
Combination Treatment with EGFR Inhibitor and Doxorubicin Synergistically Inhibits Proliferation of MCF-7 Cells and MDA-MB-231 Triple-Negative Breast Cancer Cells In Vitro


Abstract
1. Introduction
2. Results
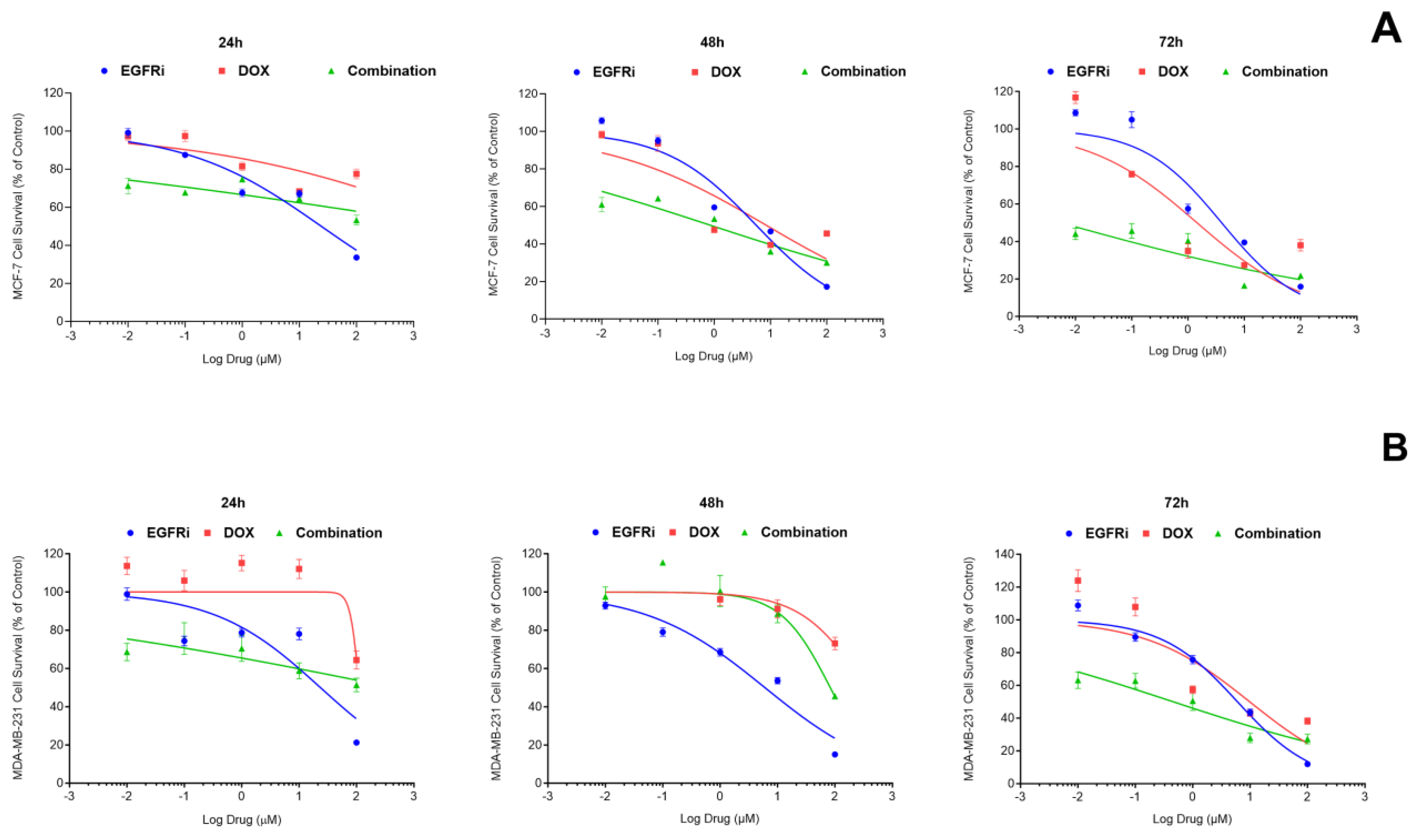
2.1. EGFRi Displays Time-Dependent Increase in Potency When Used as a Single-Agent Whilst Doxorubicin Is More Effective in Combination
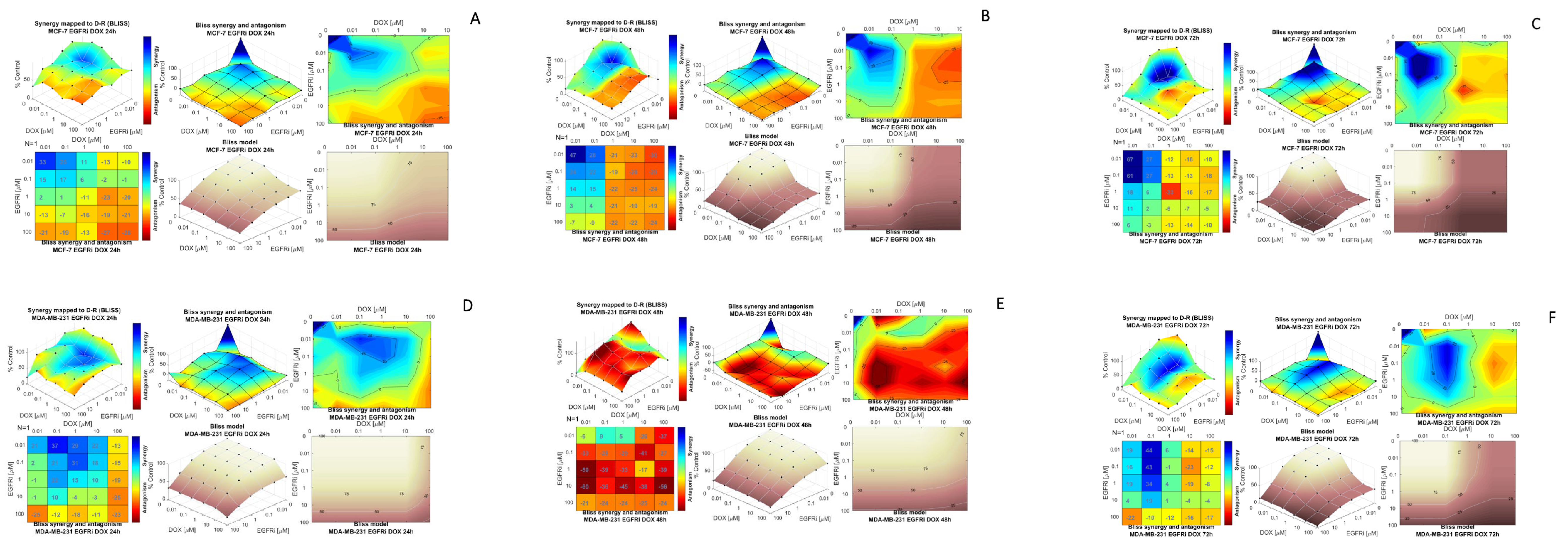
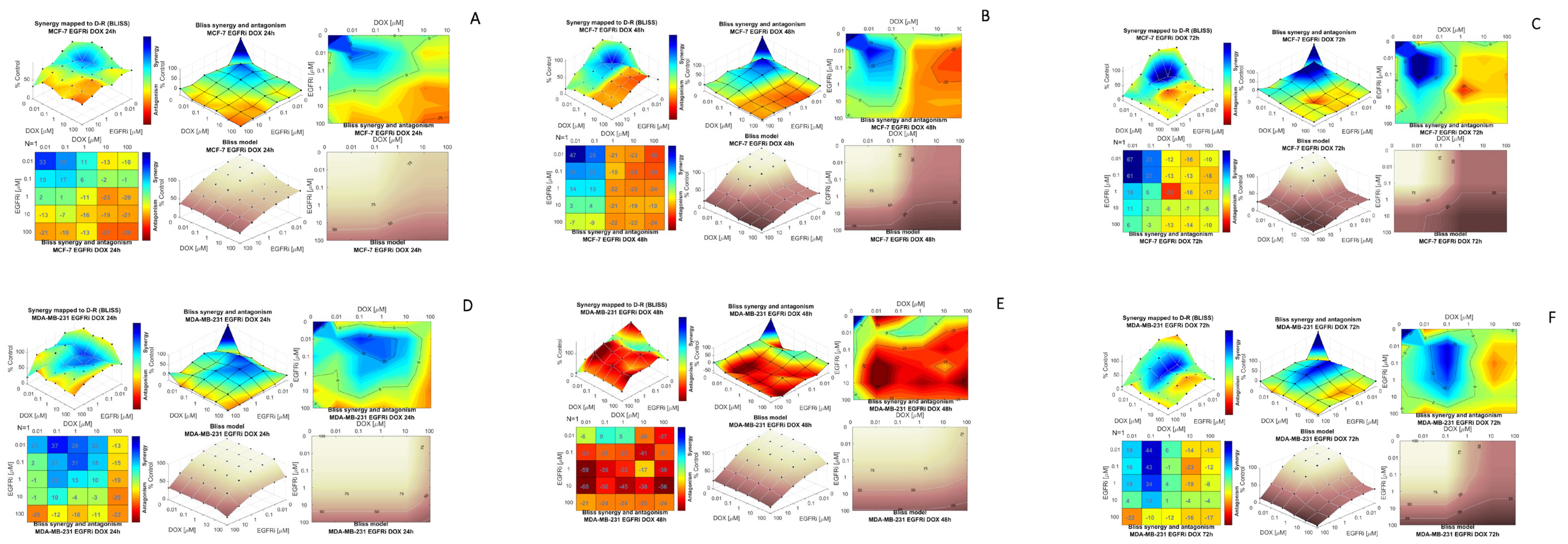
2.2. Combination Treatment of EGFRi and Dox Leads to Synergistic Growth Inhibition in MCF-7 and MDA-MB-231 Cells
2.3. Modeling Synergy Distribution of EGFRi and Dox Combination Treatment in MCF-7 and MDA-MB 231 Cells
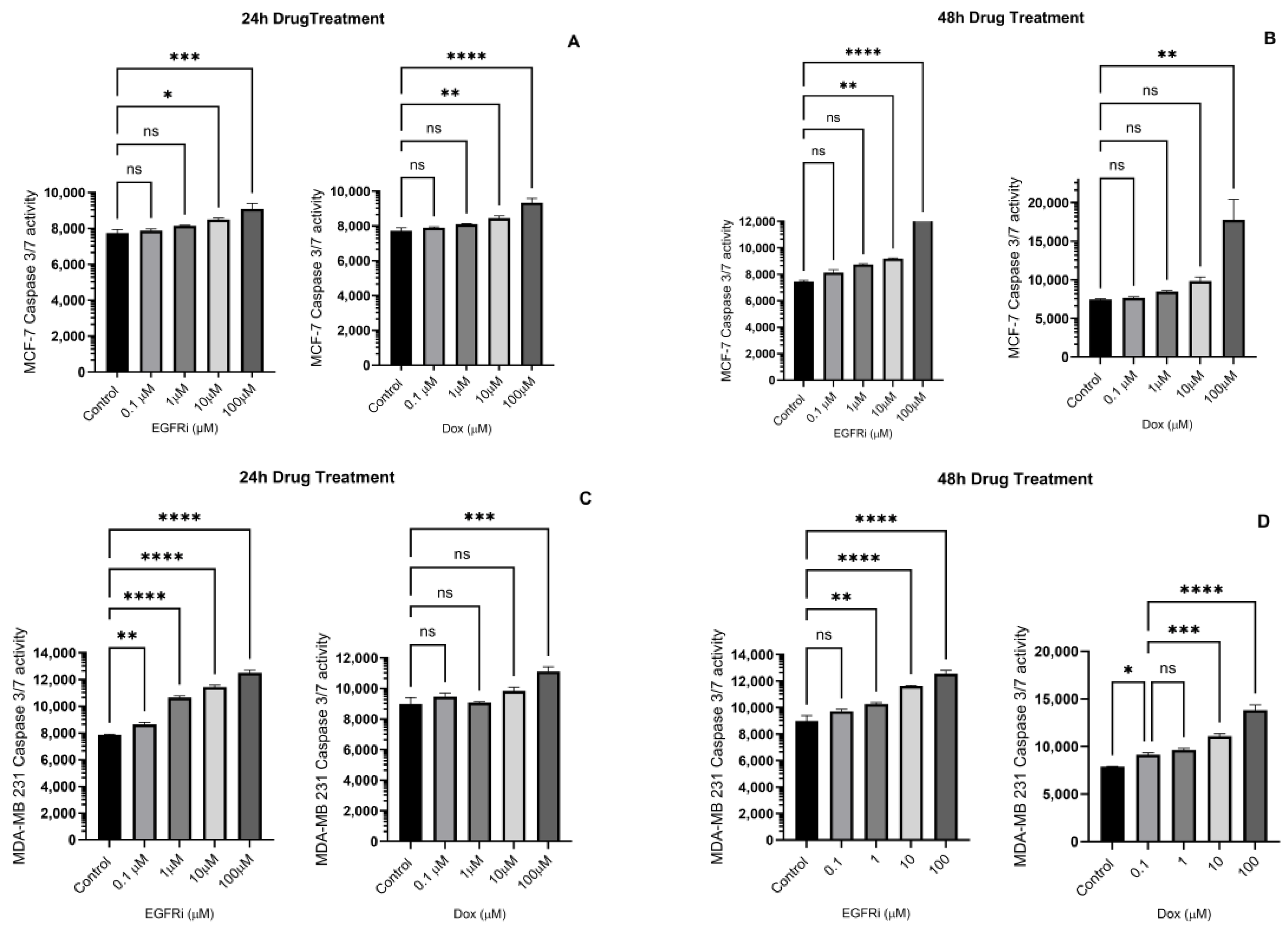
2.4. Caspase-3/7 Induction in MCF-7 and MDA-MB-231 Cells Following EGFRi and Dox Treatment
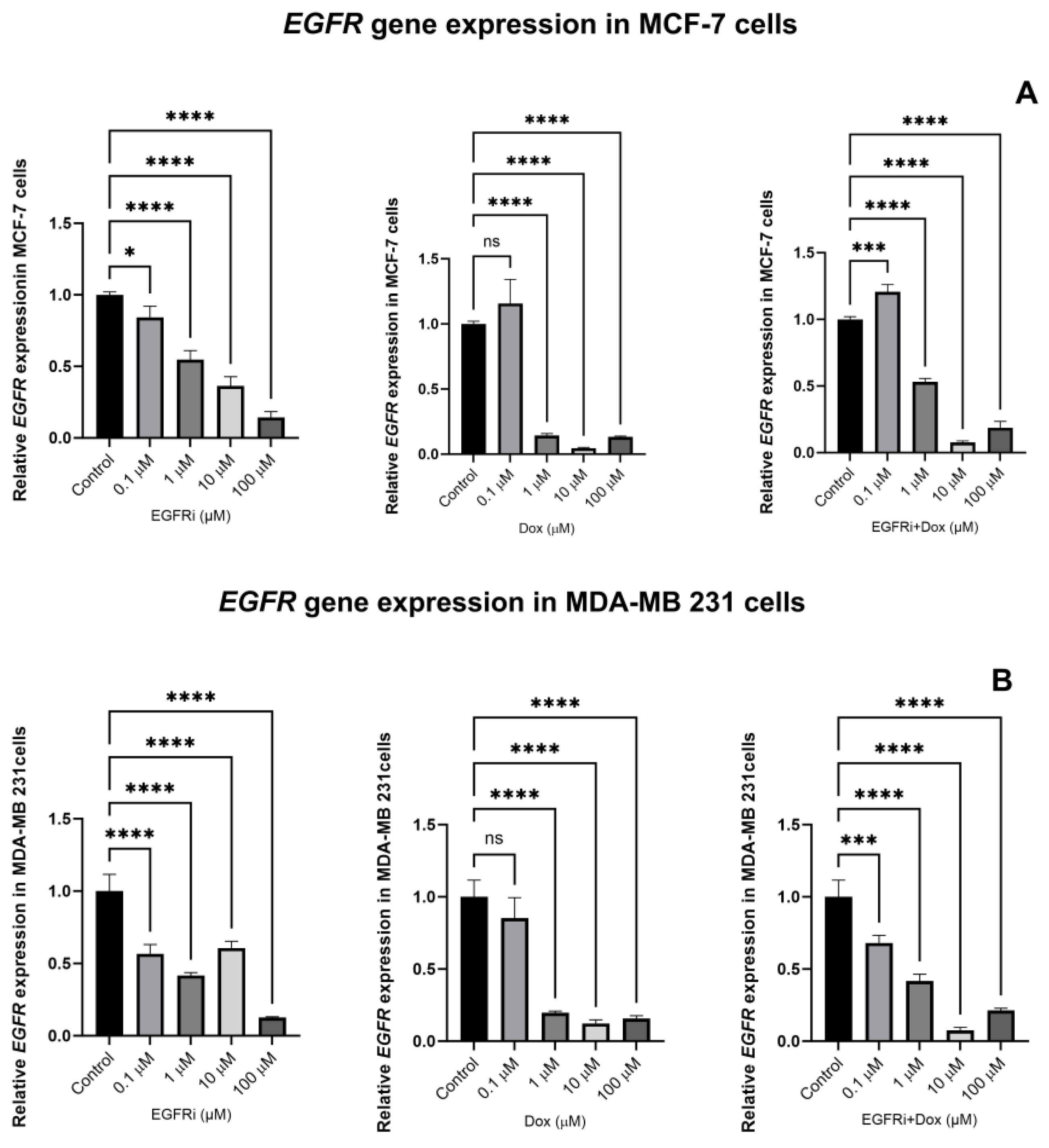
2.5. EGFRi and Dox Synergistically Downregulate the Expression of EGFR Gene
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Culture Conditions
4.2. Drug Preparation
4.3. Cytotoxicity Assay
4.4. Caspase-3/7 Assay
4.5. RT-qPCR–EGFR Gene Expression Analysis
4.6. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Wang, Y.; Kiani, M.F.; Wang, B. Classification, Treatment Strategy, and Associated Drug Resistance in Breast Cancer. Clin. Breast Cancer 2016, 16, 335–343. [Google Scholar] [CrossRef] [PubMed]
- Harbeck, N.; Gnant, M. Breast cancer. Lancet 2017, 389, 1134–1150. [Google Scholar] [CrossRef]
- Jeon, J.; Kim, S.E.; Lee, D.Y.; Choi, D. Factors associated with endometrial pathology during tamoxifen therapy in women with breast cancer: A retrospective analysis of 821 biopsies. Breast Cancer Res. Treat. 2020, 179, 125–130. [Google Scholar] [CrossRef]
- Qian, X.; Li, Z.; Ruan, G.; Tu, C.; Ding, W. Efficacy and toxicity of extended aromatase inhibitors after adjuvant aromatase inhibitors-containing therapy for hormone-receptor-positive breast cancer: A literature-based meta-analysis of randomized trials. Breast Cancer Res. Treat. 2020, 179, 275–285. [Google Scholar] [CrossRef]
- Rimawi, M.; Ferrero, J.M.; de la Haba-Rodriguez, J.; Poole, C.; De Placido, S.; Osborne, C.K.; Hegg, R.; Easton, V.; Wohlfarth, C.; Arpino, G.; et al. First-Line Trastuzumab Plus an Aromatase Inhibitor, with or without Pertuzumab, in Human Epidermal Growth Factor Receptor 2-Positive and Hormone Receptor-Positive Metastatic or Locally Advanced Breast Cancer (PERTAIN): A Randomized, Open-Label Phase II Trial. J. Clin. Oncol. 2018, 36, 2826–2835. [Google Scholar] [PubMed]
