 2011, 585, 2786–2794. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.Y.; Myung, K. PCNA Modifications for Regulation of Post-Replication Repair Pathways. Mol. Cells 2008, 26, 5–11. [Google Scholar] [CrossRef] [PubMed]
- Hoege, C.; Pfander, B.; Moldovan, G.-L.; Pyrowolakis, G.; Jentsch, S. RAD6-Dependent DNA Repair Is Linked to Modification of PCNA by Ubiquitin and SUMO. Nature 2002, 419, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Ulrich, H.D.; Walden, H. Ubiquitin Signalling in DNA Replication and Repair. Nat. Rev. Mol. Cell Biol. 2010, 11, 479–489. [Google Scholar] [CrossRef]
- Hofmann, R.M.; Pickart, C.M. Noncanonical MMS2-Encoded Ubiquitin-Conjugating Enzyme Functions in Assembly of Novel Polyubiquitin Chains for DNA Repair. Cell 1999, 96, 645–653. [Google Scholar] [CrossRef] [PubMed]
- Minca, E.C.; Kowalski, D. Multiple Rad5 Activities Mediate Sister Chromatid Recombination to Bypass DNA Damage at Stalled Replication Forks. Mol. Cell 2010, 38, 649–661. [Google Scholar] [CrossRef] [PubMed]
- Eddins, M.J.; Carlile, C.M.; Gomez, K.M.; Pickart, C.M.; Wolberger, C. Mms2-Ubc13 Covalently Bound to Ubiquitin Reveals the Structural Basis of Linkage-Specific Polyubiquitin Chain Formation. Nat. Struct. Mol. Biol. 2006, 13, 915–920. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.S.; Ryu, E.; Lee, S.W.; Park, J.; Ha, N.Y.; Ra, J.S.; Kim, Y.J.; Kim, J.; Abdel-Rahman, M.; Park, S.H.; et al. Regulation of PCNA Cycling on Replicating DNA by RFC and RFC-like Complexes. Nat. Commun. 2019, 10, 20420. [Google Scholar] [CrossRef]
- Johnson, C.; Gali, V.K.; Takahashi, T.S.; Kubota, T. PCNA Retention on DNA into G2/M Phase Causes Genome Instability in Cells Lacking Elg1. Cell Rep. 2016, 16, 684–695. [Google Scholar] [CrossRef]
- Kubota, T.; Katou, Y.; Nakato, R.; Shirahige, K.; Donaldson, A.D. Replication-Coupled PCNA Unloading by the Elg1 Complex Occurs Genome-Wide and Requires Okazaki Fragment Ligation. Cell Rep. 2015, 12, 774–787. [Google Scholar] [CrossRef]
- Bellaoui, M.; Chang, M.; Ou, J.; Xu, H.; Boone, C.; Brown, G.W. Elg1 Forms an Alternative RFC Complex Important for DNA Replication and Genome Integrity. EMBO J. 2003, 22, 4304–4313. [Google Scholar] [CrossRef]
- Kubota, T.; Nishimura, K.; Kanemaki, M.T.; Donaldson, A.D. The Elg1 Replication Factor C-like Complex Functions in PCNA Unloading during DNA Replication. Mol. Cell 2013, 50, 273–280. [Google Scholar] [CrossRef]
- Shemesh, K.; Sebesta, M.; Pacesa, M.; Sau, S.; Bronstein, A.; Parnas, O.; Liefshitz, B.; Venclovas, Č.; Krejci, L.; Kupiec, M. A Structure-Function Analysis of the Yeast Elg1 Protein Reveals the Importance of PCNA Unloading in Genome Stability Maintenance. Nucleic Acids Res. 2017, 45, 3189–3203. [Google Scholar] [CrossRef] [PubMed]
- Papouli, E.; Chen, S.; Davies, A.A.; Huttner, D.; Krejci, L.; Sung, P.; Ulrich, H.D. Crosstalk between SUMO and Ubiquitin on PCNA Is Mediated by Recruitment of the Helicase Srs2p. Mol. Cell 2005, 19, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Watts, F.Z. Sumoylation of PCNA: Wrestling with Recombination at Stalled Replication Forks. DNA Repair 2006, 5, 399–403. [Google Scholar] [CrossRef]
