
The CHD Protein Kismet Restricts the Synaptic Localization of Cell Adhesion Molecules at the Drosophila Neuromuscular Junction

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
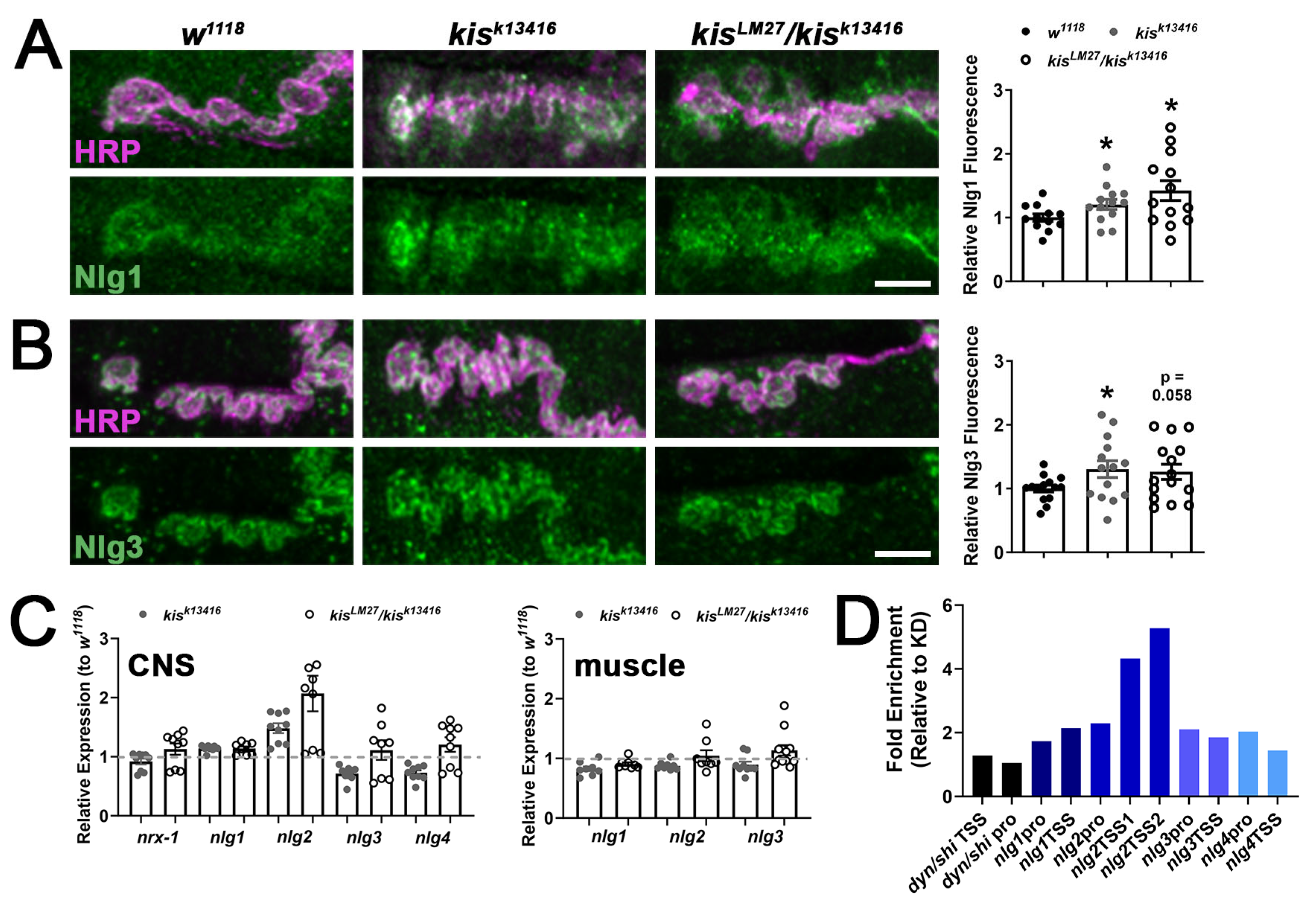
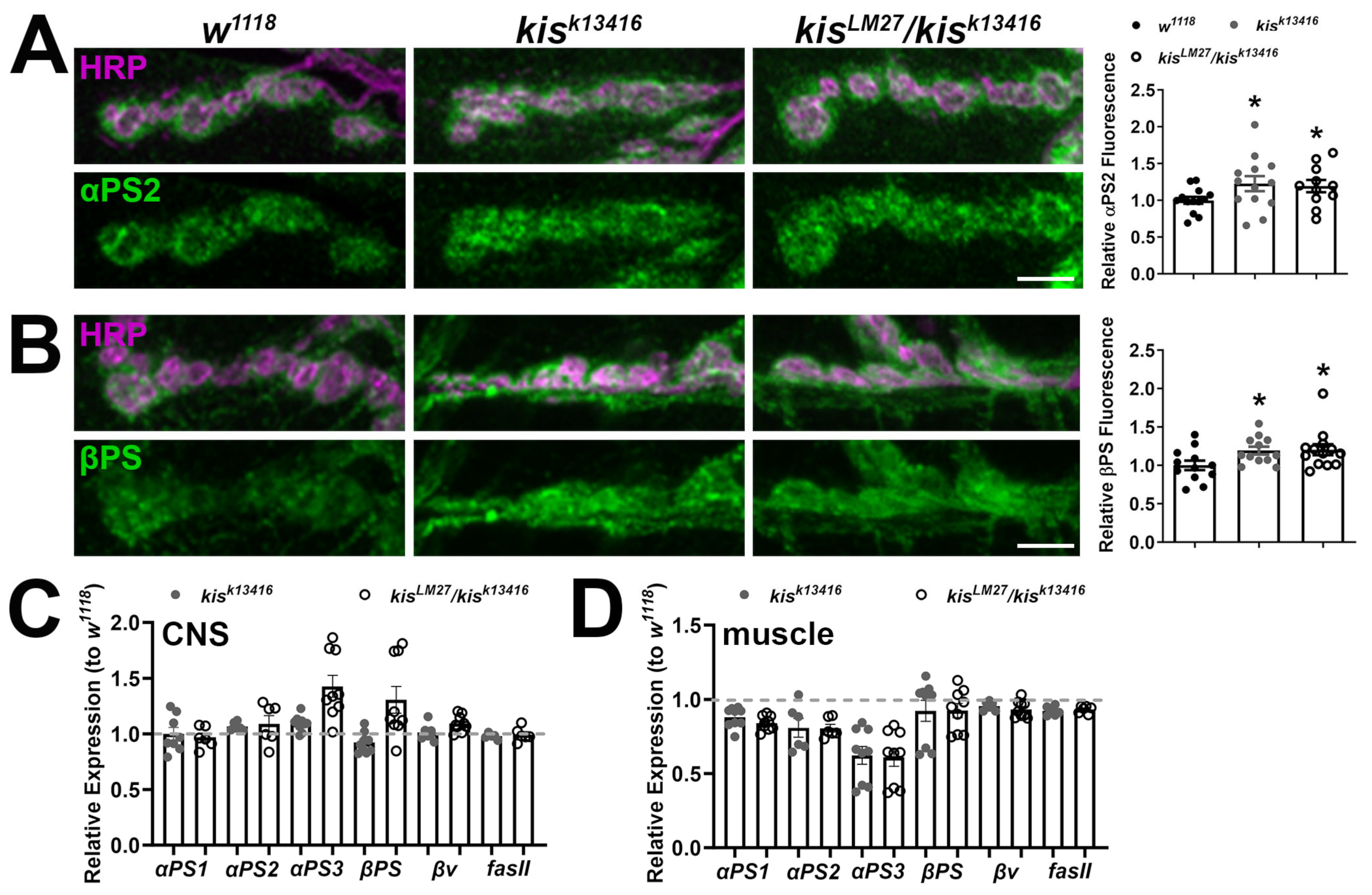
2.1. kis Restricts the Synaptic Localization of CAMs
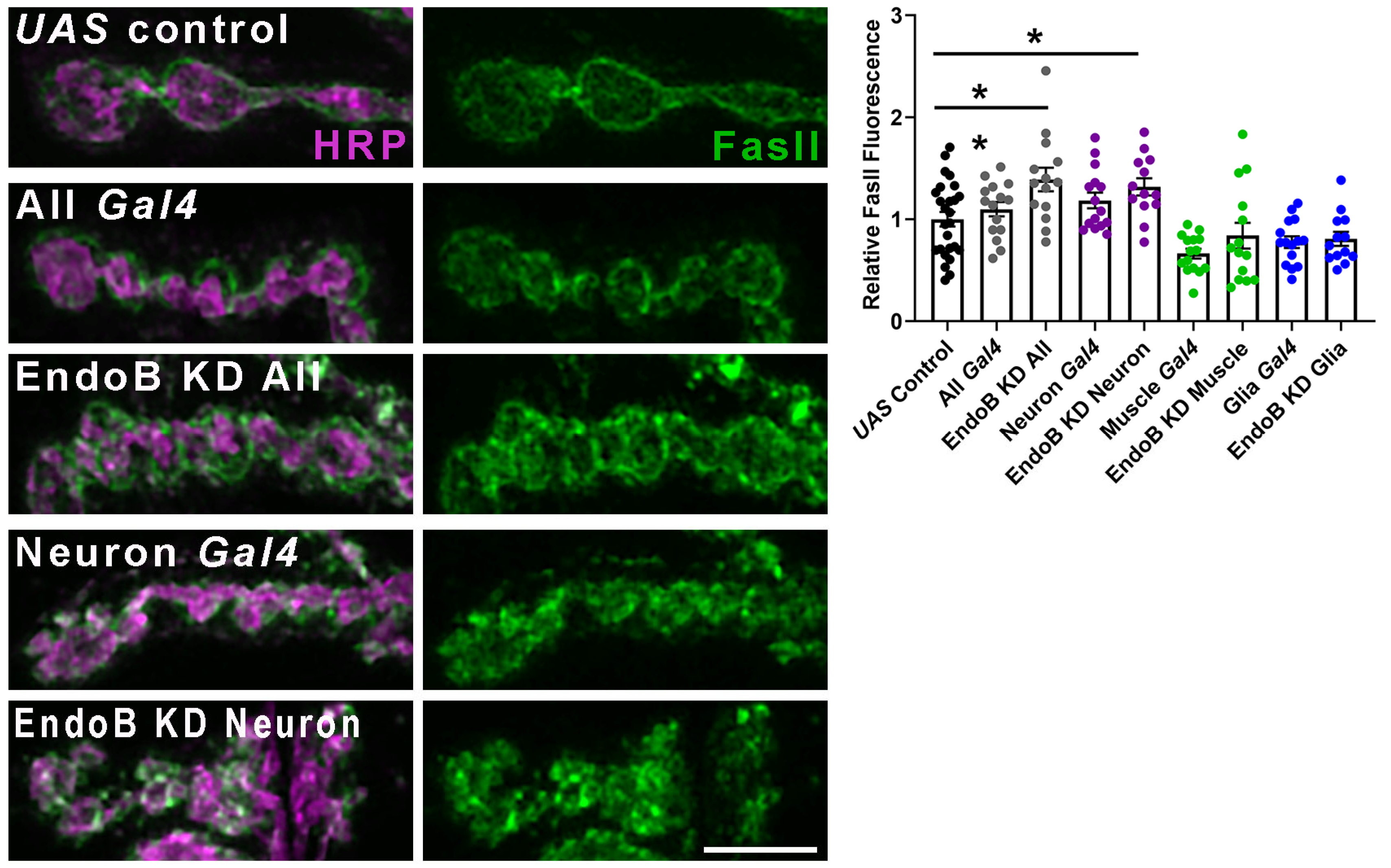
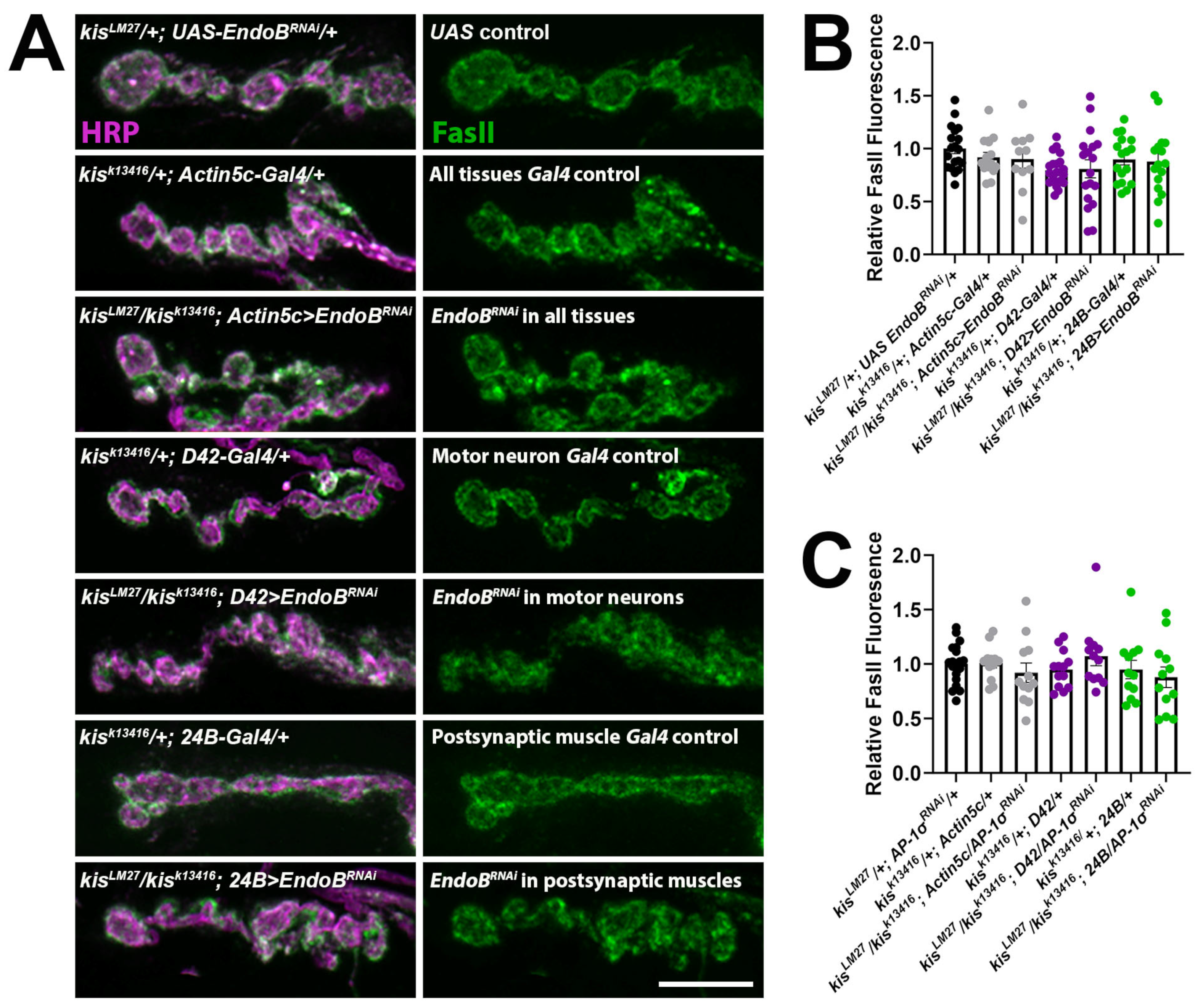
2.2. Impairing Vesicle Trafficking Increases Synaptic FasII in Wild-Type Larvae but Does Not Change Synaptic FasII in kis Mutants
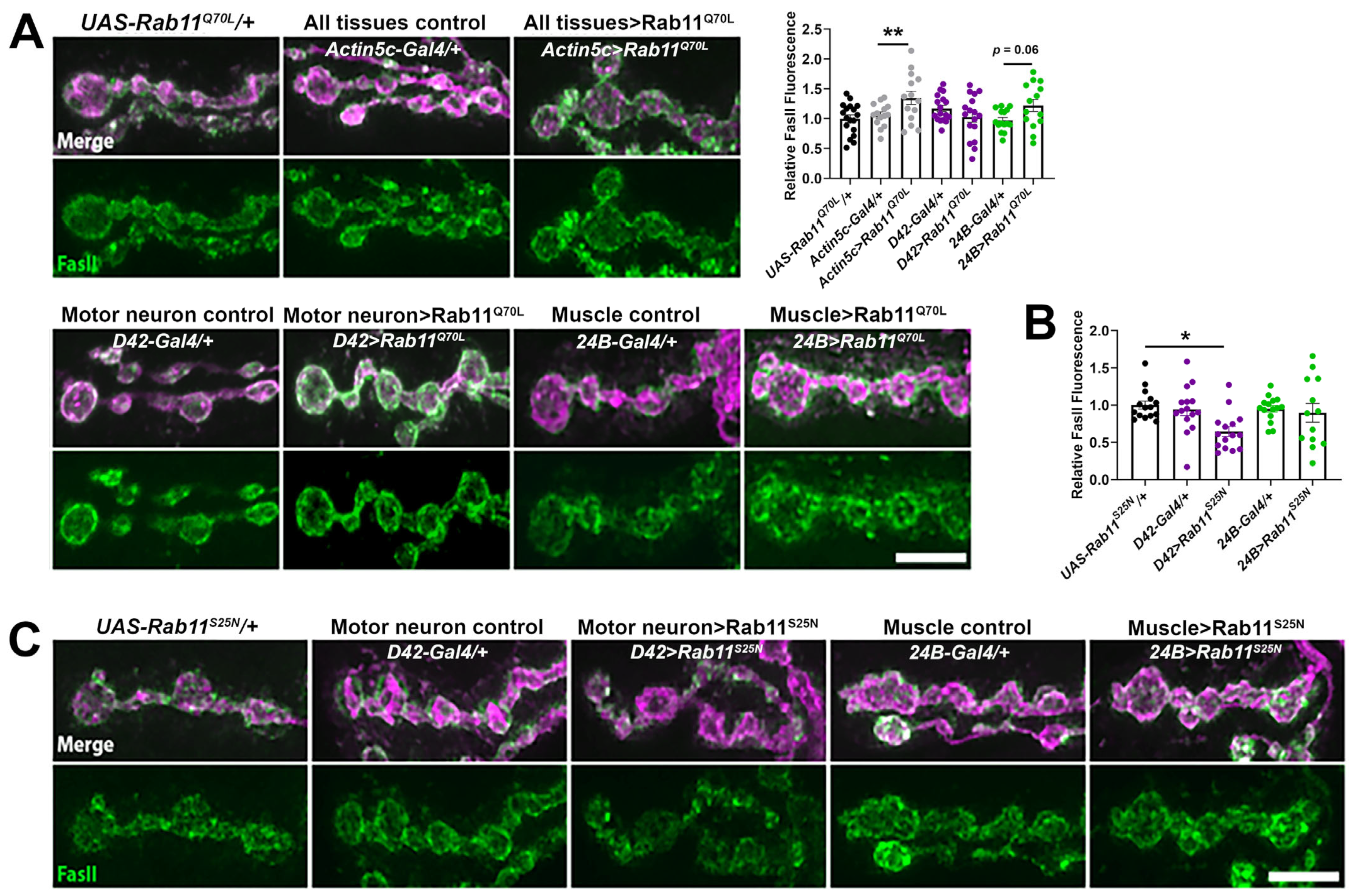
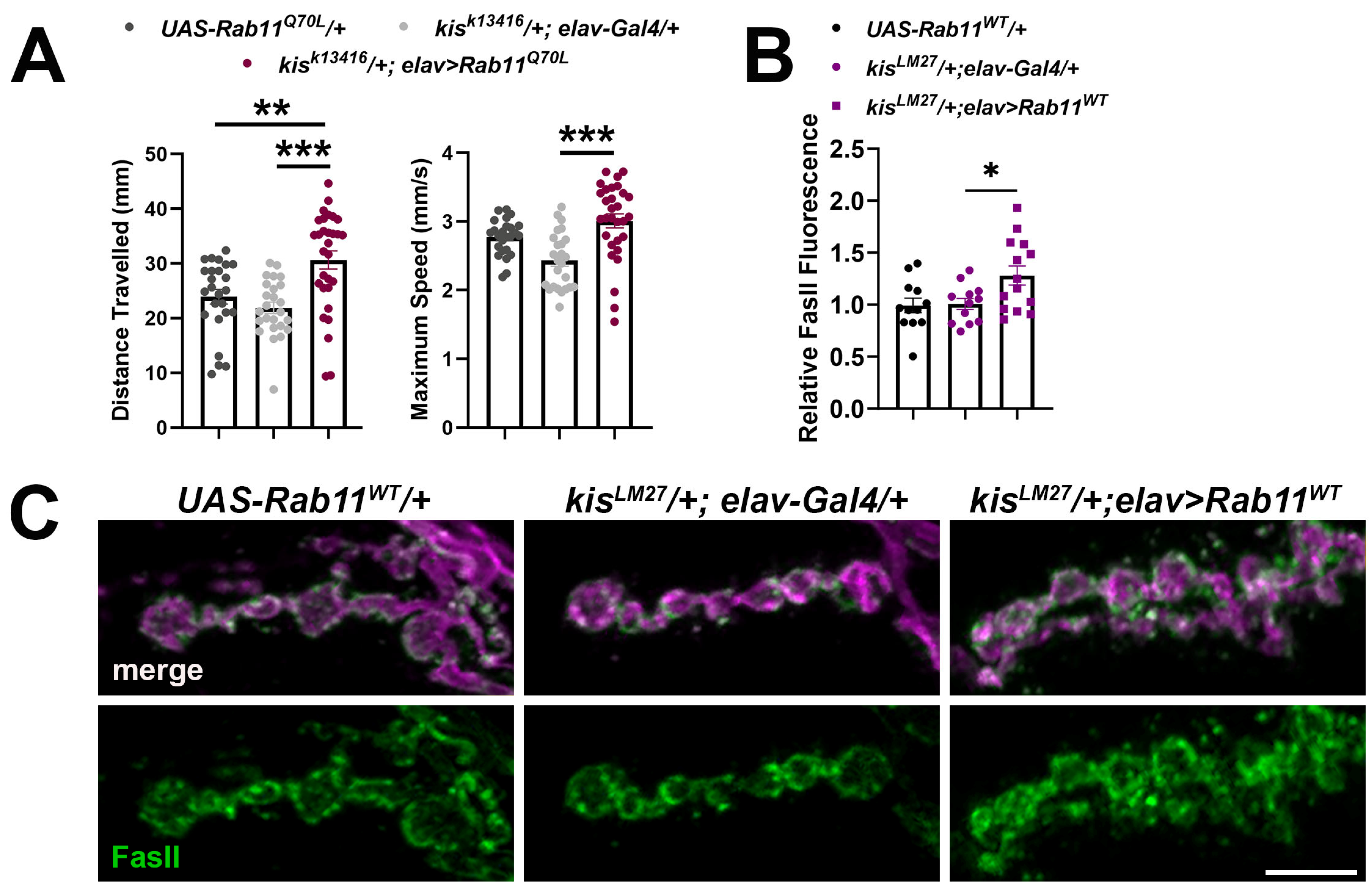
2.3. Increased Rab11 Activity Promotes the Synaptic Localization of FasII in Wild-Type Larvae and Shows an Additive Effect in kis Mutants
3. Discussion
4. Materials and Methods
4.1. Drosophila Stocks and Husbandry
4.2. Chromatin Immunoprecipitation, RNA Isolation, Reverse Transcription PCR, and qPCR
4.3. Immunocytochemistry
4.4. Larval Locomotion
4.5. Experimental Design and Statistical Analyses
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Saint-Martin, M.; Goda, Y. Astrocyte–synapse interactions and cell adhesion molecules. FEBS J. 2022, 290, 3512–3526. [Google Scholar] [CrossRef]
- Keable, R.; Leshchyns’ka, I.; Sytnyk, V. Trafficking and Activity of Glutamate and GABA Receptors: Regulation by Cell Adhesion Molecules. Neuroscientist 2020, 26, 415–437. [Google Scholar] [CrossRef]
- Shetty, A.; Sytnyk, V.; Leshchyns’Ka, I.; Puchkov, D.; Haucke, V.; Schachner, M. The Neural Cell Adhesion Molecule Promotes Maturation of the Presynaptic Endocytotic Machinery by Switching Synaptic Vesicle Recycling from Adaptor Protein 3 (AP-3)- to AP-2-Dependent Mechanisms. J. Neurosci. 2013, 33, 16828–16845. [Google Scholar] [CrossRef]
- Biederer, T.; Kaeser, P.S.; Blanpied, T.A. Transcellular Nanoalignment of Synaptic Function. Neuron 2017, 96, 680–696. [Google Scholar] [CrossRef] [PubMed]
- Guzikowski, N.J.; Kavalali, E.T. Nano-Organization at the Synapse: Segregation of Distinct Forms of Neurotransmission. Front. Synaptic Neurosci. 2021, 13, 796498. [Google Scholar] [CrossRef] [PubMed]
- Zieger, H.L.; Choquet, D. Nanoscale synapse organization and dysfunction in neurodevelopmental disorders. Neurobiol. Dis. 2021, 158, 105453. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, L.; Love, J.; Colman, D.R. Adhesion Molecules in the Nervous System: Structural Insights into Function and Diversity. Annu. Rev. Neurosci. 2007, 30, 451–474. [Google Scholar] [CrossRef] [PubMed]
- de Agustín-Durán, D.; Mateos-White, I.; Fabra-Beser, J.; Gil-Sanz, C. Stick around: Cell–Cell Adhesion Molecules during Neocortical Development. Cells 2021, 10, 118. [Google Scholar] [CrossRef] [PubMed]
- Yamada, S.; Nelson, W.J. Synapses: Sites of Cell Recognition, Adhesion, and Functional Specification. Annu. Rev. Biochem. 2007, 76, 267–294. [Google Scholar] [CrossRef] [PubMed]
- Sanes, J.R.; Zipursky, S.L. Synaptic Specificity, Recognition Molecules, and Assembly of Neural Circuits. Cell 2020, 181, 536–556. [Google Scholar] [CrossRef]
- Connor, S.A.; Siddiqui, T.J. Synapse organizers as molecular codes for synaptic plasticity. Trends Neurosci. 2023, 46, 971–985. [Google Scholar] [CrossRef]
- Dönertaş, H.M.; İzgi, H.; Kamacıoğlu, A.; He, Z.; Khaitovich, P.; Somel, M. Gene expression reversal toward pre-adult levels in the aging human brain and age-related loss of cellular identity. Sci. Rep. 2017, 7, 1–13. [Google Scholar] [CrossRef]