- Li, Y.; Yang, D.; Yin, X.; Zhang, X.; Huang, J.; Wu, Y.; Wang, M.; Yi, Z.; Li, H.; Li, H.; et al. Clinicopathological Characteristics and Breast Cancer–Specific Survival of Patients With Single Hormone Receptor–Positive Breast Cancer. JAMA Netw. Open 2020, 3, e1918160. [Google Scholar] [CrossRef]
- Waks, A.G.; Winer, E.P. Breast Cancer Treatment: A Review. JAMA 2019, 321, 288–300. [Google Scholar] [CrossRef]
- Harbeck, N.; Penault-Llorca, F.; Cortes, J.; Gnant, M.; Houssami, N.; Poortmans, P.; Ruddy, K.; Tsang, J.; Cardoso, F. Breast cancer. Nat. Rev. Dis. Primers 2019, 5, 66. [Google Scholar] [CrossRef]
- Garrido-Castro, A.C.; Lin, N.U.; Polyak, K. Insights into Molecular Classifications of Triple-Negative Breast Cancer: Improving Patient Selection for Treatment. Cancer Discov. 2019, 9, 176–198. [Google Scholar] [CrossRef]
- Harbeck, N.; Gluz, O. Neoadjuvant therapy for triple negative and HER2-positive early breast cancer. Breast 2017, 34 (Suppl. S1), S99–S103. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Mu, Q.; Fung, M.; Xu, X.; Zhu, L.; Ho, R.J.Y. Challenges and opportunities in metastatic breast cancer treatments: Nano-drug combinations delivered preferentially to metastatic cells may enhance therapeutic response. Pharmacol. Ther. 2022, 236, 108108. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.F. Structure, function and regulation of P-glycoprotein and its clinical relevance in drug disposition. Xenobiotica 2008, 38, 802–832. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.; DeSantis, C.; Virgo, K.; Stein, K.; Mariotto, A.; Smith, T.; Cooper, D.; Gansler, T.; Lerro, C.; Fedewa, S.; et al. Cancer treatment and survivorship statistics, 2012. CA Cancer J. Clin. 2012, 62, 220–241. [Google Scholar] [CrossRef] [PubMed]
- Jacobi, N.; Seeboeck, R.; Hofmann, E.; Eger, A. ErbB Family Signalling: A Paradigm for Oncogene Addiction and Personalized Oncology. Cancers 2017, 9, 33. [Google Scholar] [CrossRef]
- Wee, P.; Wang, Z. Epidermal Growth Factor Receptor Cell Proliferation Signaling Pathways. Cancers 2017, 9, 52. [Google Scholar] [CrossRef]
- Appert-Collin, A.; Hubert, P.; Cremel, G.; Bennasroune, A. Role of ErbB Receptors in Cancer Cell Migration and Invasion. Front. Pharmacol. 2015, 6, 283. [Google Scholar] [CrossRef]
- Roskoski, R., Jr. Properties of FDA-approved small molecule protein kinase inhibitors. Pharmacol. Res. 2019, 144, 19–50. [Google Scholar] [CrossRef]
