
Integrated Experimental and Mathematical Exploration of Modular Tissue Cultures for Developmental Engineering



Abstract
1. Introduction
2. Results and Discussions
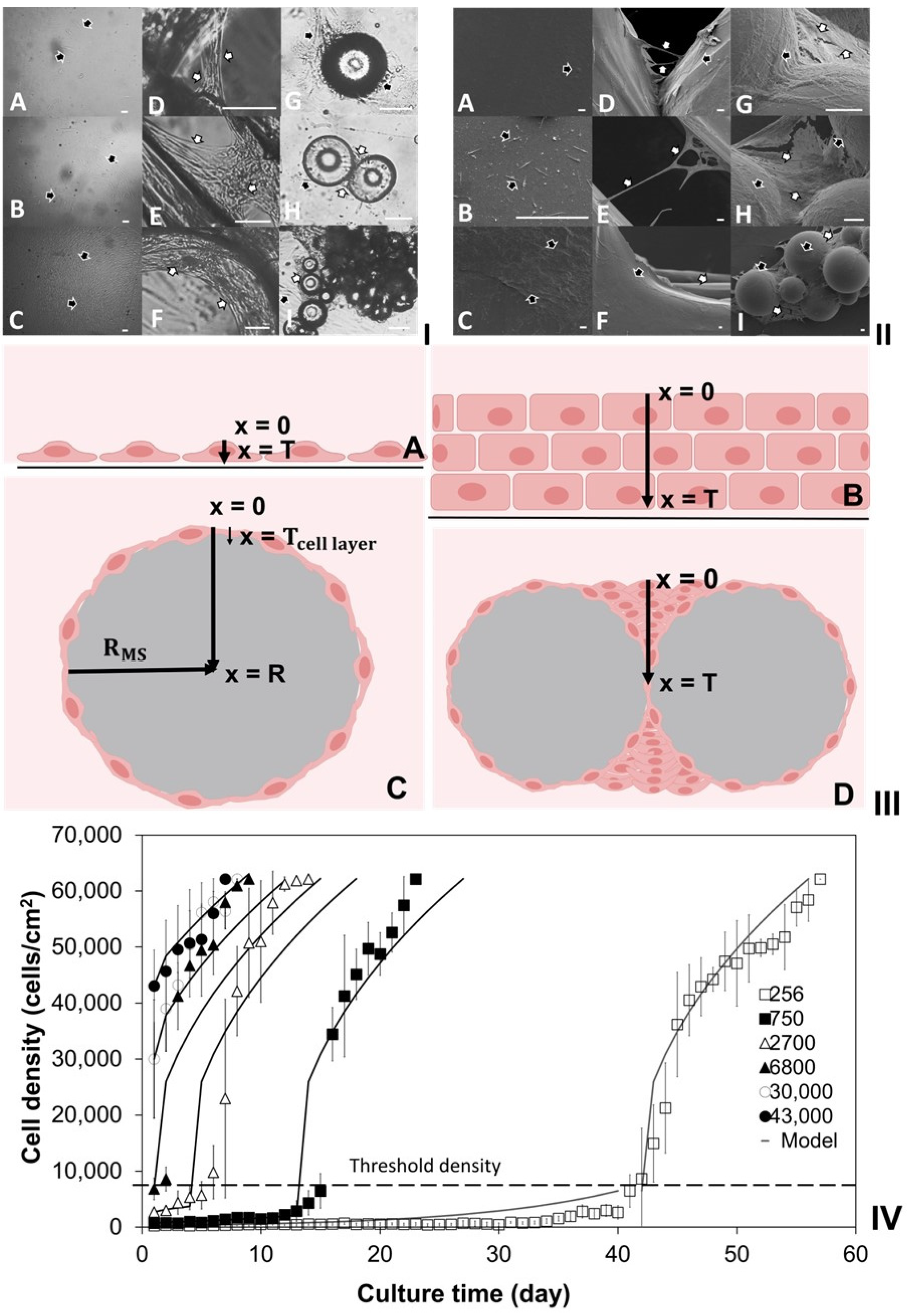
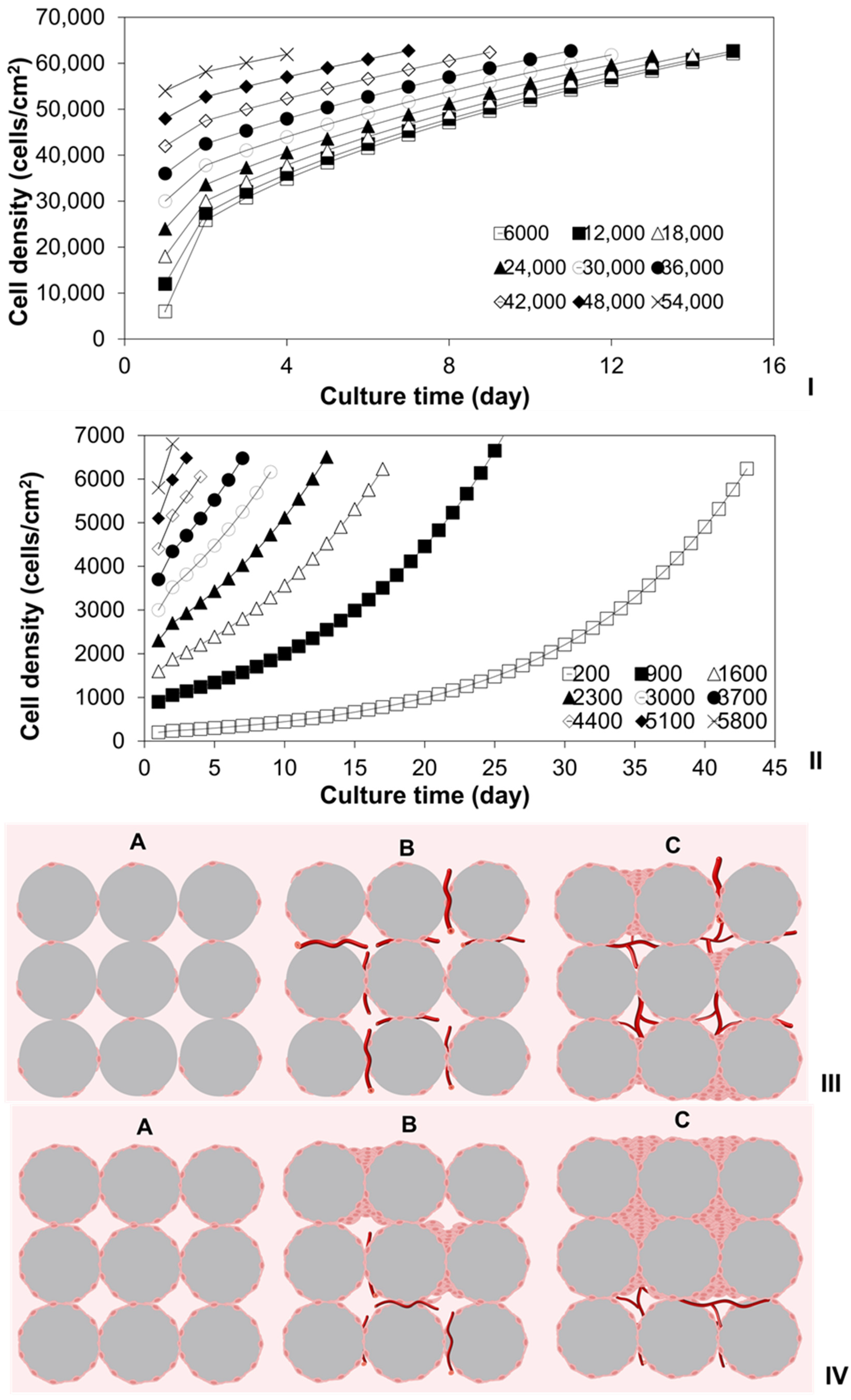
2.1. Investigation of Density-Dependent Cell Growths in 2D Cell Cultures
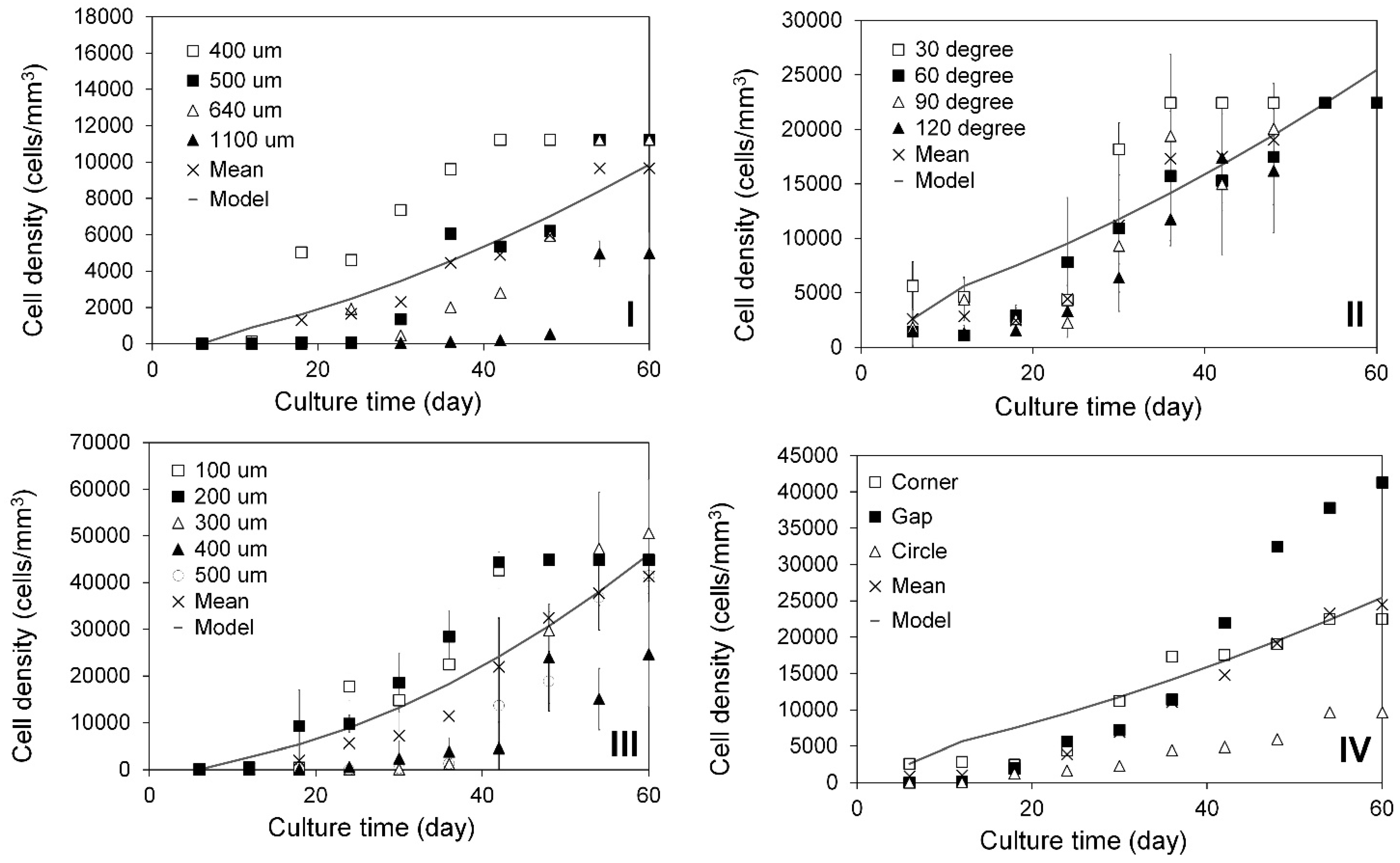
2.2. Investigation of Cell Cultures on Solid Surfaces of Modular Scaffolds
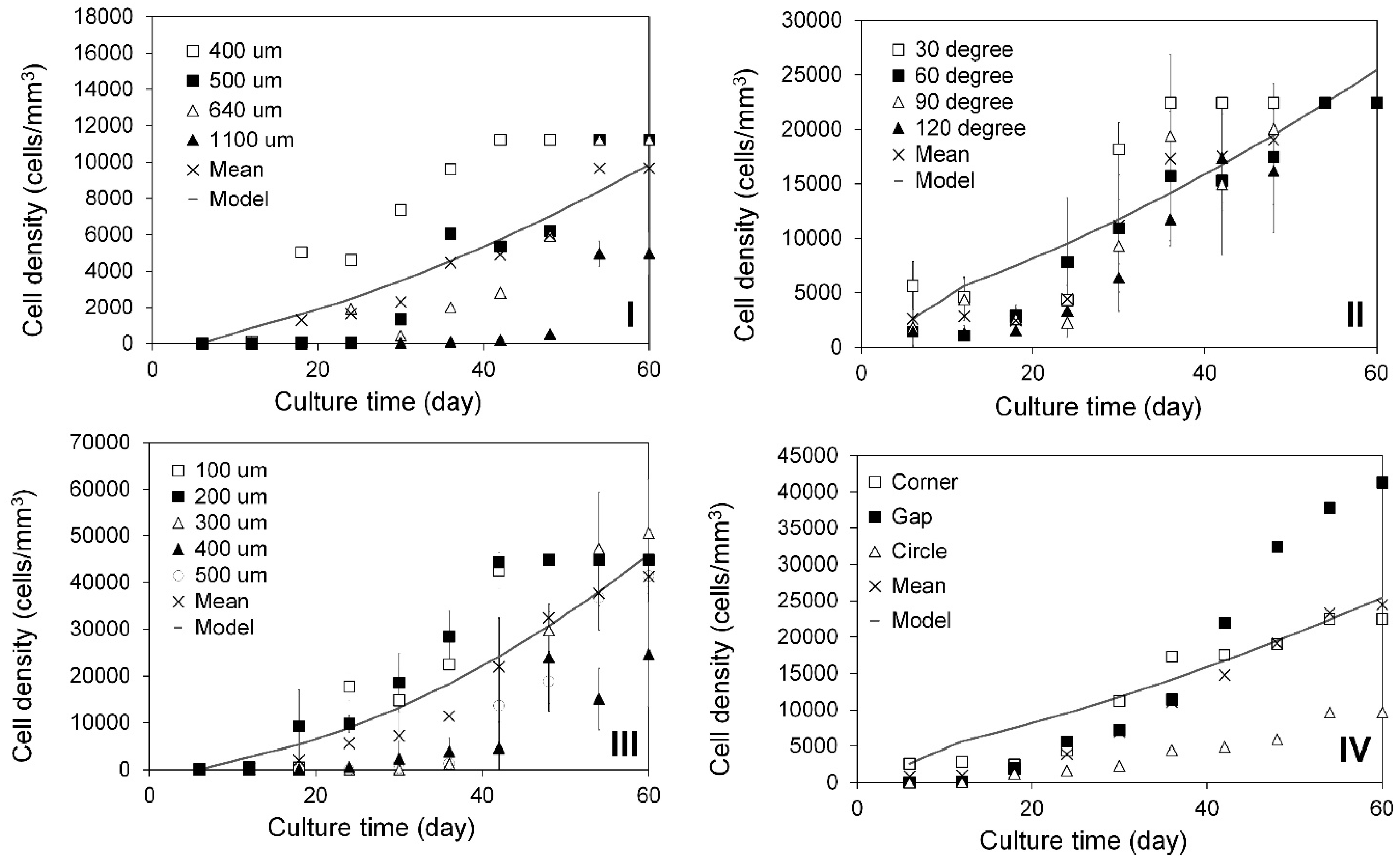
