
An Overview of Current Detection Methods for RNA Methylation


Abstract
1. Epitranscriptomics
2. Types of RNA Methylation
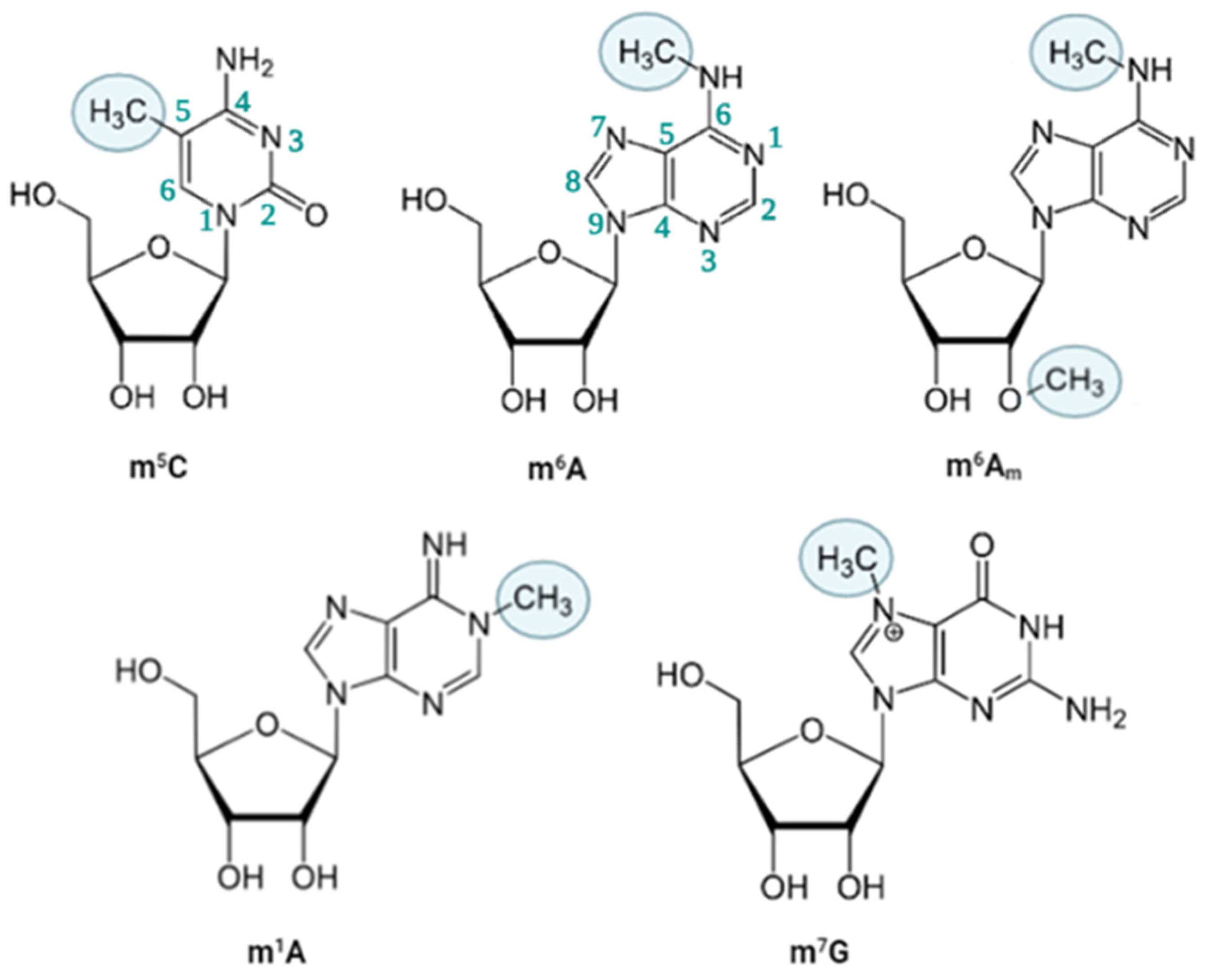
2.1. m6A
2.2. m6Am
2.3. m1A
2.4. m7G
2.5. m5C
3. Detection Technologies for RNA Methylation
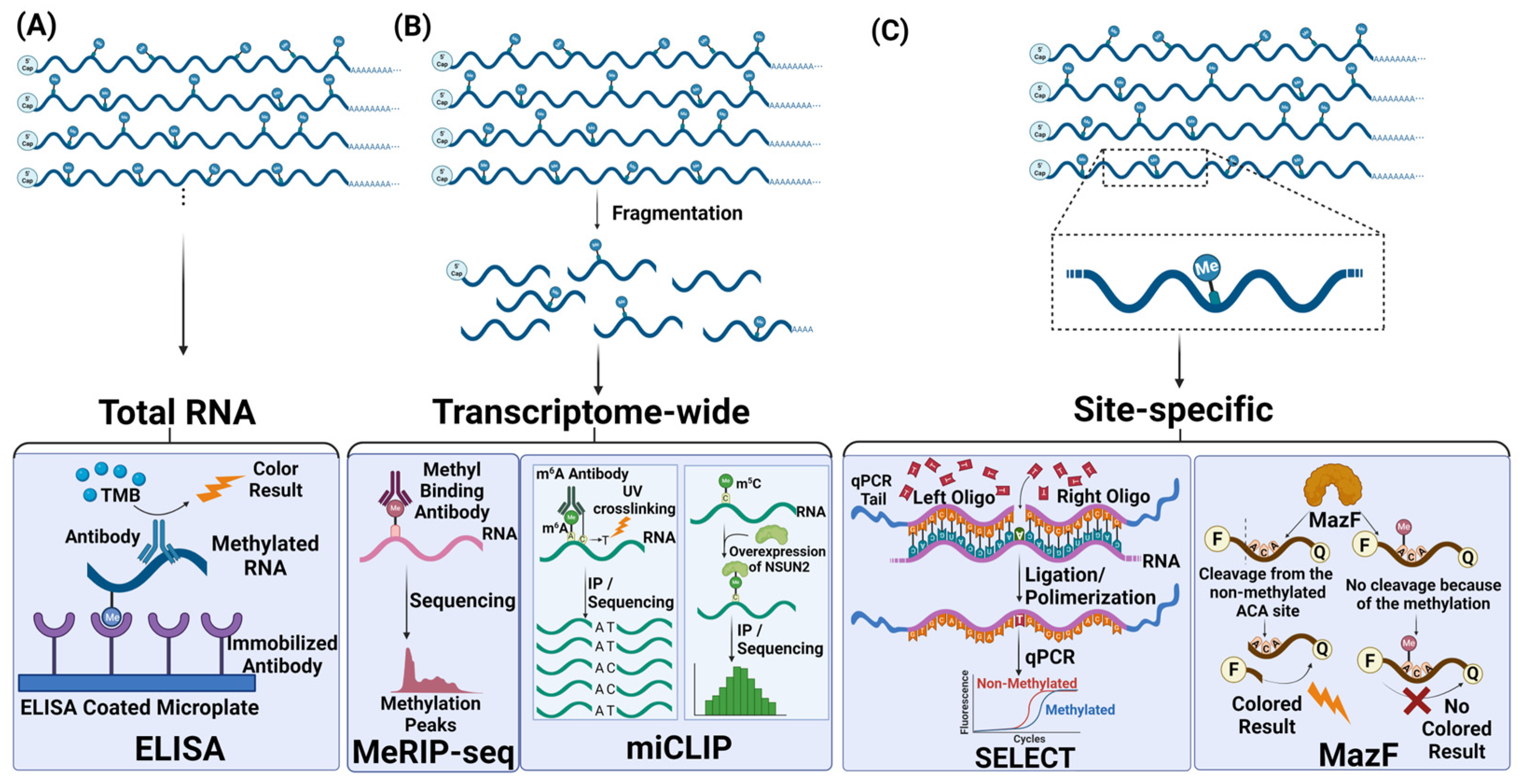
3.1. Approaches to Measure Global Changes in RNA Methylation
3.2. Transcriptomic Detection Methods

{kind=link}
{kind=link}
| Methods | Advantages | Disadvantages | References |
|---|---|---|---|
| Global RNA Methylation Detection Methods | |||
| ELISA |
|
| [97] |
| 2D-TLC |
|
| [98,101,102] |
| LC-MS |
|
| [99,100] |
| SCARLET |
|
| [103,104] |
| Dot Blot |
|
| [105,106,107] |
| DART-Seq |
|
| [108] |
| Transcriptome-wide RNA Methylation Detection Methods | |||
| Nanopore |
|
| [109] |
| SMRT |
|
| [110,111] |
| MeRIP-seq |
|
| [33,34,112] |
| miCLIP |
|
| [59,113] |
| Site-specific RNA Methylation Detection Methods | |||
| Reverse transcriptase based-qPCR assay |
|
| [117,118,119] |
| HRM |
|
| [120,121] |
| MazF |
|
| [122] |
| T3/T4 DNA ligase-qPCR |
|
| [123] |
| SELECT |
|
| [124] |
3.3. Site-Specific Detection Methods
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Pierrat, O.A.; Mikitova, V.; Bush, M.S.; Browning, K.S.; Doonan, J.H. Control of Protein Translation by Phosphorylation of the MRNA 5-Cap-Binding Complex. Biochem. Soc. Trans. 2007, 35, 1634–1637. [Google Scholar] [CrossRef]
- Wiener, D.; Schwartz, S. The Epitranscriptome beyond m6A. Nat. Rev. Genet. 2021, 22, 119–131. [Google Scholar] [CrossRef]
- Arango, D.; Sturgill, D.; Yang, R.; Kanai, T.; Bauer, P.; Roy, J.; Wang, Z.; Hosogane, M.; Schiffers, S.; Oberdoerffer, S. Direct Epitranscriptomic Regulation of Mammalian Translation Initiation through N4-Acetylcytidine. Mol. Cell 2022, 82, 2797–2814.e11. [Google Scholar] [CrossRef]
- Castello, A.; Fischer, B.; Frese, C.K.; Horos, R.; Alleaume, A.M.; Foehr, S.; Curk, T.; Krijgsveld, J.; Hentze, M.W. Comprehensive Identification of RNA-Binding Domains in Human Cells. Mol. Cell 2016, 63, 696–710. [Google Scholar] [CrossRef]
- Zhu, Z.M.; Huo, F.C.; Pei, D.S. Function and Evolution of RNA N6-Methyladenosine Modification. Int. J. Biol. Sci. 2020, 16, 1929–1940. [Google Scholar] [CrossRef]
- Bazak, L.; Haviv, A.; Barak, M.; Jacob-Hirsch, J.; Deng, P.; Zhang, R.; Isaacs, F.J.; Rechavi, G.; Li, J.B.; Eisenberg, E.; et al. A-to-I RNA Editing Occurs at over a Hundred Million Genomic Sites, Located in a Majority of Human Genes. Genome Res. 2014, 24, 365–376. [Google Scholar] [CrossRef] [PubMed]
- Saletore, Y.; Meyer, K.; Korlach, J.; Vilfan, I.D.; Jaffrey, S.; Mason, C.E. The Birth of the Epitranscriptome: Deciphering the Function of RNA Modifications. Genome Biol. 2012, 13, 175. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, W.V.; Nachtergaele, S. mRNA Regulation by RNA Modifications. Annu. Rev. Biochem. 2023, 92, 175–198. [Google Scholar] [CrossRef] [PubMed]
- Khosravi, H.M.; Jantsch, M.F. Site-Directed RNA Editing: Recent Advances and Open Challenges. RNA Biol. 2021, 18, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Understanding RNA Editing and Its Use in Gene Editing. Gene Genome Ed. 2022, 3–4, 100021. [CrossRef]
- Boccaletto, P.; Stefaniak, F.; Ray, A.; Cappannini, A.; Mukherjee, S.; Purta, E.; Kurkowska, M.; Shirvanizadeh, N.; Destefanis, E.; Groza, P.; et al. MODOMICS: A Database of RNA Modification Pathways. 2021 Update. Nucleic Acids Res. 2022, 50, D231–D235. [Google Scholar] [CrossRef]
- Farooqi, A.A.; Fayyaz, S.; Poltronieri, P.; Calin, G.; Mallardo, M. Epigenetic Deregulation in Cancer: Enzyme Players and Non-Coding RNAs. Semin. Cancer Biol. 2022, 83, 197–207. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Huang, L.; Wang, K.; Zhang, L.; Zhong, X.; Yan, Z.; Liu, B.; Zhu, P. RtcisE2F Promotes the Self-Renewal and Metastasis of Liver Tumor-Initiating Cells via N6-Methyladenosine-Dependent E2F3/E2F6 MRNA Stability. Sci. China Life Sci. 2022, 65, 1840–1854. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.H.; Zhang, G.L.; Jiang, R.F.; Hong, Y.Q.; Zhang, Q.Y.; He, J.P.; Liu, X.R.; Yang, Z.S.; Yang, L.; Jiang, X.; et al. METTL3 Is Essential for Normal Progesterone Signaling during Embryo Implantation via m6A-Mediated Translation Control of Progesterone Receptor. Proc. Natl. Acad. Sci. USA 2023, 120, e2214684120. [Google Scholar] [CrossRef]