- Li, X.; Zhao, L.; Chen, C.; Nie, J.; Jiao, B. Can EGFR be a therapeutic target in breast cancer? Biochim. Et Biophys. Acta (BBA)-Rev. Cancer 2022, 1877, 188789. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Wu, J.; Ling, R.; Li, N. Quadruple negative breast cancer. Breast Cancer 2020, 27, 527–533. [Google Scholar] [CrossRef] [PubMed]
- Chow, L.W.C.; Lie, E.F.; Toi, M. Chapter Ten—Advances in EGFR/HER2-directed clinical research on breast cancer. In Advances in Cancer Research; Kumar, R., Fisher, P.B., Eds.; Academic Press: Cambridge, MA, USA, 2020; Volume 147, pp. 375–428. [Google Scholar]
- Sabbah, D.A.; Hajjo, R.; Sweidan, K. Review on Epidermal Growth Factor Receptor (EGFR) Structure, Signaling Pathways, Interactions, and Recent Updates of EGFR Inhibitors. Curr. Top. Med. Chem. 2020, 20, 815–834. [Google Scholar] [CrossRef] [PubMed]
- Bhullar, K.S.; Lagaron, N.O.; McGowan, E.M.; Parmar, I.; Jha, A.; Hubbard, B.P.; Rupasinghe, H.P.V. Kinase-targeted cancer therapies: Progress, challenges and future directions. Mol. Cancer 2018, 17, 48. [Google Scholar] [CrossRef] [PubMed]
- Madden, R.; Kosari, S.; Peterson, G.M.; Bagheri, N.; Thomas, J. Lapatinib plus capecitabine in patients with HER2-positive metastatic breast cancer: A systematic review. Int. J. Clin. Pharmacol. Ther. 2018, 56, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Kulukian, A.; Lee, P.; Taylor, J.; Rosler, R.; de Vries, P.; Watson, D.; Forero-Torres, A.; Peterson, S. Preclinical Activity of HER2-Selective Tyrosine Kinase Inhibitor Tucatinib as a Single Agent or in Combination with Trastuzumab or Docetaxel in Solid Tumor Models. Mol. Cancer Ther. 2020, 19, 976–987. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Yan, M.; Hu, X.; Zhang, Q.; Ouyang, Q.; Feng, J.; Yin, Y.; Sun, T.; Tong, Z.; Wang, X.; et al. Pyrotinib combined with capecitabine in women with HER2+ metastatic breast cancer previously treated with trastuzumab and taxanes: A randomized phase III study. J. Clin. Oncol. 2019, 37, 1001. [Google Scholar] [CrossRef]
- Roskoski, R., Jr. Properties of FDA-approved small molecule protein kinase inhibitors: A 2022 update. Pharmacol. Res. 2022, 175, 106037. [Google Scholar] [CrossRef]
- Roskoski, R., Jr. Properties of FDA-approved small molecule protein kinase inhibitors: A 2023 update. Pharmacol. Res. 2023, 187, 106552. [Google Scholar] [CrossRef]
- Tan, Y.Q.; Chiou, Y.-S.; Guo, H.; Zhang, S.; Huang, X.; Dukanya, D.; Kumar, A.M.; Basappa, S.; Liu, S.; Zhu, T.; et al. Vertical pathway inhibition of receptor tyrosine kinases and BAD with synergistic efficacy in triple negative breast cancer. npj Precis. Oncol. 2024, 8, 8. [Google Scholar] [CrossRef]