- Pfander, B.; Moldovan, G.-L.; Sacher, M.; Hoege, C.; Jentsch, S. SUMO-Modified PCNA Recruits Srs2 to Prevent Recombination during S Phase. Nature 2005, 436, 428–433. [Google Scholar] [CrossRef] [PubMed]
- Niu, H.; Klein, H.L. Multifunctional Roles of Saccharomyces Cerevisiae Srs2 Protein in Replication, Recombination and Repair. FEMS Yeast Res. 2017, 17, fow111. [Google Scholar] [CrossRef] [PubMed]
- Candelli, A.; Holthausen, J.T.; Depken, M.; Brouwer, I.; Franker, M.A.M.; Marchetti, M.; Heller, I.; Bernard, S.; Garcin, E.B.; Modesti, M.; et al. Visualization and Quantification of Nascent RAD51 Filament Formation at Single-Monomer Resolution. Proc. Natl. Acad. Sci. USA 2014, 111, 15090–150905. [Google Scholar] [CrossRef]
- Gali, H.; Juhasz, S.; Morocz, M.; Hajdu, I.; Fatyol, K.; Szukacsov, V.; Burkovics, P.; Haracska, L. Role of SUMO Modification of Human PCNA at Stalled Replication Fork. Nucleic Acids Res. 2012, 40, 6049–6059. [Google Scholar] [CrossRef]
- Marini, V.; Krejci, L. Srs2: The “Odd-Job Man” in DNA Repair. DNA Repair 2010, 9, 268–275. [Google Scholar] [CrossRef] [PubMed]
- Krejci, L.; Van Komen, S.; Li, Y.; Villemain, J.; Reddy, M.S.; Klein, H.; Ellenberger, T.; Sung, P. DNA Helicase Srs2 Disrupts the Rad51 Presynaptic Filament. Nature 2003, 423, 305–309. [Google Scholar] [CrossRef]
- Veaute, X.; Jeusset, J.; Soustelle, C.; Kowalczykowski, S.C.; Le Cam, E.; Fahre, F. The Srs2 Helicase Prevents Recombination by Disrupting Rad51 Nucleoprotein Filaments. Nature 2003, 423, 309–312. [Google Scholar] [CrossRef]
- Arbel, M.; Bronstein, A.; Sau, S.; Liefshitz, B.; Kupiec, M. Access to Pcna by Srs2 and Elg1 Controls the Choice between Alternative Repair Pathways in Saccharomyces Cerevisiae. MBio 2020, 11, e00705–e00720. [Google Scholar] [CrossRef]
- Goellner, E.M.; Smith, C.E.; Campbell, C.S.; Hombauer, H.; Desai, A.; Putnam, C.D.; Kolodner, R.D. PCNA and Msh2-Msh6 Activate an Mlh1-Pms1 Endonuclease Pathway Required for Exo1-Independent Mismatch Repair. Mol. Cell 2014, 55, 291–304. [Google Scholar] [CrossRef]
- Amin, N.S.; Nguyen, M.-N.; Oh, S.; Kolodner, R.D. Exo1-Dependent Mutator Mutations: Model System for Studying Functional Interactions in Mismatch Repair. Mol. Cell. Biol. 2001, 21, 5142–5155. [Google Scholar] [CrossRef]
- Branzei, D.; Vanoli, F.; Foiani, M. SUMOylation Regulates Rad18-Mediated Template Switch. Nature 2008, 456, 915–920. [Google Scholar] [CrossRef]
- Symington, L.S. Role of RAD52 Epistasis Group Genes in Homologous Recombination and Double-Strand Break Repair. Microbiol. Mol. Biol. Rev. 2002, 66, 630–670. [Google Scholar] [CrossRef]
- Mortensen, U.H.; Bendixen, C.; Sunjevaric, I.; Rothstein, R. DNA Strand Annealing Is Promoted by the Yeast RaD52 Protein. Proc. Natl. Acad. Sci. USA 1996, 93, 10729–10734. [Google Scholar] [CrossRef] [PubMed]