- Lin, S.; Choe, J.; Du, P.; Triboulet, R.; Gregory, R.I. The m6A Methyltransferase METTL3 Promotes Translation in Human Cancer Cells. Mol. Cell 2016, 62, 335–345. [Google Scholar] [CrossRef] [PubMed]
- Qiao, Y.; Sun, Q.; Chen, X.; He, L.; Wang, D.; Su, R.; Xue, Y.; Sun, H.; Wang, H. Nuclear m6A Reader YTHDC1 Promotes Muscle Stem Cell Activation/Proliferation by Regulating MRNA Splicing and Nuclear Export. eLife 2023, 12, e82703. [Google Scholar] [CrossRef] [PubMed]
- Ries, R.J.; Zaccara, S.; Klein, P.; Olarerin-George, A.; Namkoong, S.; Pickering, B.F.; Patil, D.P.; Kwak, H.; Lee, J.H.; Jaffrey, S.R. m6A Enhances the Phase Separation Potential of MRNA. Nature 2019, 571, 424–428. [Google Scholar] [CrossRef] [PubMed]
- Zheng, G.; Dahl, J.A.; Niu, Y.; Fedorcsak, P.; Huang, C.M.; Li, C.J.; Vågbø, C.B.; Shi, Y.; Wang, W.L.; Song, S.H.; et al. ALKBH5 Is a Mammalian RNA Demethylase That Impacts RNA Metabolism and Mouse Fertility. Mol. Cell 2013, 49, 18–29. [Google Scholar] [CrossRef]
- Wang, Y.; Li, Y.; Toth, J.I.; Petroski, M.D.; Zhang, Z.; Zhao, J.C. N6-Methyladenosine Modification Destabilizes Developmental Regulators in Embryonic Stem Cells. Nat. Cell Biol. 2014, 16, 191–198. [Google Scholar] [CrossRef]
- Liu, L.; Song, B.; Ma, J.; Song, Y.; Zhang, S.Y.; Tang, Y.; Wu, X.; Wei, Z.; Chen, K.; Su, J.; et al. Bioinformatics Approaches for Deciphering the Epitranscriptome: Recent Progress and Emerging Topics. Comput. Struct. Biotechnol. J. 2020, 18, 1587–1604. [Google Scholar] [CrossRef]
- Zhao, C.; Xu, G.; Zhang, X.; Ye, Y.; Cai, W.; Shao, Q. RNA m6A Modification Orchestrates the Rhythm of Immune Cell Development from Hematopoietic Stem Cells to T and B Cells. Front. Immunol. 2022, 3, 839291. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Xie, H.; Ying, Y.; Chen, H.; Yan, H.; He, L.; Xu, M.; Xu, X.; Liang, Z.; Liu, B.; et al. YTHDF2 Mediates the MRNA Degradation of the Tumor Suppressors to Induce AKT Phosphorylation in N6-Methyladenosine-Dependent Way in Prostate Cancer. Mol. Cancer 2020, 19, 152. [Google Scholar] [CrossRef] [PubMed]
- Alasar, A.A.; Tüncel, Ö.; Gelmez, A.B.; Sağlam, B.; Vatansever, İ.E.; Akgül, B. Genomewide m6A Mapping Uncovers Dynamic Changes in the m6A Epitranscriptome of Cisplatin-Treated Apoptotic HeLa Cells. Cells 2022, 11, 3905. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Wang, J.; Sun, G.; Wu, Q.; Ma, J.; Zhang, X.; Huang, N.; Bian, Z.; Gu, S.; Xu, M.; et al. m6A MRNA Methylation Regulates CTNNB1 to Promote the Proliferation of Hepatoblastoma. Mol. Cancer 2019, 18, 188. [Google Scholar] [CrossRef] [PubMed]
- Zhao, F.; Xu, Y.; Gao, S.; Qin, L.; Austria, Q.; Siedlak, S.L.; Pajdzik, K.; Dai, Q.; He, C.; Wang, W.; et al. METTL3-Dependent RNA m6A Dysregulation Contributes to Neurodegeneration in Alzheimer’s Disease through Aberrant Cell Cycle Events. Mol. Neurodegener. 2021, 16, 70. [Google Scholar] [CrossRef] [PubMed]
- Akçaöz-Alasar, A.; Tüncel, Ö.; Sağlam, B.; Gazaloğlu, Y.; Atbinek, M.; Cagiral, U.; Iscan, E.; Ozhan, G.; Akgül, B. Epitranscriptomics m6A Analyses Reveal Distinct m6A Marks under Tumor Necrosis Factor α (TNF-α)-induced Apoptotic Conditions in HeLa Cells. J. Cell Physiol. 2024; in press. [Google Scholar] [CrossRef]
- Xie, S.; Chen, W.; Chen, K.; Chang, Y.; Yang, F.; Lin, A.; Shu, Q.; Zhou, T.; Yan, X. Emerging Roles of RNA Methylation in Gastrointestinal Cancers. Cancer Cell Int. 2020, 20, 585. [Google Scholar] [CrossRef]