- Morton, S.W.; Lee, M.J.; Deng, Z.J.; Dreaden, E.C.; Siouve, E.; Shopsowitz, K.E.; Shah, N.J.; Yaffe, M.B.; Hammond, P.T. A Nanoparticle-Based Combination Chemotherapy Delivery System for Enhanced Tumor Killing by Dynamic Rewiring of Signaling Pathways. Sci. Signal. 2014, 7, ra44. [Google Scholar] [CrossRef]
- Lin, S.; Zhang, X.; Yu, Z.; Huang, X.; Xu, J.; Liu, Y.; Wu, L. Synthesis of novel dual target inhibitors of PARP and EGFR and their antitumor activities in triple negative breast cancers. Biorg. Med. Chem. 2022, 61, 116739. [Google Scholar] [CrossRef] [PubMed]
- Lev, S. Targeted therapy and drug resistance in triple-negative breast cancer: The EGFR axis. Biochem. Soc. Trans. 2020, 48, 657–665. [Google Scholar] [CrossRef]
- Vagia, E.; Mahalingam, D.; Cristofanilli, M. The Landscape of Targeted Therapies in TNBC. Cancers 2020, 12, 916. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Liu, Y.; Gao, F.; Ding, Q.; Cho, C.; Hur, W.; Jin, Y.; Uno, T.; Joazeiro, C.A.P.; Gray, N. Discovery of EGFR Selective 4,6-Disubstituted Pyrimidines from a Combinatorial Kinase-Directed Heterocycle Library. J. Am. Chem. Soc. 2006, 128, 2182–2183. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.J.; Ye, A.S.; Gardino, A.K.; Heijink, A.M.; Sorger, P.K.; MacBeath, G.; Yaffe, M.B. Sequential application of anticancer drugs enhances cell death by rewiring apoptotic signaling networks. Cell 2012, 149, 780–794. [Google Scholar] [CrossRef] [PubMed]
- Di Veroli, G.Y.; Fornari, C.; Wang, D.; Mollard, S.; Bramhall, J.L.; Richards, F.M.; Jodrell, D.I. Combenefit: An interactive platform for the analysis and visualization of drug combinations. Bioinformatics 2016, 32, 2866–2868. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Yin, X.; Languino, L.R.; Altieri, D.C. Evaluation of Drug Combination Effect Using a Bliss Independence Dose–Response Surface Model. Stat. Biopharm. Res. 2018, 10, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Chun, S.Y.; Kwon, Y.S.; Nam, K.S.; Kim, S. Lapatinib enhances the cytotoxic effects of doxorubicin in MCF-7 tumorspheres by inhibiting the drug efflux function of ABC transporters. Biomed. Pharmacother. 2015, 72, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Rivankar, S. An overview of doxorubicin formulations in cancer therapy. J. Cancer Res. Ther. 2014, 10, 853–858. [Google Scholar] [CrossRef] [PubMed]