2.3. Modeling of Cell Colonization inside the Open Structures
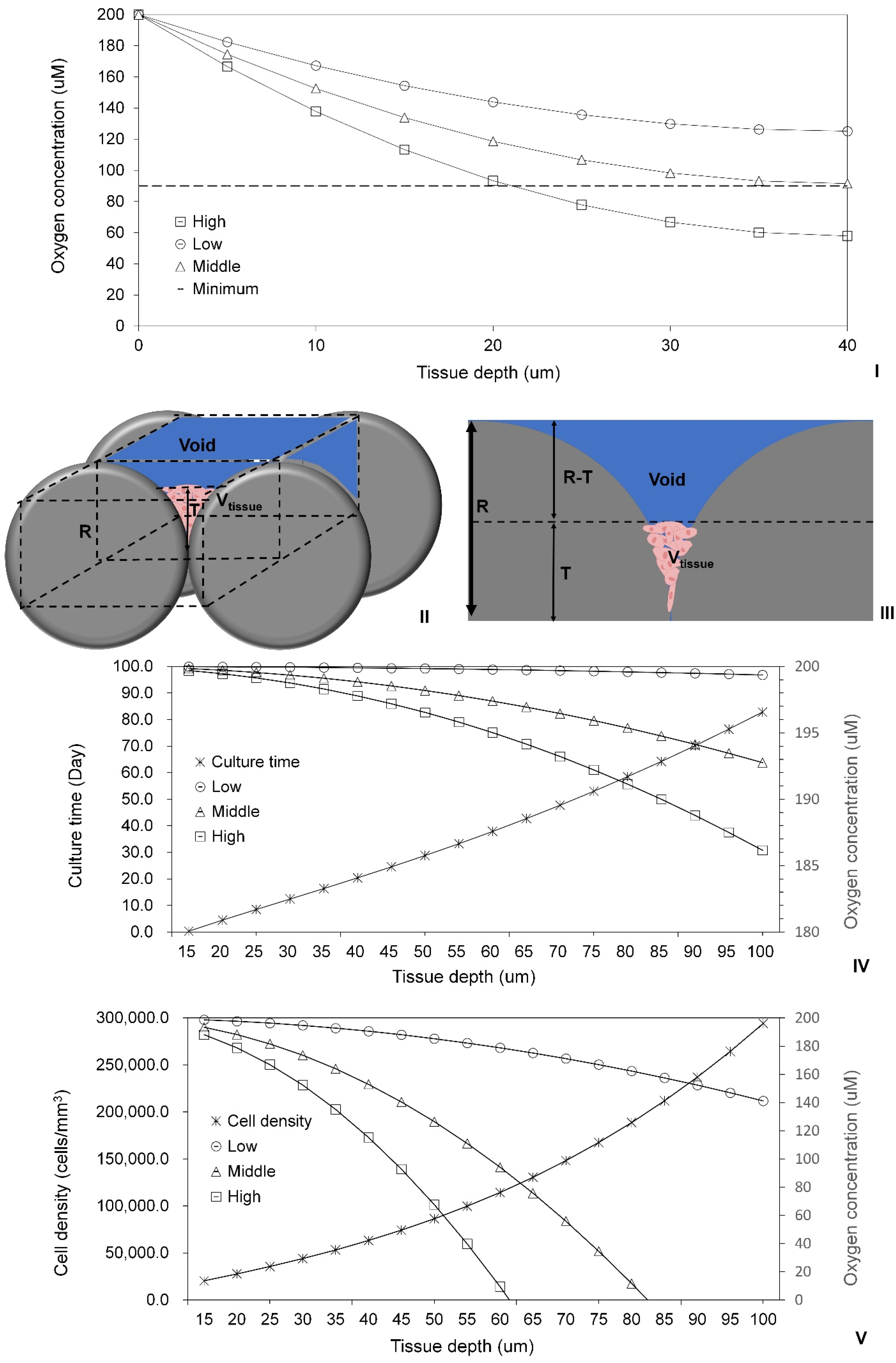
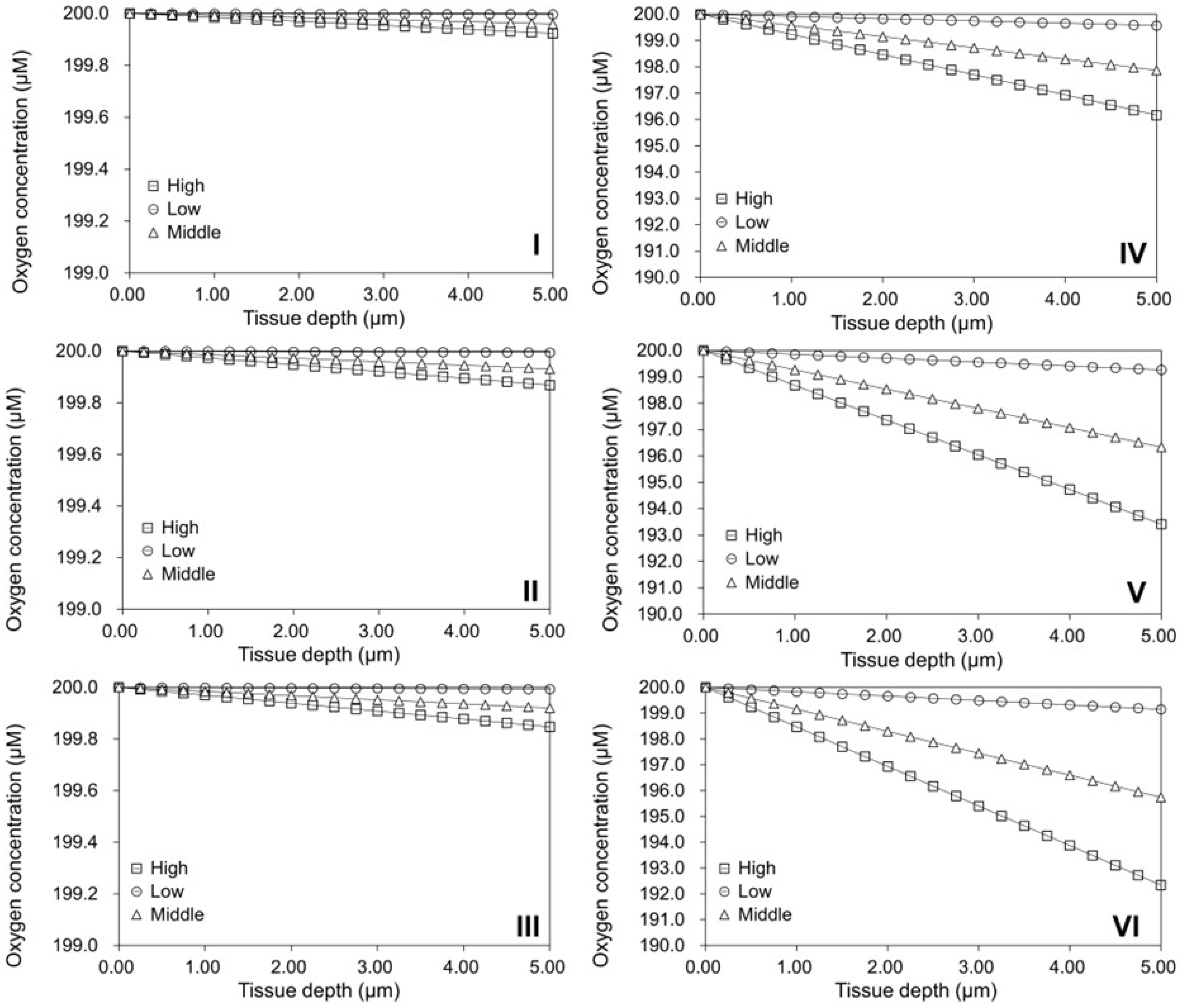
2.4. Investigation of Modular Tissue Cultures on Individual Spherical Modular Scaffolds
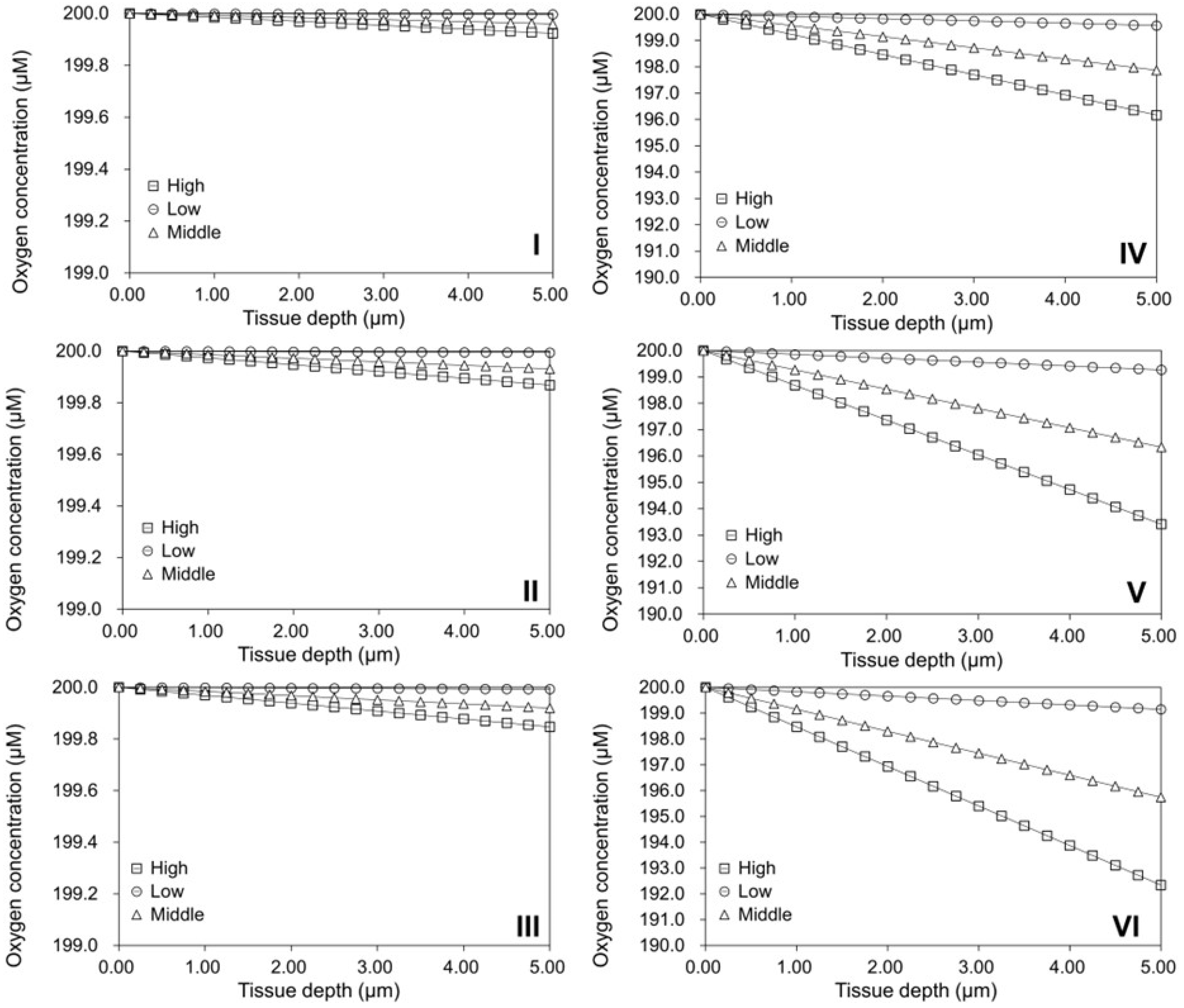
2.5. Validation of the Oxygen Diffusion Model Using Layered Tissues
2.6. Investigation of Tissue Assembly via the Bottom-Up Strategy
3. Materials and Methods
3.1. 2D Cell Culture
3.2. Modular Tissue Culture on Spherical Modular Scaffolds
3.3. Tissue Culture on PLA Discs with Finely Controlled Open Structures
3.4. Phase Contrast and Scanning Electron Microscopy
3.5. Development of 2D Cell Growth Models
3.6. Development of 3D Cell Growth Models
3.7. Oxygen Diffusion Model for Tissues Cultured on Flat TCP Surfaces
3.8. Oxygen Diffusion Model for Cells Cultured on Spherical Modular Scaffolds
3.9. Summary of The Models Used in This Research
- (1)
- 2D Cell Growth Models
- (i)
- Slow Cell Growth Model:
- (ii)
- Rapid Cell Growth Model:
where t is culture time, d is cell density, dl is the initial low cell seeding density, dh is the initial high cell seeding density, k is the cell growth constant, and n is order of cell growth. - (2)
- 3D Cell Growth Model:where t is culture time, d is cell density, dh is the cell numbers colonized in the open pores after the slow colonization phases, k is the cell growth constant, and n is order of cell growth.