- Xiang, Z.; Xu, M.; Liao, M.; Jiang, Y.; Jiang, Q.; Feng, R.; Zhang, L.; Ma, G.; Wang, G.; Chen, Z.; et al. Integrating Genome-Wide Association Study and Brain Expression Data Highlights Cell Adhesion Molecules and Purine Metabolism in Alzheimer’s Disease. Mol. Neurobiol. 2014, 52, 514–521. [Google Scholar] [CrossRef]
- Edwards, Y.J.K.; Beecham, G.W.; Scott, W.K.; Khuri, S.; Bademci, G.; Tekin, D.; Martin, E.R.; Jiang, Z.; Mash, D.C.; Ffrench-Mullen, J.; et al. Identifying Consensus Disease Pathways in Parkinson’s Disease Using an Integrative Systems Biology Approach. PLoS ONE 2011, 6, e16917. [Google Scholar] [CrossRef] [PubMed]
- Corvin, A.P. Neuronal cell adhesion genes. Cell Adhes. Migr. 2010, 4, 511–514. [Google Scholar] [CrossRef] [PubMed]
- Bao, X.; Liu, G.; Jiang, Y.; Jiang, Q.; Liao, M.; Feng, R.; Zhang, L.; Ma, G.; Zhang, S.; Chen, Z.; et al. Cell adhesion molecule pathway genes are regulated by cis-regulatory SNPs and show significantly altered expression in Alzheimer’s disease brains. Neurobiol. Aging 2015, 36, 2904.e1–2904.e7. [Google Scholar] [CrossRef] [PubMed]
- Bartholomew, B. Regulating the Chromatin Landscape: Structural and Mechanistic Perspectives. Annu. Rev. Biochem. 2014, 83, 671–696. [Google Scholar] [CrossRef] [PubMed]
- Murawska, M.; Brehm, A. CHD chromatin remodelers and the transcription cycle. Transcription 2011, 2, 244–253. [Google Scholar] [CrossRef] [PubMed]
- Sugathan, A.; Biagioli, M.; Golzio, C.; Erdin, S.; Blumenthal, I.; Manavalan, P.; Ragavendran, A.; Brand, H.; Lucente, D.; Miles, J.; et al. CHD8 regulates neurodevelopmental pathways associated with autism spectrum disorder in neural progenitors. Proc. Natl. Acad. Sci. USA 2014, 111, E4468–E4477. [Google Scholar] [CrossRef] [PubMed]
- Schnetz, M.P.; Handoko, L.; Akhtar-Zaidi, B.; Bartels, C.F.; Pereira, C.F.; Fisher, A.G.; Adams, D.J.; Flicek, P.; Crawford, G.E.; LaFramboise, T.; et al. CHD7 Targets Active Gene Enhancer Elements to Modulate ES Cell-Specific Gene Expression. PLOS Genet. 2010, 6, e1001023. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Lin, M.; Pedrosa, E.; Hrabovsky, A.; Zhang, Z.; Guo, W.; Lachman, H.M.; Zheng, D. CRISPR/Cas9-mediated heterozygous knockout of the autism gene CHD8 and characterization of its transcriptional networks in neurodevelopment. Mol. Autism 2015, 6, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Vissers, L.E.L.M.; Van Ravenswaaij, C.M.A.; Admiraal, R.; Hurst, J.A.; De Vries, B.B.A.; Janssen, I.M.; Van Der Vliet, W.A.; Huys, E.H.L.P.G.; De Jong, P.J.; Hamel, B.C.J.; et al. Mutations in a new member of the chromodomain gene family cause CHARGE syndrome. Nat. Genet. 2004, 36, 955–957. [Google Scholar] [CrossRef] [PubMed]
- Ayhan, F.; Konopka, G. Regulatory genes and pathways disrupted in autism spectrum disorders. Prog. Neuro-Psychopharmacology Biol. Psychiatry 2018, 89, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, S.; Armstrong, J.A.; Deuring, R.; Dahlsveen, I.K.; McNeill, H.; Tamkun, J.W. The Drosophila trithorax group protein Kismet facilitates an early step in transcriptional elongation by RNA Polymerase II. Development 2005, 132, 1623–1635. [Google Scholar] [CrossRef] [PubMed]
