

Antiseptic Functions of CGK012 against HMGB1-Mediated Septic Responses

 , ,
, , 
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
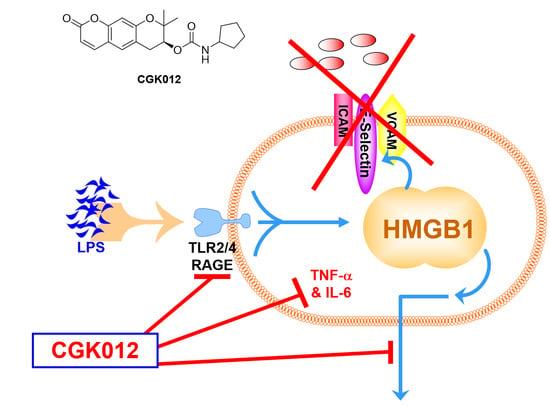
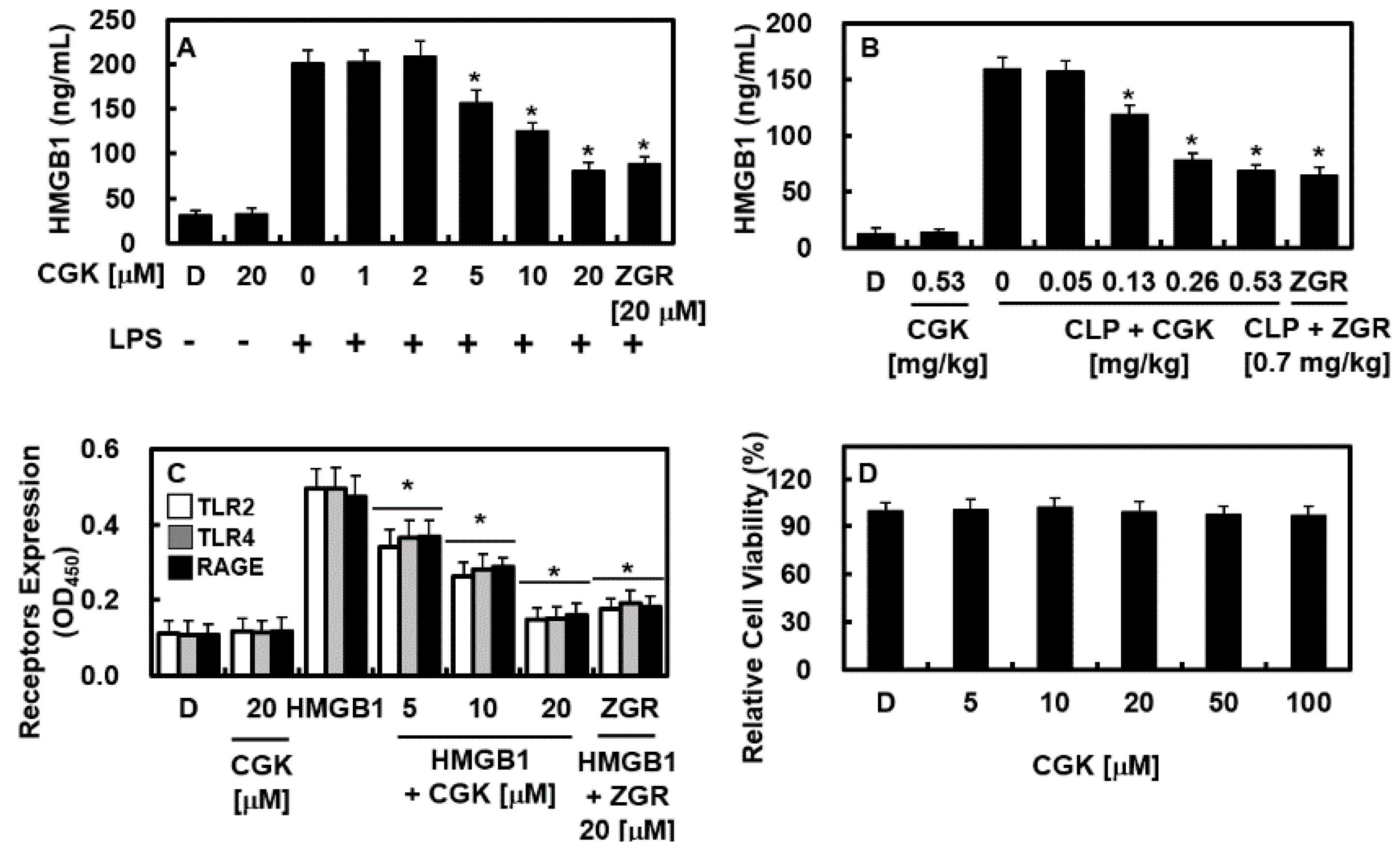