- (3)
- Oxygen Diffusion Model for Tissues Cultured on Flat TCP Surfaces:where x is the location inside the simulated tissue, T is the maximum tissue thickness, φ is the oxygen consumption rate, D is the oxygen diffusion coefficient, is the oxygen concentration at the depth of () within the simulated tissue, and is the initial oxygen concentration at the tissue surface, i.e., as .
- (4)
- Oxygen Diffusion Model for Cells Cultured on Spherical Modular Scaffolds:where R is the radius of spherical modular scaffold (or the microcarrier) plus the thickness of the cultured tissue, x is the location inside the simulated tissue, φ is the oxygen consumption rate, D is the oxygen diffusion coefficient, is the oxygen concentration at the depth of () within the simulated tissue, and is the initial oxygen concentration at the tissue surface, i.e., as .
3.10. Statistical Analysis
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Langer, R.; Vacanti, J.P. Tissue Engineering. Science 1993, 260, 920–926. [Google Scholar] [CrossRef]
- Chan, B.P.; Leong, K.W. Scaffolding in Tissue Engineering: General Approaches and Tissue-Specific Considerations. Eur. Spine J. 2008, 17, 467–479. [Google Scholar] [CrossRef]
- Lu, T.; Li, Y.; Chen, T. Techniques for Fabrication and Construction of Three-Dimensional Scaffolds for Tissue Engineering. Int. J. Nanomed. 2013, 8, 337–350. [Google Scholar] [CrossRef]
- Caddeo, S.; Boffito, M.; Sartori, S. Tissue Engineering Approaches in the Design of Healthy and Pathological In Vitro Tissue Models. Front. Bioeng. Biotechnol. 2017, 5, 40. [Google Scholar] [CrossRef]
- Das, S.; Gordián-Vélez, W.J.; Ledebur, H.C.; Mourkioti, F.; Rompolas, P.; Chen, H.I.; Serruya, M.D.; Cullen, D.K. Innervation: The Missing Link for Biofabricated Tissues and Organs. NPJ Regen. Med. 2020, 5, 11. [Google Scholar] [CrossRef] [PubMed]
- Tiruvannamalai-Annamalai, R.; Armant, D.R.; Matthew, H.W.T. A Glycosaminoglycan Based, Modular Tissue Scaffold System for Rapid Assembly of Perfusable, High Cell Density, Engineered Tissues. PLoS ONE 2014, 9, e84287. [Google Scholar] [CrossRef] [PubMed]
- Sarker, M.D.; Naghieh, S.; Sharma, N.K.; Chen, X. 3D Biofabrication of Vascular Networks for Tissue Regeneration: A Report on Recent Advances. J. Pharm. Anal. 2018, 8, 277–296. [Google Scholar] [CrossRef] [PubMed]
- Tse, H.M.; Gardner, G.; Dominguez-Bendala, J.; Fraker, C.A. The Importance of Proper Oxygenation in 3D Culture. Front. Bioeng. Biotechnol. 2021, 9, 634403. [Google Scholar] [CrossRef] [PubMed]
- Place, T.L.; Domann, F.E.; Case, A.J. Limitations of Oxygen Delivery to Cells in Culture: An Underappreciated Problem in Basic and Translational Research. Free Radic. Biol. Med. 2017, 113, 311–322. [Google Scholar] [CrossRef] [PubMed]
- Heydari, Z.; Pooyan, P.; Bikmulina, P.; Pozdnyakov, A.; Fomin, V.; Seydi, H.; Shpichka, A.; Timashev, P.; Vosough, M. Mimicking the Liver Function in Micro-Patterned Units: Challenges and Perspectives in 3D Bioprinting. Bioprinting 2022, 27, e00208. [Google Scholar] [CrossRef]
- Zeenat, L.; Zolfagharian, A.; Sriya, Y.; Sasikumar, S.; Bodaghi, M.; Pati, F. 4D Printing for Vascular Tissue Engineering: Progress and Challenges. Adv. Mater. Technol. 2023, 8, 2300200. [Google Scholar] [CrossRef]
- Dellaquila, A.; Le Bao, C.; Letourneur, D.; Simon-Yarza, T. In Vitro Strategies to Vascularize 3D Physiologically Relevant Models. Adv. Sci. 2021, 8, 20100798. [Google Scholar] [CrossRef]
- Yang, G.; Mahadik, B.; Choi, J.Y.; Fisher, J.P. Vascularization in Tissue Engineering: Fundamentals and State-of-Art. Progress. Biomed. Eng. 2020, 2, 012002. [Google Scholar] [CrossRef] [PubMed]
- Gordan, R.; Gwathmey, J.K.; Xie, L.-H. Autonomic and Endocrine Control of Cardiovascular Function. World J. Cardiol. 2015, 7, 204. [Google Scholar] [CrossRef] [PubMed]
- Rivera, K.R.; Yokus, M.A.; Erb, P.D.; Pozdin, V.A.; Daniele, M. Measuring and Regulating Oxygen Levels in Microphysiological Systems: Design, Material, and Sensor Considerations. Analyst 2019, 144, 3190–3215. [Google Scholar] [CrossRef] [PubMed]
- Ramadan, Q.; Zourob, M. Organ-on-a-Chip Engineering: Toward Bridging the Gap between Lab and Industry. Biomicrofluidics 2020, 14, 041501. [Google Scholar] [CrossRef] [PubMed]
- Mekala, N.K.; Baadhe, R.R.; Potumarthi, R. Mass Transfer Aspects of 3D Cell Cultures in Tissue Engineering. Asia-Pac. J. Chem. Eng. 2014, 9, 318–329. [Google Scholar] [CrossRef]
- Sphabmixay, P.; Raredon, M.S.B.; Wang, A.J.S.; Lee, H.; Hammond, P.T.; Fang, N.X.; Griffith, L.G. High Resolution Stereolithography Fabrication of Perfusable Scaffolds to Enable Long-Term Meso-Scale Hepatic Culture for Disease Modeling. Biofabrication 2021, 13, 045024. [Google Scholar] [CrossRef] [PubMed]
- Eghbali, H.; Nava, M.M.; Mohebbi-Kalhori, D.; Raimondi, M.T. Hollow Fiber Bioreactor Technology for Tissue Engineering Applications. Int. J. Artif. Organs 2016, 39, 1–15. [Google Scholar] [CrossRef]
- Lenas, P.; Moos, M.; Luyten, F.P. Developmental Engineering: A New Paradigm for the Design and Manufacturing of Cell-Based Products. Part I: From Three-Dimensional Cell Growth to Biomimetics of In Vivo Development. Tissue Eng. Part B 2009, 15, 381–394. [Google Scholar] [CrossRef]