- Gervais, L.; Beek, M.v.D.; Josserand, M.; Sallé, J.; Stefanutti, M.; Perdigoto, C.N.; Skorski, P.; Mazouni, K.; Marshall, O.J.; Brand, A.H.; et al. Stem Cell Proliferation Is Kept in Check by the Chromatin Regulators Kismet/CHD7/CHD8 and Trr/MLL3/4. Dev. Cell 2019, 49, 556–573.e6. [Google Scholar] [CrossRef] [PubMed]
- Chou, V.T.; Johnson, S.A.; Van Vactor, D. Synapse development and maturation at the drosophila neuromuscular junction. Neural Dev. 2020, 15, 1–17. [Google Scholar] [CrossRef]
- Liebl, F.L.; Featherstone, D.E. Identification and Investigation of Drosophila Postsynaptic Density Homologs. Bioinform. Biol. Insights 2008, 2, 369–381. [Google Scholar] [CrossRef] [PubMed]
- Menon, K.P.; Carrillo, R.A.; Zinn, K. Development and plasticity of the Drosophila larval neuromuscular junction. Wiley Interdiscip. Rev. Dev. Biol. 2013, 2, 647–670. [Google Scholar] [CrossRef]
- Rickels, R.; Hu, D.; Collings, C.K.; Woodfin, A.R.; Piunti, A.; Mohan, M.; Herz, H.-M.; Kvon, E.; Shilatifard, A. An Evolutionary Conserved Epigenetic Mark of Polycomb Response Elements Implemented by Trx/MLL/COMPASS. Mol. Cell 2016, 63, 318–328. [Google Scholar] [CrossRef]
- Latcheva, N.K.; Delaney, T.L.; Viveiros, J.M.; Smith, R.A.; Bernard, K.M.; Harsin, B.; Marenda, D.R.; Liebl, F.L.W. The CHD Protein, Kismet, is Important for the Recycling of Synaptic Vesicles during Endocytosis. Sci. Rep. 2019, 9, 1–13. [Google Scholar] [CrossRef]
- Ghosh, R.; Vegesna, S.; Safi, R.; Bao, H.; Zhang, B.; Marenda, D.R.; Liebl, F.L.W. Kismet Positively Regulates Glutamate Receptor Localization and Synaptic Transmission at the Drosophila Neuromuscular Junction. PLoS ONE 2014, 9, e113494. [Google Scholar] [CrossRef] [PubMed]
- Latcheva, N.K.; Viveiros, J.M.; Waddell, E.A.; Nguyen, P.T.; Liebl, F.L.; Marenda, D.R. Epigenetic crosstalk: Pharmacological inhibition of HDACs can rescue defective synaptic morphology and neurotransmission phenotypes associated with loss of the chromatin reader Kismet. Mol. Cell. Neurosci. 2018, 87, 77–85. [Google Scholar] [CrossRef]
- Missler, M.; Südhof, T.C.; Biederer, T. Synaptic Cell Adhesion. Cold Spring Harb. Perspect. Biol. 2012, 4, a005694. [Google Scholar] [CrossRef] [PubMed]
- Leshchyns’ka, I.; Sytnyk, V. Synaptic Cell Adhesion Molecules in Alzheimer’s Disease. Neural Plast. 2016, 2016, 1–9. [Google Scholar] [CrossRef]
- Melicharek, D.J.; Ramirez, L.C.; Singh, S.; Thompson, R.; Marenda, D.R. Kismet/CHD7 regulates axon morphology, memory and locomotion in a Drosophila model of CHARGE syndrome. Hum. Mol. Genet. 2010, 19, 4253–4264. [Google Scholar] [CrossRef] [PubMed]
- Dean, C.; Scholl, F.G.; Choih, J.; DeMaria, S.; Berger, J.; Isacoff, E.; Scheiffele, P. Neurexin mediates the assembly of presynaptic terminals. Nat. Neurosci. 2003, 6, 708–716. [Google Scholar] [CrossRef] [PubMed]
- Graf, E.R.; Zhang, X.; Jin, S.-X.; Linhoff, M.W.; Craig, A.M. Neurexins Induce Differentiation of GABA and Glutamate Postsynaptic Specializations via Neuroligins. Cell 2004, 119, 1013–1026. [Google Scholar] [CrossRef] [PubMed]
- Budreck, E.C.; Kwon, O.-B.; Jung, J.H.; Baudouin, S.; Thommen, A.; Kim, H.-S.; Fukazawa, Y.; Harada, H.; Tabuchi, K.; Shigemoto, R.; et al. Neuroligin-1 controls synaptic abundance of NMDA-type glutamate receptors through extracellular coupling. Proc. Natl. Acad. Sci. USA 2012, 110, 725–730. [Google Scholar] [CrossRef]
- Shipman, S.L.; Nicoll, R.A. A Subtype-Specific Function for the Extracellular Domain of Neuroligin 1 in Hippocampal LTP. Neuron 2012, 76, 309–316. [Google Scholar] [CrossRef]