- Pottier, C.; Fresnais, M.; Gilon, M.; Jérusalem, G.; Longuespée, R.; Sounni, N.E. Tyrosine Kinase Inhibitors in Cancer: Breakthrough and Challenges of Targeted Therapy. Cancers 2020, 12, 731. [Google Scholar] [CrossRef]
- Meredith, A.M.; Dass, C.R. Increasing role of the cancer chemotherapeutic doxorubicin in cellular metabolism. J. Pharm. Pharmacol. 2016, 68, 729–741. [Google Scholar] [CrossRef]
- Devarajan, E.; Sahin, A.A.; Chen, J.S.; Krishnamurthy, R.R.; Aggarwal, N.; Brun, A.M.; Sapino, A.; Zhang, F.; Sharma, D.; Yang, X.H.; et al. Down-regulation of caspase 3 in breast cancer: A possible mechanism for chemoresistance. Oncogene 2002, 21, 8843–8851. [Google Scholar] [CrossRef]
- Azim, H.A.; Ghosn, M.; Oualla, K.; Kassem, L. Personalized treatment in metastatic triple-negative breast cancer: The outlook in 2020. Breast J. 2020, 26, 69–80. [Google Scholar] [CrossRef]
- Cerma, K.; Piacentini, F.; Moscetti, L.; Barbolini, M.; Canino, F.; Tornincasa, A.; Caggia, F.; Cerri, S.; Molinaro, A.; Dominici, M.; et al. Targeting PI3K/AKT/mTOR Pathway in Breast Cancer: From Biology to Clinical Challenges. Biomedicines 2023, 11, 109. [Google Scholar] [CrossRef]
- Lehmann, B.D.; Bauer, J.A.; Chen, X.; Sanders, M.E.; Chakravarthy, A.B.; Shyr, Y.; Pietenpol, J.A. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J. Clin. Investig. 2011, 121, 2750–2767. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, R.I.; Gee, J.M.; Harper, M.E. EGFR and cancer prognosis. Eur. J. Cancer 2001, 37 (Suppl. S4), S9–S15. [Google Scholar] [CrossRef] [PubMed]
- De Luca, A.; Carotenuto, A.; Rachiglio, A.; Gallo, M.; Maiello, M.R.; Aldinucci, D.; Pinto, A.; Normanno, N. The role of the EGFR signaling in tumor microenvironment. J. Cell Physiol. 2008, 214, 559–567. [Google Scholar] [CrossRef] [PubMed]
- Weihua, Z.; Tsan, R.; Huang, W.C.; Wu, Q.; Chiu, C.H.; Fidler, I.J.; Hung, M.C. Survival of cancer cells is maintained by EGFR independent of its kinase activity. Cancer Cell 2008, 13, 385–393. [Google Scholar] [CrossRef]
- Mitsudomi, T.; Yatabe, Y. Epidermal growth factor receptor in relation to tumor development: EGFR gene and cancer. FEBS J. 2010, 277, 301–308. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.J.; Luan, J.; Zhang, H.S.; Ruan, C.P.; Xu, X.Y.; Li, Q.Q.; Wang, N.H. EGFR-mediated G1/S transition contributes to the multidrug resistance in breast cancer cells. Mol. Biol. Rep. 2012, 39, 5465–5471. [Google Scholar] [CrossRef] [PubMed]