- Lenas, P.; Moos, M.; Luyten, F.P. Developmental Engineering: A New Paradigm for the Design and Manufacturing of Cell-Based Products. Part II. From Genes to Networks: Tissue Engineering from the Viewpoint of Systems Biology and Network Science. Tissue Eng. Part B 2009, 15, 395–422. [Google Scholar] [CrossRef] [PubMed]
- Lenas, P.; Ikonomou, L. Developmental Engineering: Design of Clinically Efficacious Bioartificial Tissues through Developmental and Systems Biology. Sci. China Life Sci. 2018, 61, 978–981. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, T.; Xiang, Y.; Bao, X.; Sun, T. A Paradigm Shift in Tissue Engineering: From a Top–down to a Bottom–up Strategy. Processes 2021, 9, 935. [Google Scholar] [CrossRef]
- Ouyang, L.; Armstrong, J.P.K.; Salmeron-Sanchez, M.; Stevens, M.M. Assembling Living Building Blocks to Engineer Complex Tissues. Adv. Funct. Mater. 2020, 30, 1909009. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, H.M.M.A.M.; Teixeira, L.S.M. New Endeavors of (Micro)Tissue Engineering: Cells Tissues Organs on-Chip and Communication Thereof. Cells Tissues Organs 2023, 211, 721–735. [Google Scholar] [CrossRef] [PubMed]
- Gaspar, V.M.; Lavrador, P.; Borges, J.; Oliveira, M.B.; Mano, J.F. Advanced Bottom-Up Engineering of Living Architectures. Adv. Mater. 2020, 32, 1903975. [Google Scholar] [CrossRef]
- Nichol, J.W.; Khademhosseini, A. Modular Tissue Engineering: Engineering Biological Tissues from the Bottom Up. Soft Matter 2009, 5, 1312–1319. [Google Scholar] [CrossRef]
- Rademakers, T.; Horvath, J.M.; van Blitterswijk, C.A.; LaPointe, V.L.S. Oxygen and Nutrient Delivery in Tissue Engineering: Approaches to Graft Vascularization. J. Tissue Eng. Regen. Med. 2019, 13, 1815–1829. [Google Scholar] [CrossRef]
- Cui, H.; Nowicki, M.; Fisher, J.P.; Zhang, L.G. 3D Bioprinting for Organ Regeneration. Adv. Healthc. Mater. 2017, 6, 1601118. [Google Scholar] [CrossRef]
- Moroni, L.; Burdick, J.A.; Highley, C.; Lee, S.J.; Morimoto, Y.; Takeuchi, S.; Yoo, J.J. Biofabrication Strategies for 3D In Vitro Models and Regenerative Medicine. Nat. Rev. Mater. 2018, 3, 21–37. [Google Scholar] [CrossRef]
- Loai, S.; Kingston, B.R.; Wang, Z.; Philpott, D.N.; Tao, M.; Cheng, H.L.M. Clinical Perspectives on 3D Bioprinting Paradigms for Regenerative Medicine. Regen. Med. Front. 2019, 1, e190004. [Google Scholar]
- Feng, Q.; Li, D.; Li, Q.; Cao, X.; Dong, H. Microgel Assembly: Fabrication, Characteristics and Application in Tissue Engineering and Regenerative Medicine. Bioact. Mater. 2022, 9, 105–119. [Google Scholar] [CrossRef] [PubMed]
- Van Duinen, V.; Van Den Heuvel, A.; Trietsch, S.J.; Lanz, H.L.; Van Gils, J.M.; Van Zonneveld, A.J.; Vulto, P.; Hankemeier, T. 96 Perfusable Blood Vessels to Study Vascular Permeability In Vitro. Sci. Rep. 2017, 7, 18071. [Google Scholar] [CrossRef] [PubMed]
- Song, H.H.G.; Lammers, A.; Sundaram, S.; Rubio, L.; Chen, A.X.; Li, L.; Eyckmans, J.; Bhatia, S.N.; Chen, C.S. Transient Support from Fibroblasts Is Sufficient to Drive Functional Vascularization in Engineered Tissues. Adv. Funct. Mater. 2020, 30, 2003777. [Google Scholar] [CrossRef] [PubMed]
- Figueiredo, L.; Pace, R.; D’Arros, C.; Réthoré, G.; Guicheux, J.; Le Visage, C.; Weiss, P. Assessing Glucose and Oxygen Diffusion in Hydrogels for the Rational Design of 3D Stem Cell Scaffolds in Regenerative Medicine. J. Tissue Eng. Regen. Med. 2018, 12, 1238–1246. [Google Scholar] [CrossRef] [PubMed]
- Magliaro, C.; Mattei, G.; Iacoangeli, F.; Corti, A.; Piemonte, V.; Ahluwalia, A. Oxygen Consumption Characteristics in 3D Constructs Depend on Cell Density. Front. Bioeng. Biotechnol. 2019, 7, 251. [Google Scholar] [CrossRef] [PubMed]
- McMurtrey, R.J. Analytic Models of Oxygen and Nutrient Diffusion, Metabolism Dynamics, and Architecture Optimization in Three-Dimensional Tissue Constructs with Applications and Insights in Cerebral Organoids. Tissue Eng. Part C Methods 2016, 22, 221–249. [Google Scholar] [CrossRef] [PubMed]
- Duval, K.; Grover, H.; Han, L.H.; Mou, Y.; Pegoraro, A.F.; Fredberg, J.; Chen, Z. Modeling Physiological Events in 2D vs. 3D Cell Culture. Physiology 2017, 32, 266–277. [Google Scholar] [CrossRef]
- Temple, J.; Velliou, E.; Shehata, M.; Lévy, R. Current Strategies with Implementation of Three-Dimensional Cell Culture: The Challenge of Quantification. Interface Focus 2022, 12, 20220019. [Google Scholar] [CrossRef]
- Jensen, C.; Teng, Y. Is It Time to Start Transitioning From 2D to 3D Cell Culture? Front. Mol. Biosci. 2020, 7, 33. [Google Scholar] [CrossRef]
- Gabbott, C.M.; Sun, T. Comparison of Human Dermal Fibroblasts and Hacat Cells Cultured in Medium with or without Serum via a Generic Tissue Engineering Research Platform. Int. J. Mol. Sci. 2018, 19, 388. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Patil, S.; Gao, Y.G.; Qian, A. The Bone Extracellular Matrix in Bone Formation and Regeneration. Front. Pharmacol. 2020, 11, 757. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Peng, R.; Ding, J. The Regulation of Stem Cell Differentiation by Cell-Cell Contact on Micropatterned Material Surfaces. Biomaterials 2010, 31, 2470–2476. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.Y.M.; Walsh, T.; Borys, B.S.; Kallos, M.S.; Rancourt, D.E. An Integrated Approach toward the Biomanufacturing of Engineered Cell Therapy Products in a Stirred-Suspension Bioreactor. Mol. Ther. Methods Clin. Dev. 2018, 9, 376–389. [Google Scholar] [CrossRef] [PubMed]