- Mozer, B.A.; Sandstrom, D.J. Drosophila neuroligin 1 regulates synaptic growth and function in response to activity and phosphoinositide-3-kinase. Mol. Cell. Neurosci. 2012, 51, 89–100. [Google Scholar] [CrossRef]
- Parkinson, W.; Dear, M.L.; Rushton, E.; Broadie, K. N-glycosylation requirements in neuromuscular synaptogenesis. Development 2013, 140, 4970–4981. [Google Scholar] [CrossRef]
- Buszczak, M.; Paterno, S.; Lighthouse, D.; Bachman, J.; Planck, J.; Owen, S.; Skora, A.D.; Nystul, T.G.; Ohlstein, B.; Allen, A.; et al. The Carnegie Protein Trap Library: A Versatile Tool for Drosophila Developmental Studies. Genetics 2007, 175, 1505–1531. [Google Scholar] [CrossRef]
- Latcheva, N.K.; Viveiros, J.M.; Marenda, D.R. The Drosophila Chromodomain Protein Kismet Activates Steroid Hormone Receptor Transcription to Govern Axon Pruning and Memory In Vivo. iScience 2019, 16, 79–93. [Google Scholar] [CrossRef]
- Jaudon, F.; Thalhammer, A.; Cingolani, L.A. Integrin adhesion in brain assembly: From molecular structure to neuropsychiatric disorders. Eur. J. Neurosci. 2020, 53, 3831–3850. [Google Scholar] [CrossRef]
- Wang, Q.; Han, T.H.; Nguyen, P.; Jarnik, M.; Serpe, M. Tenectin recruits integrin to stabilize bouton architecture and regulate vesicle release at the Drosophila neuromuscular junction. eLife 2018, 7, e35518. [Google Scholar] [CrossRef]
- Tsai, P.-I.; Wang, M.; Kao, H.-H.; Cheng, Y.-J.; Lin, Y.-J.; Chen, R.-H.; Chien, C.-T. Activity-dependent retrograde laminin A signaling regulates synapse growth at Drosophila neuromuscular junctions. Proc. Natl. Acad. Sci. USA 2012, 109, 17699–17704. [Google Scholar] [CrossRef]
- Lee, J.Y.; Geng, J.; Lee, J.; Wang, A.R.; Chang, K.T. Activity-Induced Synaptic Structural Modifications by an Activator of Integrin Signaling at the Drosophila Neuromuscular Junction. J. Neurosci. 2017, 37, 3246–3263. [Google Scholar] [CrossRef] [PubMed]
- Bajpai, R.; Chen, D.A.; Rada-Iglesias, A.; Zhang, J.; Xiong, Y.; Helms, J.; Chang, C.-P.; Zhao, Y.; Swigut, T.; Wysocka, J. CHD7 cooperates with PBAF to control multipotent neural crest formation. Nature 2010, 463, 958–962. [Google Scholar] [CrossRef] [PubMed]
- Bukalo, O.; Fentrop, N.; Lee, A.Y.W.; Salmen, B.; Law, J.W.S.; Wotjak, C.T.; Schweizer, M.; Dityatev, A.; Schachner, M. Conditional Ablation of the Neural Cell Adhesion Molecule Reduces Precision of Spatial Learning, Long-Term Potentiation, and Depression in the CA1 Subfield of Mouse Hippocampus. J. Neurosci. 2004, 24, 1565–1577. [Google Scholar] [CrossRef] [PubMed]
- Schuster, C.M.; Davis, G.W.; Fetter, R.D.; Goodman, C.S. Genetic Dissection of Structural and Functional Components of Synaptic Plasticity. II. Fasciclin II Controls Presynaptic Structural Plasticity. Neuron 1996, 17, 655–667. [Google Scholar] [CrossRef] [PubMed]
- Schuster, C.M.; Davis, G.W.; Fetter, R.D.; Goodman, C.S. Genetic Dissection of Structural and Functional Components of Synaptic Plasticity. I. Fasciclin II Controls Synaptic Stabilization and Growth. Neuron 1996, 17, 641–654. [Google Scholar] [CrossRef] [PubMed]
- Bonanomi, D.; Benfenati, F.; Valtorta, F. Protein sorting in the synaptic vesicle life cycle. Prog. Neurobiol. 2006, 80, 177–217. [Google Scholar] [CrossRef] [PubMed]