- Dominissini, D.; Nachtergaele, S.; Moshitch-Moshkovitz, S.; Peer, E.; Kol, N.; Ben-Haim, M.S.; Dai, Q.; Di Segni, A.; Salmon-Divon, M.; Clark, W.C.; et al. The Dynamic N1-Methyladenosine Methylome in Eukaryotic Messenger RNA. Nature 2016, 530, 441–446. [Google Scholar] [CrossRef]
- Esteller, M.; Pandolfi, P.P. The Epitranscriptome of Noncoding RNAs in Cancer. Cancer Discov. 2017, 7, 359–368. [Google Scholar] [CrossRef]
- Squires, J.E.; Patel, H.R.; Nousch, M.; Sibbritt, T.; Humphreys, D.T.; Parker, B.J.; Suter, C.M.; Preiss, T. Widespread Occurrence of 5-Methylcytosine in Human Coding and Non-Coding RNA. Nucleic Acids Res. 2012, 40, 5023–5033. [Google Scholar] [CrossRef]
- Motorin, Y.; Helm, M. RNA Nucleotide Methylation. Wiley Interdiscip. Rev. RNA 2011, 2, 611–631. [Google Scholar] [CrossRef]
- Desrosiers, R.; Friderici, K.; Rottman, F. Identification of Methylated Nucleosides in Messenger RNA from Novikoff Hepatoma Cells (RNA Methylation/RNA Processing/Methylnucleoside Composition). Proc. Natl. Acad. Sci. USA 1974, 71, 3971–3975. [Google Scholar] [CrossRef] [PubMed]
- Meyer, K.D.; Saletore, Y.; Zumbo, P.; Elemento, O.; Mason, C.E.; Jaffrey, S.R. Comprehensive Analysis of MRNA Methylation Reveals Enrichment in 3′ UTRs and near Stop Codons. Cell 2012, 149, 1635–1646. [Google Scholar] [CrossRef] [PubMed]
- Dominissini, D.; Moshitch-Moshkovitz, S.; Schwartz, S.; Salmon-Divon, M.; Ungar, L.; Osenberg, S.; Cesarkas, K.; Jacob-Hirsch, J.; Amariglio, N.; Kupiec, M.; et al. Topology of the Human and Mouse m6A RNA Methylomes Revealed by m6A-Seq. Nature 2012, 485, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Jonkhout, N.; Tran, J.; Smith, M.A.; Schonrock, N.; Mattick, J.S.; Novoa, E.M. The RNA Modification Landscape in Human Disease. RNA 2017, 23, 1754–1769. [Google Scholar] [CrossRef] [PubMed]
- Bokar, J.A.; Shambaugh, M.E.; Polayes, D.; Matera, A.G.; Pottman, F.M. Purification and CDNA Cloning of the AdoMet-Binding Subunit of the Human MRNA (N6-Adenosine)-Methyltransferase. RNA 1997, 3, 1233–1247. [Google Scholar] [PubMed]
- Wang, X.; Huang, J.; Zou, T.; Yin, P. Human m6A Writers: Two Subunits, 2 Roles. RNA Biol. 2017, 14, 300–304. [Google Scholar] [CrossRef] [PubMed]
- Geula, S.; Moshitch-Moshkovitz, S.; Dominissini, D.; AlFatah Mansour, A.; Kol, N.; Salmon-Divon, M.; Hershkovitz, V.; Peer, E.; Mor, N.; Manor, Y.S.; et al. m6A MRNA Methylation Facilitates Resolution of Naïve Pluripotency toward Differentiation. Science 2015, 347, 1002–1006. [Google Scholar] [CrossRef] [PubMed]
- Jinek, M.; Śledz, P. Structural Insights into the Molecular Mechanism of the m 6 A Writer Complex. eLife 2016, 5, e18434. [Google Scholar] [CrossRef]
- Knuckles, P.; Lence, T.; Haussmann, I.U.; Jacob, D.; Kreim, N.; Carl, S.H.; Masiello, I.; Hares, T.; Villaseñor, R.; Hess, D.; et al. Zc3h13/Flacc Is Required for Adenosine Methylation by Bridging the MRNA-Binding Factor RbM15/Spenito to the m6A Machinery Component Wtap/Fl(2)d. Genes Dev. 2018, 32, 415–429. [Google Scholar] [CrossRef]
- Ping, X.L.; Sun, B.F.; Wang, L.; Xiao, W.; Yang, X.; Wang, W.J.; Adhikari, S.; Shi, Y.; Lv, Y.; Chen, Y.S.; et al. Mammalian WTAP Is a Regulatory Subunit of the RNA N6-Methyladenosine Methyltransferase. Cell Res. 2014, 24, 177–189. [Google Scholar] [CrossRef]
- Gao, Y.; Pei, G.; Li, D.; Li, R.; Shao, Y.; Zhang, Q.C.; Li, P. Multivalent m6A Motifs Promote Phase Separation of YTHDF Proteins. Cell Res. 2019, 29, 767–769. [Google Scholar] [CrossRef]
- Zhang, H.; Shi, X.; Huang, T.; Zhao, X.; Chen, W.; Gu, N.; Zhang, R. Dynamic Landscape and Evolution of m6A Methylation in Human. Nucleic Acids Res. 2020, 48, 6251–6264. [Google Scholar] [CrossRef]
- Song, J.; Yi, C. Reading Chemical Modifications in the Transcriptome. J. Mol. Biol. 2020, 432, 1824–1839. [Google Scholar] [CrossRef]
- Jia, G.; Fu, Y.; Zhao, X.; Dai, Q.; Zheng, G.; Yang, Y.; Yi, C.; Lindahl, T.; Pan, T.; Yang, Y.G.; et al. N6-Methyladenosine in Nuclear RNA Is a Major Substrate of the Obesity-Associated FTO. Nat. Chem. Biol. 2011, 7, 885–887. [Google Scholar] [CrossRef]