2.1. CGK012 Inhibits HMGB1 Release in the LPS-Stimulated Human Umbilical Vein Endothelial Cells (HUVECs) and Cecal Ligation and Puncture (CLP)-Induced Sepsis Mouse Model
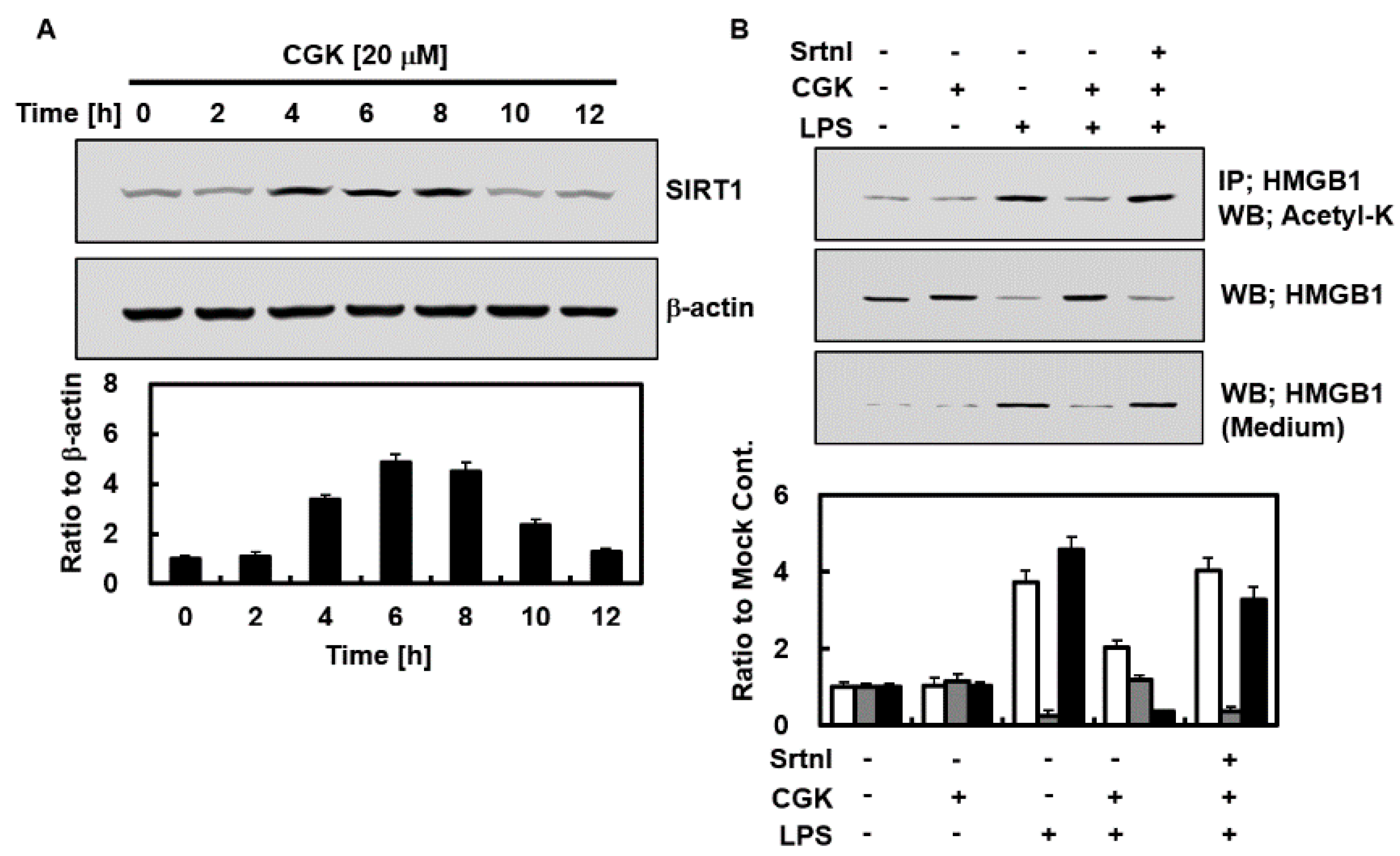