- Tracy, L.E.; Minasian, R.A.; Caterson, E.J. Extracellular Matrix and Dermal Fibroblast Function in the Healing Wound. Adv. Wound Care 2016, 5, 119–136. [Google Scholar] [CrossRef] [PubMed]
- Manou, D.; Caon, I.; Bouris, P.; Triantaphyllidou, I.-E.; Giaroni, C.; Passi, A.; Karamanos, N.K.; Vigetti, D.; Theocharis, A.D. The Complex Interplay Between Extracellular Matrix and Cells in Tissues. Extracell. Matrix Methods Protoc. 2019, 1952, 1–20. [Google Scholar]
- Nelson, C.M.; Chen, C.S. Cell-Cell Signaling by Direct Contact Increases Cell Proliferation via a PI3K-Dependent Signal. FEBS Lett. 2002, 514, 238–242. [Google Scholar] [CrossRef]
- Bulati, M.; Miceli, V.; Gallo, A.; Amico, G.; Carcione, C.; Pampalone, M.; Conaldi, P.G. The Immunomodulatory Properties of the Human Amnion-Derived Mesenchymal Stromal/Stem Cells Are Induced by INF-γ Produced by Activated Lymphomonocytes and Are Mediated by Cell-To-Cell Contact and Soluble Factors. Front. Immunol. 2020, 11, 54. [Google Scholar] [CrossRef]
- Johnson, B.E.; Kelley, M.J. Autocrine Growth Factors and Neuroendocrine Markers in the Development of Small-Cell Lung Cancer. Oncology 1998, 12, 11–14. [Google Scholar] [CrossRef]
- Li, R.; Li, D.; Zhang, H.; Wang, J.; Li, X.; Xiao, J. Growth Factors-Based Therapeutic Strategies and Their Underlying Signaling Mechanisms for Peripheral Nerve Regeneration. Acta Pharmacol. Sin. 2020, 41, 1289–1300. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Dalm, C.; Wijffels, R.H.; Martens, D.E. Metabolic Characterization of a CHO Cell Size Increase Phase in Fed-Batch Cultures. Appl. Microbiol. Biotechnol. 2017, 101, 8101–8113. [Google Scholar] [CrossRef]
- Schinn, S.M.; Morrison, C.; Wei, W.; Zhang, L.; Lewis, N.E. Systematic Evaluation of Parameters for Genome-Scale Metabolic Models of Cultured Mammalian Cells. Metab. Eng. 2021, 66, 21–30. [Google Scholar] [CrossRef]
- Ben Yahia, B.; Malphettes, L.; Heinzle, E. Macroscopic Modeling of Mammalian Cell Growth and Metabolism. Appl. Microbiol. Biotechnol. 2015, 99, 7009–7024. [Google Scholar] [CrossRef]
- Cadart, C.; Monnier, S.; Grilli, J.; Sáez, P.J.; Srivastava, N.; Attia, R.; Terriac, E.; Baum, B.; Cosentino-Lagomarsino, M.; Piel, M. Size Control in Mammalian Cells Involves Modulation of Both Growth Rate and Cell Cycle Duration. Nat. Commun. 2018, 9, 3275. [Google Scholar] [CrossRef]
- Schnyder, S.K.; Molina, J.J.; Yamamoto, R. Control of Cell Colony Growth by Contact Inhibition. Sci. Rep. 2020, 10, 6713. [Google Scholar] [CrossRef]
- Singh, J.; Pagulayan, A.; Camley, B.A.; Nain, A.S.; Weitz, D.A. Rules of Contact Inhibition of Locomotion for Cells on Suspended Nanofibers. Biophys. Comput. Biol. Cell Biol. 2021, 118, e2011815118. [Google Scholar] [CrossRef]
- Onoe, H.; Takeuchi, S. Cell-Laden Microfibers for Bottom-up Tissue Engineering. Drug Discov. Today 2015, 20, 236–246. [Google Scholar] [CrossRef] [PubMed]
- Gwyther, T.A.; Hu, J.Z.; Christakis, A.G.; Skorinko, J.K.; Shaw, S.M.; Billiar, K.L.; Rolle, M.W. Engineered Vascular Tissue Fabricated from Aggregated Smooth Muscle Cells. Cells Tissues Organs 2011, 194, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Neff, L.P.; Tillman, B.W.; Yazdani, S.K.; MacHingal, M.A.; Yoo, J.J.; Soker, S.; Bernish, B.W.; Geary, R.L.; Christ, G.J. Vascular Smooth Muscle Enhances Functionality of Tissue-Engineered Blood Vessels In Vivo. J. Vasc. Surg. 2011, 53, 426–434. [Google Scholar] [CrossRef] [PubMed]
- Białkowska, K.; Komorowski, P.; Bryszewska, M.; Miłowska, K. Spheroids as a Type of Three-Dimensional Cell Cultures—Examples of Methods of Preparation and the Most Important Application. Int. J. Mol. Sci. 2020, 21, 6225. [Google Scholar] [CrossRef] [PubMed]
- Anthon, S.G.; Valente, K.P. Vascularization Strategies in 3D Cell Culture Models: From Scaffold-Free Models to 3D Bioprinting. Int. J. Mol. Sci. 2022, 23, 14582. [Google Scholar] [CrossRef]
- Bettega, D.; Calzolari, P.; Doglia, S.M.; Dulio, B.; Tallone, L.; Villa, A.M. Technical Report: Cell Thickness Measurements by Confocal Fluorescence Microscopy on C3H10T1/2 and V79 Cells. Int. J. Radiat. Biol. 1998, 74, 397–403. [Google Scholar] [CrossRef]
- Andrique, L.; Recher, G.; Alessandri, K.; Pujol, N.; Feyeux, M.; Bon, P.; Cognet, L.; Nassoy, P.; Bikfalvi, A. A Model of Guided Cell Self-Organization for Rapid and Spontaneous Formation of Functional Vessels. Sci. Adv. 2019, 5, eaau6562. [Google Scholar] [CrossRef]
- Sekine, W.; Haraguchi, Y. Thickness Limitation and Cell Viability of Multi-Layered Cell Sheets and Overcoming the Diffusion Limit by a Porous-Membrane Culture Insert. J. Biochips Tissue Chips 2011, 1 (Suppl. S1), 7. [Google Scholar] [CrossRef]
- Yang, J.; Guertin, P.; Jia, G.; Lv, Z.; Yang, H.; Ju, D. Large-Scale Microcarrier Culture of HEK293T Cells and Vero Cells in Single-Use Bioreactors. AMB Express 2019, 9, 70. [Google Scholar] [CrossRef]