- Cohen, R.B. Current challenges and clinical investigations of epidermal growth factor receptor (EGFR)- and ErbB family-targeted agents in the treatment of head and neck squamous cell carcinoma (HNSCC). Cancer Treat. Rev. 2014, 40, 567–577. [Google Scholar] [CrossRef]
- Singh, A.; Najmi, A.K.; Mishra, P.; Talikoti, M. Abstract 671: Prognostic importance of EGFR expression and Kras gene mutations in gallbladder carcinoma. Cancer Res. 2017, 77 (Suppl. S13), 671. [Google Scholar] [CrossRef]
- Xu, M.J.; Johnson, D.E.; Grandis, J.R. EGFR-targeted therapies in the post-genomic era. Cancer Metastasis Rev. 2017, 36, 463–473. [Google Scholar] [CrossRef] [PubMed]
- Cappetta, D.; Rossi, F.; Piegari, E.; Quaini, F.; Berrino, L.; Urbanek, K.; De Angelis, A. Doxorubicin targets multiple players: A new view of an old problem. Pharmacol. Res. 2018, 127, 4–14. [Google Scholar] [CrossRef] [PubMed]
- Park, H.-K.; Lee, J.-E.; Lim, J.; Jo, D.-E.; Park, S.-A.; Suh, P.-G.; Kang, B.H. Combination treatment with doxorubicin and gamitrinib synergistically augments anticancer activity through enhanced activation of Bim. BMC Cancer 2014, 14, 431. [Google Scholar] [CrossRef] [PubMed]
- Fleisher, B.; Lezeau, J.; Werkman, C.; Jacobs, B.; Ait-Oudhia, S. In vitro to Clinical Translation of Combinatorial Effects of Doxorubicin and Abemaciclib in Rb-Positive Triple Negative Breast Cancer: A Systems-Based Pharmacokinetic/Pharmacodynamic Modeling Approach. Breast Cancer 2021, 13, 87–105. [Google Scholar] [CrossRef]
- Khandelwal Gilman, K.A.; Han, S.; Won, Y.-W.; Putnam, C.W. Complex interactions of lovastatin with 10 chemotherapeutic drugs: A rigorous evaluation of synergism and antagonism. BMC Cancer 2021, 21, 356. [Google Scholar] [CrossRef]
- Tap, W.D.; Wagner, A.J.; Schöffski, P.; Martin-Broto, J.; Krarup-Hansen, A.; Ganjoo, K.N.; Yen, C.-C.; Abdul Razak, A.R.; Spira, A.; Kawai, A.; et al. Effect of Doxorubicin Plus Olaratumab vs Doxorubicin Plus Placebo on Survival in Patients With Advanced Soft Tissue Sarcomas: The ANNOUNCE Randomized Clinical Trial. JAMA 2020, 323, 1266–1276. [Google Scholar] [CrossRef]
- Huang, Y.; Jiang, D.; Sui, M.; Wang, X.; Fan, W. Fulvestrant reverses doxorubicin resistance in multidrug-resistant breast cell lines independent of estrogen receptor expression. Oncol. Rep. 2017, 37, 705–712. [Google Scholar] [CrossRef]
- Zeng, C.; Fan, D.; Xu, Y.; Li, X.; Yuan, J.; Yang, Q.; Zhou, X.; Lu, J.; Zhang, C.; Han, J.; et al. Curcumol enhances the sensitivity of doxorubicin in triple-negative breast cancer via regulating the miR-181b-2-3p-ABCC3 axis. Biochem. Pharmacol. 2020, 174, 113795. [Google Scholar] [CrossRef]