- Patil, D.P.; Chen, C.K.; Pickering, B.F.; Chow, A.; Jackson, C.; Guttman, M.; Jaffrey, S.R. m6A RNA Methylation Promotes XIST-Mediated Transcriptional Repression. Nature 2016, 537, 369–373. [Google Scholar] [CrossRef]
- Wojtas, M.N.; Pandey, R.R.; Mendel, M.; Homolka, D.; Sachidanandam, R.; Pillai, R.S. Regulation of m6A Transcripts by the 3′→5′ RNA Helicase YTHDC2 Is Essential for a Successful Meiotic Program in the Mammalian Germline. Mol. Cell 2017, 68, 374–387.e12. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Lv, W.; Li, T.; Zhang, S.; Wang, H.; Li, X.; Wang, L.; Ma, D.; Zang, Y.; Shen, J.; et al. Dynamic Regulation and Functions of MRNA m6A Modification. Cancer Cell Int. 2022, 22, 48. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.; Tong, L. Molecular Basis for the Recognition of Methylated Adenines in RNA by the Eukaryotic YTH Domain. Proc. Natl. Acad. Sci. USA 2014, 111, 13834–13839. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Z.; Lv, Q.; Zhang, Y.; Chen, X.; Guo, R.; Liu, S.; Peng, X. Long Non-Coding RNA DDX11-AS1 Promotes the Proliferation and Migration of Glioma Cells by Combining with HNRNPC. Mol. Ther. Nucleic Acids 2022, 28, 601–612. [Google Scholar] [CrossRef] [PubMed]
- Zhou, K.I.; Parisien, M.; Dai, Q.; Liu, N.; Diatchenko, L.; Sachleben, J.R.; Pan, T. N6-Methyladenosine Modification in a Long Noncoding RNA Hairpin Predisposes Its Conformation to Protein Binding. J. Mol. Biol. 2016, 428, 822–833. [Google Scholar] [CrossRef] [PubMed]
- Coker, H.; Wei, G.; Brockdorff, N. m6A Modification of Non-Coding RNA and the Control of Mammalian Gene Expression. Biochim. Biophys. Acta Gene Regul. Mech. 2019, 1862, 310–318. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Kang, Y.; Wang, M.; Li, Y.; Xu, T.; Yang, W.; Song, H.; Wu, H.; Shu, Q.; Jin, P. Fragile X Mental Retardation Protein Modulates the Stability of Its m6A-Marked Messenger RNA Targets. Hum. Mol. Genet. 2018, 27, 3936–3950. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Sun, H.; Li, K.; Xiao, Y.; Yi, C. m6Am RNA Modification Detection by m6Am-Seq. Methods 2022, 203, 242–248. [Google Scholar] [CrossRef]
- Cesaro, B.; Tarullo, M.; Fatica, A. Regulation of Gene Expression by m6Am RNA Modification. Int. J. Mol. Sci. 2023, 3, 2277. [Google Scholar] [CrossRef] [PubMed]
- Nabeel-Shah, S.; Pu, S.; Burke, G.L.; Ahmed, N.; Braunschweig, U.; Farhangmehr, S.; Lee, H.; Wu, M.; Ni, Z.; Tang, H.; et al. Recruitment of the m6A/Am Demethylase FTO to Target RNAs by the Telomeric Zinc Finger Protein ZBTB48. bioRxiv 2024. [Google Scholar] [CrossRef]
- Ben-Haim, M.S.; Pinto, Y.; Moshitch-Moshkovitz, S.; Hershkovitz, V.; Kol, N.; Diamant-Levi, T.; Beeri, M.S.; Amariglio, N.; Cohen, H.Y.; Rechavi, G. Dynamic Regulation of N6,2′-O-Dimethyladenosine (m6Am) in Obesity. Nat. Commun. 2021, 12, 7185. [Google Scholar] [CrossRef]
- Cao, X.; Wang, M.; Huang, Y.; Zhang, M.; Zheng, F.; Zhang, G.; Su, J.; Yuan, Y.; Guo, C. Determination of Dimethylated Nucleosides in Serum from Colorectal Cancer Patients by Hydrophilic Interaction Liquid Chromatography-Tandem Mass Spectrometry. J. Chromatogr. B 2024, 1232, 123973. [Google Scholar] [CrossRef]
- Linder, B.; Grozhik, A.V.; Olarerin-George, A.O.; Meydan, C.; Mason, C.E.; Jaffrey, S.R. Single-Nucleotide-Resolution Mapping of m6A and m6Am throughout the Transcriptome. Nat. Methods 2015, 12, 767–772. [Google Scholar] [CrossRef]
- Boulias, K.; Toczydłowska-Socha, D.; Hawley, B.R.; Liberman, N.; Takashima, K.; Zaccara, S.; Guez, T.; Vasseur, J.J.; Debart, F.; Aravind, L.; et al. Identification of the m6Am Methyltransferase PCIF1 Reveals the Location and Functions of m6Am in the Transcriptome. Mol. Cell 2019, 75, 631–643.e8. [Google Scholar] [CrossRef]