- Gazy, I.; Liefshitz, B.; Bronstein, A.; Parnas, O.; Atias, N.; Sharan, R.; Kupiec, M. A Genetic Screen for High Copy Number Suppressors of the Synthetic Lethality between Elg1Δ and Srs2Δ in Yeast. G3 Genes Genomes Genet. 2013, 3, 917–926. [Google Scholar] [CrossRef]
- Schiestl, R.H.; Prakash, S.; Prakash, L. The SRS2 Suppressor of Rad6 Mutations of Saccharomyces Cerevisiae Acts by Channeling DNA Lesions into the RAD52 DNA Repair Pathway. Genetics 1990, 124, 817–832. [Google Scholar] [CrossRef] [PubMed]
- Ulrich, H.D. The Srs2 Suppressor of UV Sensitivity Acts Specifically on the RAD5- and MMS2-Dependent Branch of the RAD6 Pathway. Nucleic Acids Res. 2001, 29, 3487–3494. [Google Scholar] [CrossRef]
- Singh, P.; Gazy, I.; Kupiec, M. Control of Telomere Length in Yeast by SUMOylated PCNA and the Elg1 PCNA Unloader. eLife 2023, 12, RP86990. [Google Scholar] [CrossRef] [PubMed]
- Liefshitz, B.; Steinlauf, R.; Friedl, A.; Eckardt-Schupp, F.; Kupiec, M. Genetic Interactions between Mutants of the “error-Prone” Repair Group of Saccharomyces Cerevisiae and Their Effect on Recombination and Mutagenesis. Mutat. Res./DNA Repair. 1998, 407, 135–145. [Google Scholar] [CrossRef]
- Ben-Aroya, S.; Koren, A.; Liefshitz, B.; Steinlauf, R.; Kupiec, M. ELG1, a Yeast Gene Required for Genome Stability, Forms a Complex Related to Replication Factor C. Proc. Natl. Acad. Sci. USA 2003, 100, 9906–9911. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Genotype | Source |
|---|---|---|
| 18076 | Mat@ ade5-1,lys2::InsEa14,trp1-289,his7-2,leu2-3,112,ura3-52 | [39] |
| 19523 | Mat@ ade5-1,lys2::InsEa14,trp1-289,his7-2,leu2-3,112,ura3-52, Pol30 with C’ terminal flag with KanMX marker | This study |
| 19787 | MatA ade5-1,lys2::InsEa14,trp1-289,his7-2,leu2-3,112,ura3-52, pol30-E143K:LEU22+flag:KanMX | [30] |
| 19785 | MatA ade5-1,lys2::InsEa14,trp1-289,his7-2,leu2-3,112,ura3-52, pol30-V180D:leu2+flag:KanMX | [30] |
| 19524 | Mat@ ade5-1,lys2::InsEa14,trp1-289,his7-2,leu2-3,112,ura3-52, Elg1::Hyg, Pol30 with C’ terminal flag with KanMX marker | [40] |
| 17822 | Mat@ ade5-1,lys2::InsEa14,trp1-289,his7-2,leu2-3,112,ura3-52, pol30-V180D:LEU2 | [30] |
| 17823 | Mat@ ade5-1,lys2::InsEa14,trp1-289,his7-2,leu2-3,112,ura3-52, pol30-E143K:LEU2 | [30] |
| 19682 | Mat@ ade5-1,lys2::InsEa14,trp1-289,his7-2,leu2-3,112,ura3-52,pol30-K164R, K127R:LEU2 | [29] |
| 19808 | MatA,ade5-1,lys2::InsEa14,trp1-289,his7-2,leu2-3,112,ura3-52, pol30-K164R::LEU2 | [29] |
| 20573 | MatA; ade5-1,lys2::InsEa14,trp1-289,his7-2,leu2-3,112,ura3-52, pol30-K164R;V180D:LEU2 | This study |
| 19525 | Mat@ ade5-1,lys2::InsEa14,trp1-289,his7-2,leu2-3,112,ura3-52,pol30-E143K,K164R:LEU2 | This study |
| 20033 | E134 MatA ade5-1,lys2::InsEa14,trp1-289,his7-2,leu2-3,112,ura3-52, srs2::KanMX | [29] |
| 20464 | E134 MatA ade5-1,lys2::InsEa14,trp1-289,his7-2,leu2-3,112,ura3-52, srs2::KanMX pol30-V180D: LEU2 | This study |