- Liu, H.C.; Chiang, C.C.; Lin, C.H.; Chen, C.S.; Wei, C.W.; Lin, S.Y.; Yiang, G.T.; Yu, Y.L. Anti-cancer therapeutic benefit of red guava extracts as a potential therapy in combination with doxorubicin or targeted therapy for triple-negative breast cancer cells. Int. J. Med. Sci. 2020, 17, 1015–1022. [Google Scholar] [CrossRef]
- Fan, Y.; Ma, K.; Jing, J.; Wang, C.; Hu, Y.; Shi, Y.; Li, E.; Geng, Q. Recombinant Dual-target MDM2/MDMX Inhibitor Reverses Doxorubicin Resistance through Activation of the TAB1/TAK1/p38 MAPK Pathway in Wild-type p53 Multidrug-resistant Breast Cancer Cells. J. Cancer 2020, 11, 25–40. [Google Scholar] [CrossRef]
- Twarog, N.R.; Connelly, M.; Shelat, A.A. A critical evaluation of methods to interpret drug combinations. Sci. Rep. 2020, 10, 5144. [Google Scholar] [CrossRef] [PubMed]
- Masood, I.; Kiani, M.H.; Ahmad, M.; Masood, M.I.; Sadaquat, H. Major contributions towards finding a cure for cancer through chemotherapy: A historical review. Tumori 2016, 102, 6–17. [Google Scholar] [CrossRef]
- Sauter, E.R. Cancer prevention and treatment using combination therapy with natural compounds. Expert Rev. Clin. Pharmacol. 2020, 13, 265–285. [Google Scholar] [CrossRef] [PubMed]
- Geldof, T.; Rawal, S.; Dyck, W.V.; Huys, I. Comparative and combined effectiveness of innovative therapies in cancer: A literature review. J. Comp. Eff. Res. 2019, 8, 205–216. [Google Scholar] [CrossRef] [PubMed]
- Nair, P.R. Delivering Combination Chemotherapies and Targeting Oncogenic Pathways via Polymeric Drug Delivery Systems. Polymer 2019, 11, 630. [Google Scholar] [CrossRef] [PubMed]
- van Hasselt, J.G.C.; Iyengar, R. Systems Pharmacology: Defining the Interactions of Drug Combinations. Annu. Rev. Pharmacol. Toxicol. 2019, 59, 21–40. [Google Scholar] [CrossRef] [PubMed]
- Dry, J.R.; Yang, M.; Saez-Rodriguez, J. Looking beyond the cancer cell for effective drug combinations. Genome Med. 2016, 8, 125. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.-L.; Zhao, H.; Ma, T.-F.; Ge, F.; Chen, C.-S.; Zhang, Y.-P. Identification of Valid Reference Genes for the Normalization of RT-qPCR Expression Studies in Human Breast Cancer Cell Lines Treated with and without Transient Transfection. PLoS ONE 2015, 10, e0117058. [Google Scholar] [CrossRef]
- Jin, W.; Chen, B.B.; Li, J.Y.; Zhu, H.; Huang, M.; Gu, S.M.; Wang, Q.Q.; Chen, J.Y.; Yu, S.; Wu, J.; et al. TIEG1 inhibits breast cancer invasion and metastasis by inhibition of epidermal growth factor receptor (EGFR) transcription and the EGFR signaling pathway. Mol. Cell Biol. 2012, 32, 50–63. [Google Scholar] [CrossRef]