- Akichika, S.; Hirano, S.; Shichino, Y.; Suzuki, T.; Nishimasu, H.; Ishitani, R.; Sugita, A.; Hirose, Y.; Iwasaki, S.; Nureki, O.; et al. Cap-Specific Terminal N6-Methylation of RNA by an RNA Polymerase II–Associated Methyltransferase. Science 2019, 363, 6423. [Google Scholar] [CrossRef] [PubMed]
- Mauer, J.; Luo, X.; Blanjoie, A.; Jiao, X.; Grozhik, A.V.; Patil, D.P.; Linder, B.; Pickering, B.F.; Vasseur, J.J.; Chen, Q.; et al. Reversible Methylation of m6Am in the 5′ Cap Controls MRNA Stability. Nature 2017, 541, 371–375. [Google Scholar] [CrossRef] [PubMed]
- Mauer, J.; Jaffrey, S.R. FTO, m6Am, and the hypothesis of reversible epitranscriptomic mRNA modifications. FEBS Lett. 2018, 592, 2012–2022. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Zhang, M.; Zhang, Q.; Yu, X.; Sun, Z.; He, Y.; Guo, W. Role of Main RNA Methylation in Hepatocellular Carcinoma: N6-Methyladenosine, 5-Methylcytosine, and N1-Methyladenosine. Front. Cell Dev. Biol. 2021, 9, 767668. [Google Scholar] [CrossRef] [PubMed]
- Roundtree, I.A.; Evans, M.E.; Pan, T.; He, C. Dynamic RNA Modifications in Gene Expression Regulation. Cell 2017, 169, 1187–1200. [Google Scholar] [CrossRef]
- Richter, U.; Evans, M.E.; Clark, W.C.; Marttinen, P.; Shoubridge, E.A.; Suomalainen, A.; Wredenberg, A.; Wedell, A.; Pan, T.; Battersby, B.J. RNA Modification Landscape of the Human Mitochondrial TRNALys Regulates Protein Synthesis. Nat. Commun. 2018, 9, 3966. [Google Scholar] [CrossRef]
- Li, X.; Xiong, X.; Wang, K.; Wang, L.; Shu, X.; Ma, S.; Yi, C. Transcriptome-Wide Mapping Reveals Reversible and Dynamic N1-Methyladenosine Methylome. Nat. Chem. Biol. 2016, 12, 311–316. [Google Scholar] [CrossRef]
- Chujo, T.; Suzuki, T. Trmt61B Is a Methyltransferase Responsible for 1-Methyladenosine at Position 58 of Human Mitochondrial TRNAs. RNA 2012, 18, 2269–2276. [Google Scholar] [CrossRef]
- Safra, M.; Sas-Chen, A.; Nir, R.; Winkler, R.; Nachshon, A.; Bar-Yaacov, D.; Erlacher, M.; Rossmanith, W.; Stern-Ginossar, N.; Schwartz, S. The M1A Landscape on Cytosolic and Mitochondrial MRNA at Single-Base Resolution. Nature 2017, 551, 251–255. [Google Scholar] [CrossRef]
- Li, X.; Xiong, X.; Zhang, M.; Wang, K.; Chen, Y.; Zhou, J.; Mao, Y.; Lv, J.; Yi, D.; Chen, X.W.; et al. Base-Resolution Mapping Reveals Distinct m1A Methylome in Nuclear- and Mitochondrial-Encoded Transcripts. Mol. Cell 2017, 68, 993–1005.e9. [Google Scholar] [CrossRef]
- Wei, J.; Liu, F.; Lu, Z.; Fei, Q.; Ai, Y.; He, P.C.; Shi, H.; Cui, X.; Su, R.; Klungland, A.; et al. Differential m6A, m6Am, and m1A Demethylation Mediated by FTO in the Cell Nucleus and Cytoplasm. Mol. Cell 2018, 71, 973–985.e5. [Google Scholar] [CrossRef]
- Yang, B.; Wang, J.Q.; Tan, Y.; Yuan, R.; Chen, Z.S.; Zou, C. RNA Methylation and Cancer Treatment. Pharmacol. Res. 2021, 174, 105937. [Google Scholar] [CrossRef]
- Zheng, Q.; Gan, H.; Yang, F.; Yao, Y.; Hao, F.; Hong, L.; Jin, L. Cytoplasmic M1A Reader YTHDF3 Inhibits Trophoblast Invasion by Downregulation of m1A-Methylated IGF1R. Cell Discov. 2020, 6, 12. [Google Scholar] [CrossRef]
- Tomikawa, C. 7-Methylguanosine Modifications in Transfer RNA (TRNA). Int. J. Mol. Sci. 2018, 19, 4080. [Google Scholar] [CrossRef]
- Wang, M.K.; Gao, C.C.; Yang, Y.G. Emerging Roles of RNA Methylation in Development. Acc. Chem. Res. 2023, 56, 3417–3427. [Google Scholar] [CrossRef]
- Zhao, Z.; Qing, Y.; Deng, X.; Su, R.; Chen, J. The Roles and Therapeutic Implications of Messenger RNA Internal N7-methylguanosine and N6-methyladenosine Modifications in Chemoresistance. Clin. Transl. Med. 2023, 13, e1400. [Google Scholar] [CrossRef]
- Lin, S.; Liu, Q.; Lelyveld, V.S.; Choe, J.; Szostak, J.W.; Gregory, R.I. Mettl1/Wdr4-Mediated m7G TRNA Methylome Is Required for Normal MRNA Translation and Embryonic Stem Cell Self-Renewal and Differentiation. Mol. Cell 2018, 71, 244–255.e5. [Google Scholar] [CrossRef]
- Luo, Y.; Yao, Y.; Wu, P.; Zi, X.; Sun, N.; He, J. The Potential Role of N7-Methylguanosine (M7G) in Cancer. J. Hematol. Oncol. 2022, 15, 63. [Google Scholar] [CrossRef]