- Saheki, Y.; De Camilli, P. Synaptic Vesicle Endocytosis. Cold Spring Harb. Perspect. Biol. 2012, 4, a005645. [Google Scholar] [CrossRef] [PubMed]
- Alabi, A.A.; Tsien, R.W. Synaptic Vesicle Pools and Dynamics. Cold Spring Harb. Perspect. Biol. 2012, 4, a013680. [Google Scholar] [CrossRef] [PubMed]
- Morgan, J.R.; Comstra, H.S.; Cohen, M.; Faundez, V. Presynaptic Membrane Retrieval and Endosome Biology: Defining Molecularly Heterogeneous Synaptic Vesicles. Cold Spring Harb. Perspect. Biol. 2013, 5, a016915. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, T.; Kaibuchi, K. Numb Controls Integrin Endocytosis for Directional Cell Migration with aPKC and PAR-3. Dev. Cell 2007, 13, 15–28. [Google Scholar] [CrossRef]
- Miñana, R.; Duran, J.M.; Tomas, M.; Renau-Piqueras, J.; Guerri, C. Neural cell adhesion molecule is endocytosed via a clathrin-dependent pathway. Eur. J. Neurosci. 2001, 13, 749–756. [Google Scholar] [CrossRef]
- Kjaerulff, O.; Brodin, L.; Jung, A. The Structure and Function of Endophilin Proteins. Cell Biochem. Biophys. 2010, 60, 137–154. [Google Scholar] [CrossRef]
- Tsai, Y.-C.; Chiang, W.; Liou, W.; Lee, W.-H.; Chang, Y.-W.; Wang, P.-Y.; Li, Y.-C.; Tanaka, T.; Nakamura, A.; Pai, L.-M. Endophilin B is required for the Drosophila oocyte to endocytose yolk downstream of Oskar. Development 2014, 141, 563–573. [Google Scholar] [CrossRef][Green Version]
- Hernandez-Diaz, S.; Ghimire, S.; Sanchez-Mirasierra, I.; Montecinos-Oliva, C.; Swerts, J.; Kuenen, S.; Verstreken, P.; Soukup, S.-F. Endophilin-B regulates autophagy during synapse development and neurodegeneration. Neurobiol. Dis. 2021, 163, 105595. [Google Scholar] [CrossRef]
- Yu, S.-C.; Jánosi, B.; Liewald, J.F.; Wabnig, S.; Gottschalk, A. Endophilin A and B Join Forces With Clathrin to Mediate Synaptic Vesicle Recycling in Caenorhabditis elegans. Front. Mol. Neurosci. 2018, 11, 196. [Google Scholar] [CrossRef]
- Li, J.; Barylko, B.; Eichorst, J.P.; Mueller, J.D.; Albanesi, J.P.; Chen, Y. Association of Endophilin B1 with Cytoplasmic Vesicles. Biophys. J. 2016, 111, 565–576. [Google Scholar] [CrossRef]
- Nakatsu, F.; Hase, K.; Ohno, H. The Role of the Clathrin Adaptor AP-1: Polarized Sorting and Beyond. Membranes 2014, 4, 747–763. [Google Scholar] [CrossRef] [PubMed]
- Cheung, G.; Cousin, M.A. Adaptor Protein Complexes 1 and 3 Are Essential for Generation of Synaptic Vesicles from Activity-Dependent Bulk Endosomes. J. Neurosci. 2012, 32, 6014–6023. [Google Scholar] [CrossRef] [PubMed]
- Glyvuk, N.; Tsytsyura, Y.; Geumann, C.; D’Hooge, R.; Hüve, J.; Kratzke, M.; Baltes, J.; Böning, D.; Klingauf, J.; Schu, P. AP-1/σ1B-adaptin mediates endosomal synaptic vesicle recycling, learning and memory. EMBO J. 2010, 29, 1318–1330. [Google Scholar] [CrossRef] [PubMed]
- Candiello, E.; Kratzke, M.; Wenzel, D.; Cassel, D.; Schu, P. AP-1/σ1A and AP-1/σ1B adaptor-proteins differentially regulate neuronal early endosome maturation via the Rab5/Vps34-pathway. Sci. Rep. 2016, 6, 29950. [Google Scholar] [CrossRef] [PubMed]
- Kiral, F.R.; Kohrs, F.E.; Jin, E.J.; Hiesinger, P.R. Rab GTPases and Membrane Trafficking in Neurodegeneration. Curr. Biol. 2018, 28, R471–R486. [Google Scholar] [CrossRef] [PubMed]
- Nieuwenhuis, B.; Eva, R. ARF6 and Rab11 as intrinsic regulators of axon regeneration. Small GTPases 2018, 11, 392–401. [Google Scholar] [CrossRef] [PubMed]