| 20508 | Mat@ ade5-1,lys2::InsEa14,trp1-289,his7-2,leu2-3,112,ura3-52, pol30-K164R:LEU2, srs2::KanMX | This study |
| 20463 | E134 MatA ade5-1,lys2::InsEa14,trp1-289,his7-2,leu2-3,112,ura3-52, srs2::KanMX Pol30-V180D,K164R:LEU2 | This study |
| 20461 | E134 matA ade5-1,lys2::InsEa14,trp1-289,his7-2,leu2-3,112,ura3-52, Srs2::KanMX, pol30-K164R, K127R:LEU2 | This study |
| 19678 | Mat@ ade5-1,lys2::InsEa14,trp1-289,his7-2,leu2-3,112,ura3-52, pol30-V180D, K164R, K127R:LEU2-#2 | This study |
| 20462 | E134 MatA de5-1,lys2::InsEa14,trp1-289,his7-2,leu2-3,112,ura3-52, Srs2::KanMX pol30-V180D, K164R, K127R:LEU2 | This study |
| 20609 | E134 MatA ade5-1,lys2::InsEa14,trp1-289,his7-2,leu2-3,112,ura3-52, rad52::ura3-1 | This study |
| 20656 | Mat@ ade5-1,lys2::InsEa14,trp1-289,his7-2,leu2-3,112,ura3-52, pol30-V180D:LEU2, rad52::URA3 | This study |
| 20589 | Mat@ ade5-1,lys2::InsEa14,trp1-289,his7-2,leu2-3,112,ura3-52, pol30-K164R:LEU2, rad52::URA3 | This study |
| 20660 | MatA ade5-1,lys2::InsEa14,trp1-289,his7-2,leu2-3,112,ura3-52 Pol30-V180D,K164R:LEU2, rad52::URA3 | this study |
| 20708 | MatA; ade5-1,lys2::InsEa14,trp1-289,his7-2,leu2-3,112,ura3-52, pol30-K164R,K127R::LEU2 rad52::URA3 | This study |
| 18045 | Mat@ ade5-1,lys2::InsEa14,trp1-289,his7-2,leu2-3,112,ura3-52 elg1::HygMX | [40] |
| 20545 | Mat@; ade5-1,lys2::InsEa14,trp1-289,his7-2,leu2-3,112,ura3-52, elg1::HygMX; srs2:KanMX; | [29] |
| 20544 | Mat@; ade5-1,lys2::InsEa14,trp1-289,his7-2,leu2-3,112,ura3-52, pol30-V180D:LEU2; elg1::HygMX; srs2:KanMX; | This study |
| 20543 | Diploid; ade5-1,lys2::InsEa14,trp1-289,his7-2,leu2-3,112,ura3-52, (heterozygous) elg1::HygMX; (heterozygous) srs2:KanMX; (heterozygous) pol30-V180D:LEU2 | This study |
| 20541 | Diploid; ade5-1,lys2::InsEa14,trp1-289,his7-2,leu2-3,112,ura3-52, (heterozygous) elg1::HygMX; (heterozygous) srs2:KanMX; (heterozygous) Pol30-E143K:LEU2 | This study |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arbel-Groissman, M.; Liefshitz, B.; Katz, N.; Kuryachiy, M.; Kupiec, M. PCNA Unloading Is Crucial for the Bypass of DNA Lesions Using Homologous Recombination. Int. J. Mol. Sci. 2024, 25, 3359. https://doi.org/10.3390/ijms25063359
Arbel-Groissman M, Liefshitz B, Katz N, Kuryachiy M, Kupiec M. PCNA Unloading Is Crucial for the Bypass of DNA Lesions Using Homologous Recombination. International Journal of Molecular Sciences. 2024; 25(6):3359. https://doi.org/10.3390/ijms25063359
Chicago/Turabian StyleArbel-Groissman, Matan, Batia Liefshitz, Nir Katz, Maxim Kuryachiy, and Martin Kupiec. 2024. "PCNA Unloading Is Crucial for the Bypass of DNA Lesions Using Homologous Recombination" International Journal of Molecular Sciences 25, no. 6: 3359. https://doi.org/10.3390/ijms25063359
APA StyleArbel-Groissman, M., Liefshitz, B., Katz, N., Kuryachiy, M., & Kupiec, M. (2024). PCNA Unloading Is Crucial for the Bypass of DNA Lesions Using Homologous Recombination. International Journal of Molecular Sciences, 25(6), 3359. https://doi.org/10.3390/ijms25063359