- Martin, S.A.; Moss, B. Modification of RNA by MRNA Guanylyltransferase and MRNA (Guanine 7) Methyltransferase from Vaccinia Virions. J. Biol. Chem. 1975, 250, 9330–9335. [Google Scholar] [CrossRef]
- Schaefer, M.; Pollex, T.; Hanna, K.; Lyko, F. RNA Cytosine Methylation Analysis by Bisulfite Sequencing. Nucleic Acids Res. 2009, 37, e12. [Google Scholar] [CrossRef]
- Gao, Y.; Wang, Z.; Zhu, Y.; Zhu, Q.; Yang, Y.; Jin, Y.; Zhang, F.; Jiang, L.; Ye, Y.; Li, H.; et al. NOP2/Sun RNA Methyltransferase 2 Promotes Tumor Progression via Its Interacting Partner RPL6 in Gallbladder Carcinoma. Cancer Sci. 2019, 110, 3510–3519. [Google Scholar] [CrossRef] [PubMed]
- García-Vílchez, R.; Sevilla, A.; Blanco, S. Post-Transcriptional Regulation by Cytosine-5 Methylation of RNA. Biochim. Biophys. Acta Gene Regul. Mech. 2019, 1862, 240–252. [Google Scholar] [CrossRef]
- Gao, Y.; Fang, J. RNA 5-Methylcytosine Modification and Its Emerging Role as an Epitranscriptomic Mark. RNA Biol. 2021, 18, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Chi, L.; Delgado-Olguín, P. Expression of NOL1/NOP2/Sun Domain (Nsun) RNA Methyltransferase Family Genes in Early Mouse Embryogenesis. Gene Expr. Patterns 2013, 13, 319–327. [Google Scholar] [CrossRef]
- Genenncher, B.; Durdevic, Z.; Hanna, K.; Zinkl, D.; Mobin, M.B.; Senturk, N.; Da Silva, B.; Legrand, C.; Carré, C.; Lyko, F.; et al. Mutations in Cytosine-5 TRNA Methyltransferases Impact Mobile Element Expression and Genome Stability at Specific DNA Repeats. Cell Rep. 2018, 22, 1861–1874. [Google Scholar] [CrossRef]
- Bohnsack, K.E.; Höbartner, C.; Bohnsack, M.T. Eukaryotic 5-Methylcytosine (m5C) RNA Methyltransferases: Mechanisms, Cellular Functions, and Links to Disease. Genes 2019, 10, 102. [Google Scholar] [CrossRef]
- Li, H.; Zhu, D.; Wu, J.; Ma, Y.; Cai, C.; Chen, Y.; Qin, M.; Dai, H. New Substrates and Determinants for TRNA Recognition of RNA Methyltransferase DNMT2/TRDMT1. RNA Biol. 2021, 18, 2531–2545. [Google Scholar] [CrossRef] [PubMed]
- Van Haute, L.; Dietmann, S.; Kremer, L.; Hussain, S.; Pearce, S.F.; Powell, C.A.; Rorbach, J.; Lantaff, R.; Blanco, S.; Sauer, S.; et al. Deficient Methylation and Formylation of Mt-TRNAMet Wobble Cytosine in a Patient Carrying Mutations in NSUN3. Nat. Commun. 2016, 7, 12039. [Google Scholar] [CrossRef]
- Alagia, A.; Gullerova, M. The Methylation Game: Epigenetic and Epitranscriptomic Dynamics of 5-Methylcytosine. Front. Cell Dev. Biol. 2022, 10, 915685. [Google Scholar] [CrossRef]
- Cheng, J.X.; Chen, L.; Li, Y.; Cloe, A.; Yue, M.; Wei, J.; Watanabe, K.A.; Shammo, J.M.; Anastasi, J.; Shen, Q.J.; et al. RNA Cytosine Methylation and Methyltransferases Mediate Chromatin Organization and 5-Azacytidine Response and Resistance in Leukaemia. Nat. Commun. 2018, 9, 1163. [Google Scholar] [CrossRef]
- He, Z.; Xu, J.; Shi, H.; Wu, S. M5CRegpred: Epitranscriptome Target Prediction of 5-Methylcytosine (m5C) Regulators Based on Sequencing Features. Genes 2022, 13, 677. [Google Scholar] [CrossRef]
- Swan, B.K.; Martinez-Garcia, M.; Preston, C.M.; Sczyrba, A.; Woyke, T.; Lamy, D.; Reinthaler, T.; Poulton, N.J.; Masland, E.D.P.; Gomez, M.L.; et al. Potential for Chemolithoautotrophy among Ubiquitous Bacteria Lineages in the Dark Ocean. Science 2011, 333, 1296–1300. [Google Scholar] [CrossRef]
- Lan, J.; Rajan, N.; Bizet, M.; Penning, A.; Singh, N.K.; Guallar, D.; Calonne, E.; Li Greci, A.; Bonvin, E.; Deplus, R.; et al. Functional Role of Tet-Mediated RNA Hydroxymethylcytosine in Mouse ES Cells and during Differentiation. Nat. Commun. 2020, 11, 4956. [Google Scholar] [CrossRef]
- Song, H.; Zhang, J.; Liu, B.; Xu, J.; Cai, B.; Yang, H.; Straube, J.; Yu, X.; Ma, T. Biological Roles of RNA m5C Modification and Its Implications in Cancer Immunotherapy. Biomark. Res. 2022, 10, 15. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Yang, Y.; Sun, B.F.; Chen, Y.S.; Xu, J.W.; Lai, W.Y.; Li, A.; Wang, X.; Bhattarai, D.P.; Xiao, W.; et al. 5-Methylcytosine Promotes MRNA Export-NSUN2 as the Methyltransferase and ALYREF as an m5C Reader. Cell Res. 2017, 27, 606–625. [Google Scholar] [CrossRef]