- Mitsui, Y.; Schneider, E.L. Relationship between Cell Replication and Volume in Senescent Human Diploid Fibroblasts. Mech. Ageing Dev. 1976, 5, 45–56. [Google Scholar] [CrossRef]
- Lodish, H. Molecular Cell Biology, 5th ed.; W. H. Freeman: New York, NY, USA, 2003. [Google Scholar]
- Melgarejo-Ramírez, Y.; Sánchez-Sánchez, R.; García-López, J.; Brena-Molina, A.M.; Gutiérrez-Gómez, C.; Ibarra, C.; Velasquillo, C. Characterization of Pediatric Microtia Cartilage: A Reservoir of Chondrocytes for Auricular Reconstruction Using Tissue Engineering Strategies. Cell Tissue Bank. 2016, 17, 481–489. [Google Scholar] [CrossRef]
- Cai, L.; Liu, W.; Cui, Y.; Liu, Y.; Du, W.; Zheng, L.; Pi, C.; Zhang, D.; Xie, J.; Zhou, X. Biomaterial Stiffness Guides Cross-Talk between Chondrocytes: Implications for a Novel Cellular Response in Cartilage Tissue Engineering. ACS Biomater. Sci. Eng. 2020, 6, 4476–4489. [Google Scholar] [CrossRef] [PubMed]
- Tao, X.; Shaolin, L.; Yaoting, Y. Preparation and Culture of Hepatocyte on Gelatin Microcarriers. J. Biomed. Mater. Res. A 2003, 65, 306–310. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Y.; Yan, J.; Bao, X.; Gleadall, A.; Roach, P.; Sun, T. Evaluation of Polymeric Particles for Modular Tissue Cultures in Developmental Engineering. Int. J. Mol. Sci. 2023, 24, 5234. [Google Scholar] [CrossRef] [PubMed]
- van Wenum, M.; Adam, A.A.A.; van der Mark, V.A.; Chang, J.C.; Wildenberg, M.E.; Hendriks, E.J.; Jongejan, A.; Moerland, P.D.; van Gulik, T.M.; Oude Elferink, R.P.; et al. Oxygen Drives Hepatocyte Differentiation and Phenotype Stability in Liver Cell Lines. J. Cell Commun. Signal 2018, 12, 575–588. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Niu, Y.; Fu, X.; Lin, Q.; Liang, H.; Liu, L.; Li, N. Large-Scale Microcarrier Culture of Chinese Perch Brain Cell for Viral Vaccine Production in a Stirred Bioreactor. Vaccines 2021, 9, 1003. [Google Scholar] [CrossRef] [PubMed]
- Badenes, S.M.; Fernandes, T.G.; Rodrigues, C.A.V.; Diogo, M.M.; Cabral, J.M.S. Microcarrier-Based Platforms for in Vitro Expansion and Differentiation of Human Pluripotent Stem Cells in Bioreactor Culture Systems. J. Biotechnol. 2016, 234, 71–82. [Google Scholar] [CrossRef] [PubMed]
- Rafiq, Q.A.; Coopman, K.; Nienow, A.W.; Hewitt, C.J. Systematic Microcarrier Screening and Agitated Culture Conditions Improves Human Mesenchymal Stem Cell Yield in Bioreactors. Biotechnol. J. 2016, 11, 473–486. [Google Scholar] [CrossRef]
- Woodley, J.P.; Lambert, D.W.; Asencio, I.O. Understanding Fibroblast Behavior in 3D Biomaterials. Tissue Eng. Part B Rev. 2022, 28, 569–578. [Google Scholar] [CrossRef]
- Seifu, D.G.; Mequanint, K. Fabrication of Vascular Tissue Engineering Scaffolds with Enhanced Oxygen Diffusivity and Cell Infiltration. J. Biomater. Tissue Eng. 2012, 2, 280–291. [Google Scholar] [CrossRef]
- Atashrouz, S.; Hatampoor, A.; Yadegari, A.; Ghasemi, H.; Tayebi, L.; Rasoulianboroujeni, M. Mathematical Modeling of Oxygen Transfer in Porous Scaffolds for Stem Cell Growth: The Effects of Porosity, Cell Type, Scaffold Architecture and Cell Distribution. Mater. Chem. Phys. 2019, 222, 377–383. [Google Scholar] [CrossRef]
- Paul, A.; Laurila, T.; Vuorinen, V.; Divinski, S.V. Fick’s Laws of Diffusion. In Thermodynamics, Diffusion and the Kirkendall Effect in Solids; Springer International Publishing: Cham, Switzerland, 2014; pp. 115–139. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Constant | Value | Reference |
|---|---|---|
| D 1 | 2.5 × 10−9 m2/s | [37] |
| C0 2 | 2.0 × 10−4 M | [37] |
| Cmin(hMSCs) 3 | 9.0 × 10−5 M | [64] |
| OCR (hMSCs) 4 | 2.0 × 10−17 to 3.8 × 10−17 moles cell−1 s−1 | [37] |
| OCR (human fibroblasts) | 8.3 × 10−19 to 1.8 × 10−17 moles cell−1 s−1 | [37] |
| OCR (human hepatocyte) | 1.0 × 10−16 to 9.0 × 10−16 moles cell−1 s−1 | [37] |
| R 5 | 100 × 10−6 m | [65] |
| Thickness of a fibroblast | 4.7 μm | [62] |
| Volume of senescent human diploid fibroblast | 2600 μm3 | [66] |
| Volume of a hepatocyte | 3400 μm3 | [67] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, T.; Xiang, Y.; Turner, F.; Bao, X. Integrated Experimental and Mathematical Exploration of Modular Tissue Cultures for Developmental Engineering. Int. J. Mol. Sci. 2024, 25, 2987. https://doi.org/10.3390/ijms25052987
Sun T, Xiang Y, Turner F, Bao X. Integrated Experimental and Mathematical Exploration of Modular Tissue Cultures for Developmental Engineering. International Journal of Molecular Sciences. 2024; 25(5):2987. https://doi.org/10.3390/ijms25052987
Chicago/Turabian StyleSun, Tao, Yu Xiang, Freya Turner, and Xujin Bao. 2024. "Integrated Experimental and Mathematical Exploration of Modular Tissue Cultures for Developmental Engineering" International Journal of Molecular Sciences 25, no. 5: 2987. https://doi.org/10.3390/ijms25052987
APA StyleSun, T., Xiang, Y., Turner, F., & Bao, X. (2024). Integrated Experimental and Mathematical Exploration of Modular Tissue Cultures for Developmental Engineering. International Journal of Molecular Sciences, 25(5), 2987. https://doi.org/10.3390/ijms25052987