- Rozés-Salvador, V.; González-Billault, C.; Conde, C. The Recycling Endosome in Nerve Cell Development: One Rab to Rule Them All? Front. Cell Dev. Biol. 2020, 8, 603794. [Google Scholar] [CrossRef] [PubMed]
- Kokotos, A.C.; Peltier, J.; Davenport, E.C.; Trost, M.; Cousin, M.A. Activity-dependent bulk endocytosis proteome reveals a key presynaptic role for the monomeric GTPase Rab11. Proc. Natl. Acad. Sci. USA 2018, 115, E10177–E10186. [Google Scholar] [CrossRef]
- Heckscher, E.S.; Lockery, S.R.; Doe, C.Q. Characterization of Drosophila Larval Crawling at the Level of Organism, Segment, and Somatic Body Wall Musculature. J. Neurosci. 2012, 32, 12460–12471. [Google Scholar] [CrossRef] [PubMed]
- Gjorgjieva, J.; Berni, J.; Evers, J.F.; Eglen, S.J. Neural circuits for peristaltic wave propagation in crawling Drosophila larvae: Analysis and modeling. Front. Comput. Neurosci. 2013, 7, 24. [Google Scholar] [CrossRef] [PubMed]
- Sokpor, G.; Castro-Hernandez, R.; Rosenbusch, J.; Staiger, J.F.; Tuoc, T. ATP-Dependent Chromatin Remodeling During Cortical Neurogenesis. Front. Neurosci. 2018, 12, 226. [Google Scholar] [CrossRef] [PubMed]
- Weng, M.K.; Zimmer, B.; Pöltl, D.; Broeg, M.P.; Ivanova, V.; Gaspar, J.A.; Sachinidis, A.; Wüllner, U.; Waldmann, T.; Leist, M. Extensive Transcriptional Regulation of Chromatin Modifiers during Human Neurodevelopment. PLoS ONE 2012, 7, e36708. [Google Scholar] [CrossRef]
- Jiménez, J.A.; Ptacek, T.S.; Tuttle, A.H.; Schmid, R.S.; Moy, S.S.; Simon, J.M.; Zylka, M.J. Chd8 haploinsufficiency impairs early brain development and protein homeostasis later in life. Mol. Autism 2020, 11, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Schulz, Y.; Freese, L.; Mänz, J.; Zoll, B.; Völter, C.; Brockmann, K.; Bögershausen, N.; Becker, J.; Wollnik, B.; Pauli, S. CHARGE and Kabuki syndromes: A phenotypic and molecular link. Hum. Mol. Genet. 2014, 23, 4396–4405. [Google Scholar] [CrossRef] [PubMed]
- Wade, A.A.; Lim, K.; Catta-Preta, R.; Nord, A.S. Common CHD8 Genomic Targets Contrast With Model-Specific Transcriptional Impacts of CHD8 Haploinsufficiency. Front. Mol. Neurosci. 2019, 11, 481. [Google Scholar] [CrossRef]
- Zhao, C.; Dong, C.; Frah, M.; Deng, Y.; Marie, C.; Zhang, F.; Xu, L.; Ma, Z.; Dong, X.; Lin, Y.; et al. Dual Requirement of CHD8 for Chromatin Landscape Establishment and Histone Methyltransferase Recruitment to Promote CNS Myelination and Repair. Dev. Cell 2018, 45, 753–768.e8. [Google Scholar] [CrossRef]
- Ashley, J.; Packard, M.; Ataman, B.; Budnik, V. Fasciclin II Signals New Synapse Formation through Amyloid Precursor Protein and the Scaffolding Protein dX11/Mint. J. Neurosci. 2005, 25, 5943–5955. [Google Scholar] [CrossRef]
- Robbins, E.M.; Krupp, A.J.; de Arce, K.P.; Ghosh, A.K.; Fogel, A.I.; Boucard, A.; Südhof, T.C.; Stein, V.; Biederer, T. SynCAM 1 Adhesion Dynamically Regulates Synapse Number and Impacts Plasticity and Learning. Neuron 2010, 68, 894–906. [Google Scholar] [CrossRef]
- Leshchyns’Ka, I.; Tanaka, M.M.; Schachner, M.; Sytnyk, V. Immobilized Pool of NCAM180 in the Postsynaptic Membrane Is Homeostatically Replenished by the Flux of NCAM180 from Extrasynaptic Regions. J. Biol. Chem. 2011, 286, 23397–23406. [Google Scholar] [CrossRef] [PubMed]
- Stan, A.; Pielarski, K.N.; Brigadski, T.; Wittenmayer, N.; Fedorchenko, O.; Gohla, A.; Lessmann, V.; Dresbach, T.; Gottmann, K. Essential cooperation of N-cadherin and neuroligin-1 in the transsynaptic control of vesicle accumulation. Proc. Natl. Acad. Sci. USA 2010, 107, 11116–11121. [Google Scholar] [CrossRef] [PubMed]