- Fieller, E.C. A fundamental formula in the statistics of biological assay, and some applications. Quart. J. Pharm. Pharmacol. 1944, 17, 117–123. [Google Scholar]
- Bliss, C. Confidence limits for measuring the precision of bioassays. Biometrics 1956, 12, 491–526. [Google Scholar] [CrossRef]
- Loewe, S. The problem of synergism and antagonism of combined drugs. Arzneimittelforschung 1953, 3, 285–290. [Google Scholar] [PubMed]
- Loewe, S. Antagonism and antagonists. Pharmacol. Rev. 1957, 9, 237–242. [Google Scholar]
- Bliss, C. The toxicity of poisons applied jointly. Ann. Appl. Biol. 1939, 26, 585–615. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MCF-7: Potency Ratios of EGFRi and DOX in EGFRi + DOX Combination | Time (h) | PR |
| Potency Ratio of EGFRi in EGFRi + DOX* | 48 | 6.55 |
| 72 | 396 | |
| Potency Ratio of DOX in EGFRi + DOX | 48 | 9.77 |
| 72 | 140 | |
| MDA-MB 231: Potency Ratios of EGFRi and DOX in EGFRi + DOX Combination | Time (h) | PR |
| Potency Ratio of EGFRi in EGFRi + DOX* | 48 | 0.29 |
| 72 | 13.11 | |
| Potency Ratio of DOX in EGFRi + DOX | 48 | 4.16 |
| 72 | 21.02 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abrahams, B.; Gerber, A.; Hiss, D.C. Combination Treatment with EGFR Inhibitor and Doxorubicin Synergistically Inhibits Proliferation of MCF-7 Cells and MDA-MB-231 Triple-Negative Breast Cancer Cells In Vitro. Int. J. Mol. Sci. 2024, 25, 3066. https://doi.org/10.3390/ijms25053066
Abrahams B, Gerber A, Hiss DC. Combination Treatment with EGFR Inhibitor and Doxorubicin Synergistically Inhibits Proliferation of MCF-7 Cells and MDA-MB-231 Triple-Negative Breast Cancer Cells In Vitro. International Journal of Molecular Sciences. 2024; 25(5):3066. https://doi.org/10.3390/ijms25053066
Chicago/Turabian StyleAbrahams, Beynon, Anthonie Gerber, and Donavon Charles Hiss. 2024. "Combination Treatment with EGFR Inhibitor and Doxorubicin Synergistically Inhibits Proliferation of MCF-7 Cells and MDA-MB-231 Triple-Negative Breast Cancer Cells In Vitro" International Journal of Molecular Sciences 25, no. 5: 3066. https://doi.org/10.3390/ijms25053066
APA StyleAbrahams, B., Gerber, A., & Hiss, D. C. (2024). Combination Treatment with EGFR Inhibitor and Doxorubicin Synergistically Inhibits Proliferation of MCF-7 Cells and MDA-MB-231 Triple-Negative Breast Cancer Cells In Vitro. International Journal of Molecular Sciences, 25(5), 3066. https://doi.org/10.3390/ijms25053066