- Chen, X.; Li, A.; Sun, B.F.; Yang, Y.; Han, Y.N.; Yuan, X.; Chen, R.X.; Wei, W.S.; Liu, Y.; Gao, C.C.; et al. 5-Methylcytosine Promotes Pathogenesis of Bladder Cancer through Stabilizing MRNAs. Nat. Cell Biol. 2019, 21, 978–990. [Google Scholar] [CrossRef] [PubMed]
- Shinde, H.; Dudhate, A.; Kadam, U.S.; Hong, J.C. RNA Methylation in Plants: An Overview. Front. Plant Sci. 2023, 14, 1132959. [Google Scholar] [CrossRef] [PubMed]
- Keith, G. Mobilities of Modified Ribonucleotides on Two-Dimensiona|Cellulose Thin-Layer Chromatography. Biochimie 1995, 77, 142–144. [Google Scholar] [CrossRef]
- Liu, J.; Yue, Y.; Han, D.; Wang, X.; Fu, Y.; Zhang, L.; Jia, G.; Yu, M.; Lu, Z.; Deng, X.; et al. A METTL3-METTL14 Complex Mediates Mammalian Nuclear RNA N6-Adenosine Methylation. Nat. Chem. Biol. 2014, 10, 93–95. [Google Scholar] [CrossRef] [PubMed]
- Thüring, K.; Schmid, K.; Keller, P.; Helm, M. LC-MS Analysis of Methylated RNA. In Methods in Molecular Biology; Humana Press Inc.: Totowa, NJ, USA, 2017; Volume 1562, pp. 3–18. [Google Scholar] [CrossRef]
- Mongan, N.P.; Emes, R.D.; Archer, N. Detection and Analysis of RNA Methylation. F1000Research 2019, 8, 559. [Google Scholar] [CrossRef]
- Bodi, Z.; Fray, R.G. Detection and Quantification of N6-Methyladenosine in Messenger RNA by TLC. In Methods in Molecular Biology; Humana Press Inc.: Totowa, NJ, USA, 2017; Volume 1562, pp. 79–87. [Google Scholar] [CrossRef]
- Liu, N.; Parisien, M.; Dai, Q.; Zheng, G.; He, C.; Pan, T. Probing N6-Methyladenosine RNA Modification Status at Single Nucleotide Resolution in MRNA and Long Noncoding RNA. RNA 2013, 19, 1848–1856. [Google Scholar] [CrossRef]
- Liu, N.; Pan, T. Probing RNA Modification Status at Single-Nucleotide Resolution in Total RNA. Methods Enzymol. 2015, 560, 149–159. [Google Scholar] [CrossRef]
- Nagarajan, A.; Janostiak, R.; Wajapeyee, N. Dot Blot Analysis for Measuring Global N6-Methyladenosine Modification of RNA. In Methods in Molecular Biology; Humana Press Inc.: Totowa, NJ, USA, 2019; Volume 1870, pp. 263–271. [Google Scholar] [CrossRef]
- Miao, Z.; Xin, N.; Wei, B.; Hua, X.; Zhang, G.; Leng, C.; Zhao, C.; Wu, D.; Li, J.; Ge, W.; et al. 5-Hydroxymethylcytosine Is Detected in RNA from Mouse Brain Tissues. Brain Res. 2016, 1642, 546–552. [Google Scholar] [CrossRef]
- Róna, G.; Scheer, I.; Nagy, K.; Pálinkás, H.L.; Tihanyi, G.; Borsos, M.; Békési, A.; Vértessy, B.G. Detection of Uracil within DNA Using a Sensitive Labeling Method for in Vitro and Cellular Applications. Nucleic Acids Res. 2016, 44, e28. [Google Scholar] [CrossRef] [PubMed]
- Meyer, K.D. DART-Seq: An Antibody-Free Method for Global m6A Detection. Nat. Methods 2019, 16, 1275–1280. [Google Scholar] [CrossRef] [PubMed]
- Garalde, D.R.; Snell, E.A.; Jachimowicz, D.; Sipos, B.; Lloyd, J.H.; Bruce, M.; Pantic, N.; Admassu, T.; James, P.; Warland, A.; et al. Highly Parallel Direct RN A Sequencing on an Array of Nanopores. Nat. Methods 2018, 15, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wu, Y.; Liu, L.; Feng, J.; Zhang, T.; Qin, S.; Zhao, X.; Wang, C.; Li, D.; Han, W.; et al. Study of the Whole Genome, Methylome and Transcriptome of Cordyceps Militaris. Sci. Rep. 2019, 9, 898. [Google Scholar] [CrossRef] [PubMed]
- Zaccara, S.; Ries, R.J.; Jaffrey, S.R. Reading, Writing and Erasing MRNA Methylation. Nat. Rev. Mol. Cell Biol. 2019, 20, 608–624. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, S.; Motorin, Y. Next-Generation Sequencing Technologies for Detection of Modified Nucleotides in RNAs. RNA Biol. 2017, 14, 1124–1137. [Google Scholar] [CrossRef]