- Aiga, M.; Levinson, J.N.; Bamji, S.X. N-cadherin and Neuroligins Cooperate to Regulate Synapse Formation in Hippocampal Cultures. J. Biol. Chem. 2011, 286, 851–858. [Google Scholar] [CrossRef] [PubMed]
- Mortillo, S.; Elste, A.; Ge, Y.; Patil, S.B.; Hsiao, K.; Huntley, G.W.; Davis, R.L.; Benson, D.L. Compensatory redistribution of neuroligins and N-cadherin following deletion of synaptic β1-integrin. J. Comp. Neurol. 2011, 520, 2041–2052. [Google Scholar] [CrossRef] [PubMed]
- Grochowska, K.M.; Andres-Alonso, M.; Karpova, A.; Kreutz, M.R. The needs of a synapse—How local organelles serve synaptic proteostasis. EMBO J. 2022, 41, e110057. [Google Scholar] [CrossRef] [PubMed]
- Gan, Q.; Watanabe, S. Synaptic Vesicle Endocytosis in Different Model Systems. Front. Cell. Neurosci. 2018, 12, 171. [Google Scholar] [CrossRef]
- Kaksonen, M.; Roux, A. Mechanisms of clathrin-mediated endocytosis. Nat. Rev. Mol. Cell Biol. 2018, 19, 313–326. [Google Scholar] [CrossRef]
- Batchelder, E.M.; Yarar, D. Differential Requirements for Clathrin-dependent Endocytosis at Sites of Cell–Substrate Adhesion. Mol. Biol. Cell 2010, 21, 3070–3079. [Google Scholar] [CrossRef]
- van Stegen, B.; Dagar, S.; Gottmann, K. Release activity-dependent control of vesicle endocytosis by the synaptic adhesion molecule N-cadherin. Sci. Rep. 2017, 7, 40865. [Google Scholar] [CrossRef]
- Luo, J.K.; Melland, H.; Nithianantharajah, J.; Gordon, S.L. Postsynaptic Neuroligin-1 Mediates Presynaptic Endocytosis During Neuronal Activity. Front. Mol. Neurosci. 2021, 14, 744845. [Google Scholar] [CrossRef]
- Polo-Parada, L.; Bose, C.M.; Plattner, F.; Landmesser, L.T. Distinct Roles of Different Neural Cell Adhesion Molecule (NCAM) Isoforms in Synaptic Maturation Revealed by Analysis of NCAM 180 kDa Isoform-Deficient Mice. J. Neurosci. 2004, 24, 1852–1864. [Google Scholar] [CrossRef] [PubMed]
- Gompers, A.L.; Su-Feher, L.; Ellegood, J.; A Copping, N.; Riyadh, M.A.; Stradleigh, T.W.; Pride, M.C.; Schaffler, M.D.; Wade, A.A.; Catta-Preta, R.; et al. Germline Chd8 haploinsufficiency alters brain development in mouse. Nat. Neurosci. 2017, 20, 1062–1073. [Google Scholar] [CrossRef] [PubMed]
- Suetterlin, P.; Hurley, S.; Mohan, C.; Riegman, K.L.H.; Pagani, M.; Caruso, A.; Ellegood, J.; Galbusera, A.; Crespo-Enriquez, I.; Michetti, C.; et al. Altered Neocortical Gene Expression, Brain Overgrowth and Functional Over-Connectivity in Chd8 Haploinsufficient Mice. Cereb. Cortex 2018, 28, 2192–2206. [Google Scholar] [CrossRef] [PubMed]
- Ivanova, D.; Cousin, M.A. Synaptic Vesicle Recycling and the Endolysosomal System: A Reappraisal of Form and Function. Front. Synaptic Neurosci. 2022, 14, 826098. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.-Y.; Xiong, Y.; Zhang, W.; Wan, J. Endophilin B1 regulates EGFR endocytic degradation in prostate cancer cell. Cell Mol. Biol. 2016, 62, 37–42. [Google Scholar] [PubMed]
- Duncan, M.C. New directions for the clathrin adaptor AP-1 in cell biology and human disease. Curr. Opin. Cell Biol. 2022, 76, 102079. [Google Scholar] [CrossRef]
- Tarpey, P.S.; Stevens, C.; Teague, J.; Edkins, S.; O’meara, S.; Avis, T.; Barthorpe, S.; Buck, G.; Butler, A.; Cole, J.; et al. Mutations in the Gene Encoding the Sigma 2 Subunit of the Adaptor Protein 1 Complex, AP1S2, Cause X-Linked Mental Retardation. Am. J. Hum. Genet. 2006, 79, 1119–1124. [Google Scholar] [CrossRef]
- Kratzke, M.; Candiello, E.; Schmidt, B.; Jahn, O.; Schu, P. AP-1/σ1B-Dependent SV Protein Recycling Is Regulated in Early Endosomes and Is Coupled to AP-2 Endocytosis. Mol. Neurobiol. 2014, 52, 142–161. [Google Scholar] [CrossRef]
- Wilkinson, B.; Grepo, N.; Thompson, B.L.; Kim, J.; Wang, K.; Evgrafov, O.V.; Lu, W.; A Knowles, J.; Campbell, D.B. The autism-associated gene chromodomain helicase DNA-binding protein 8 (CHD8) regulates noncoding RNAs and autism-related genes. Transl. Psychiatry 2015, 5, e568. [Google Scholar] [CrossRef]
- Rajgor, D.; Welle, T.M.; Smith, K.R. The Coordination of Local Translation, Membranous Organelle Trafficking, and Synaptic Plasticity in Neurons. Front. Cell Dev. Biol. 2021, 9, 711446. [Google Scholar] [CrossRef]
- Sun, C.; Nold, A.; Fusco, C.M.; Rangaraju, V.; Tchumatchenko, T.; Heilemann, M.; Schuman, E.M. The prevalence and specificity of local protein synthesis during neuronal synaptic plasticity. Sci. Adv. 2021, 7, eabj0790. [Google Scholar] [CrossRef]
- Glock, C.; Biever, A.; Tushev, G.; Nassim-Assir, B.; Kao, A.; Bartnik, I.; Dieck, S.T.; Schuman, E.M. The translatome of neuronal cell bodies, dendrites, and axons. Proc. Natl. Acad. Sci. USA 2021, 118, e2113929118. [Google Scholar] [CrossRef] [PubMed]
- Biever, A.; Donlin-Asp, P.G.; Schuman, E.M. Local translation in neuronal processes. Curr. Opin. Neurobiol. 2019, 57, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Verma, P.; Cohen, S.M. miR-965 controls cell proliferation and migration during tissue morphogenesis in the Drosophila abdomen. eLife 2015, 4, e07389. [Google Scholar] [CrossRef]
- Agarwal, V.; Bell, G.W.; Nam, J.-W.; Bartel, D.P. Predicting effective microRNA target sites in mammalian mRNAs. eLife 2015, 4, e05005. [Google Scholar] [CrossRef] [PubMed]
- Parenti, I.; Rabaneda, L.G.; Schoen, H.; Novarino, G. Neurodevelopmental Disorders: From Genetics to Functional Pathways. Trends Neurosci. 2020, 43, 608–621. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Smith, I.R.; Hendricks, E.L.; Latcheva, N.K.; Marenda, D.R.; Liebl, F.L.W. The CHD Protein Kismet Restricts the Synaptic Localization of Cell Adhesion Molecules at the Drosophila Neuromuscular Junction. Int. J. Mol. Sci. 2024, 25, 3074. https://doi.org/10.3390/ijms25053074
Smith IR, Hendricks EL, Latcheva NK, Marenda DR, Liebl FLW. The CHD Protein Kismet Restricts the Synaptic Localization of Cell Adhesion Molecules at the Drosophila Neuromuscular Junction. International Journal of Molecular Sciences. 2024; 25(5):3074. https://doi.org/10.3390/ijms25053074
Chicago/Turabian StyleSmith, Ireland R., Emily L. Hendricks, Nina K. Latcheva, Daniel R. Marenda, and Faith L. W. Liebl. 2024. "The CHD Protein Kismet Restricts the Synaptic Localization of Cell Adhesion Molecules at the Drosophila Neuromuscular Junction" International Journal of Molecular Sciences 25, no. 5: 3074. https://doi.org/10.3390/ijms25053074
APA StyleSmith, I. R., Hendricks, E. L., Latcheva, N. K., Marenda, D. R., & Liebl, F. L. W. (2024). The CHD Protein Kismet Restricts the Synaptic Localization of Cell Adhesion Molecules at the Drosophila Neuromuscular Junction. International Journal of Molecular Sciences, 25(5), 3074. https://doi.org/10.3390/ijms25053074