- Van Nostrand, E.L.; Pratt, G.A.; Shishkin, A.A.; Gelboin-Burkhart, C.; Fang, M.Y.; Sundararaman, B.; Blue, S.M.; Nguyen, T.B.; Surka, C.; Elkins, K.; et al. Robust Transcriptome-Wide Discovery of RNA-Binding Protein Binding Sites with Enhanced CLIP (ECLIP). Nat. Methods 2016, 13, 508–514. [Google Scholar] [CrossRef] [PubMed]
- Ke, S.; Alemu, E.A.; Mertens, C.; Gantman, E.C.; Fak, J.J.; Mele, A.; Haripal, B.; Zucker-Scharff, I.; Moore, M.J.; Park, C.Y.; et al. A Majority of m6A Residues Are in the Last Exons, Allowing the Potential for 3′ UTR Regulation. Genes Dev. 2015, 29, 2037–2053. [Google Scholar] [CrossRef] [PubMed]
- Bringmann, P.; Lührmann, R. Antibodies Specific for N6-Methyladenosine React with Intact SnRNPs U2 and U4/U6. Biochem. Biophys. Res. Commun. 1987, 213, 309–315. [Google Scholar] [CrossRef] [PubMed]
- Selmi, T.; Hussain, S.; DIetmann, S.; Heiß, M.; Borland, K.; Flad, S.; Carter, J.M.; Dennison, R.; Huang, Y.L.; Kellner, S.; et al. Sequence- And Structure-Specific Cytosine-5 MRNA Methylation by NSUN6. Nucleic Acids Res. 2021, 49, 1006–1022. [Google Scholar] [CrossRef] [PubMed]
- Castellanos-Rubio, A.; Santin, I.; Olazagoitia-Garmendia, A.; Romero-Garmendia, I.; Jauregi-Miguel, A.; Legarda, M.; Bilbao, J.R. A Novel RT-QPCR-Based Assay for the Relative Quantification of Residue Specific m6A RNA Methylation. Sci. Rep. 2019, 9, 4220. [Google Scholar] [CrossRef] [PubMed]
- Harcourt, E.M.; Ehrenschwender, T.; Batista, P.J.; Chang, H.Y.; Kool, E.T. Identification of a Selective Polymerase Enables Detection of N6-Methyladenosine in RNA. J. Am. Chem. Soc. 2013, 135, 19079–19082. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Wang, J.; Zhang, X.; Fu, B.; Song, Y.; Ma, P.; Gu, K.; Zhou, X.; Zhang, X.; Tian, T.; et al. N6-Methyladenine Hinders RNA- and DNA-Directed DNA Synthesis: Application in Human RRNA Methylation Analysis of Clinical Specimens. Chem. Sci. 2016, 7, 1440–1446. [Google Scholar] [CrossRef] [PubMed]
- Golovina, A.Y.; Dzama, M.M.; Petriukov, K.S.; Zatsepin, T.S.; Sergiev, P.V.; Bogdanov, A.A.; Dontsova, O.A. Method for Site-Specific Detection of m6A Nucleoside Presence in RNA Based on High-Resolution Melting (HRM) Analysis. Nucleic Acids Res. 2014, 42, e27. [Google Scholar] [CrossRef]
- Wojdacz, T.K.; Dobrovic, A. Methylation-Sensitive High Resolution Melting (MS-HRM): A New Approach for Sensitive and High-Throughput Assessment of Methylation. Nucleic Acids Res. 2007, 35, e41. [Google Scholar] [CrossRef]
- Imanishi, M.; Tsuji, S.; Suda, A.; Futaki, S. Detection of: N6-Methyladenosine Based on the Methyl-Sensitivity of MazF RNA Endonuclease. Chem. Commun. 2017, 53, 12930–12933. [Google Scholar] [CrossRef]
- Liu, W.; Yan, J.; Zhang, Z.; Pian, H.; Liu, C.; Li, Z. Identification of a Selective DNA Ligase for Accurate Recognition and Ultrasensitive Quantification of: N6-Methyladenosine in RNA at One-Nucleotide Resolution. Chem. Sci. 2018, 9, 3354–3359. [Google Scholar] [CrossRef]
- Xiao, Y.; Wang, Y.; Tang, Q.; Wei, L.; Zhang, X.; Jia, G. An Elongation- and Ligation-Based QPCR Amplification Method for the Radiolabeling-Free Detection of Locus-Specific N6-Methyladenosine Modification. Angew. Chem. 2018, 130, 16227–16232. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sağlam, B.; Akgül, B. An Overview of Current Detection Methods for RNA Methylation. Int. J. Mol. Sci. 2024, 25, 3098. https://doi.org/10.3390/ijms25063098
Sağlam B, Akgül B. An Overview of Current Detection Methods for RNA Methylation. International Journal of Molecular Sciences. 2024; 25(6):3098. https://doi.org/10.3390/ijms25063098
Chicago/Turabian StyleSağlam, Buket, and Bünyamin Akgül. 2024. "An Overview of Current Detection Methods for RNA Methylation" International Journal of Molecular Sciences 25, no. 6: 3098. https://doi.org/10.3390/ijms25063098
APA StyleSağlam, B., & Akgül, B. (2024). An Overview of Current Detection Methods for RNA Methylation. International Journal of Molecular Sciences, 25(6), 3098. https://doi.org/10.3390/ijms25063098