2.2. Effects of CGK012 on the Activation of Silent Information Regulator Sirtuin 1 (SIRT1) and HMGB1 Acetylation
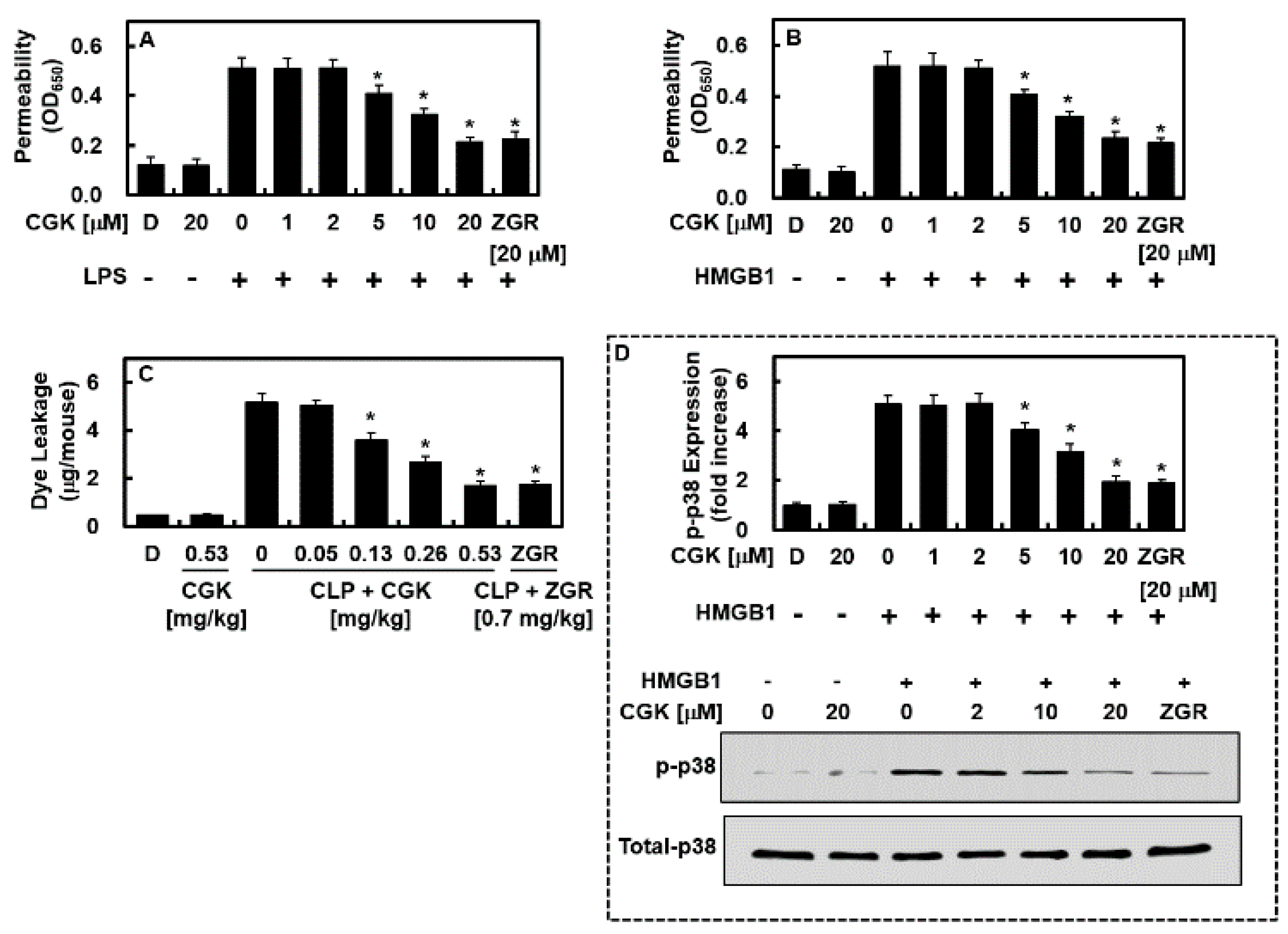
2.3. CGK012 Inhibits HMGB1-Mediated Disruption of the Vascular Barrier Integrity
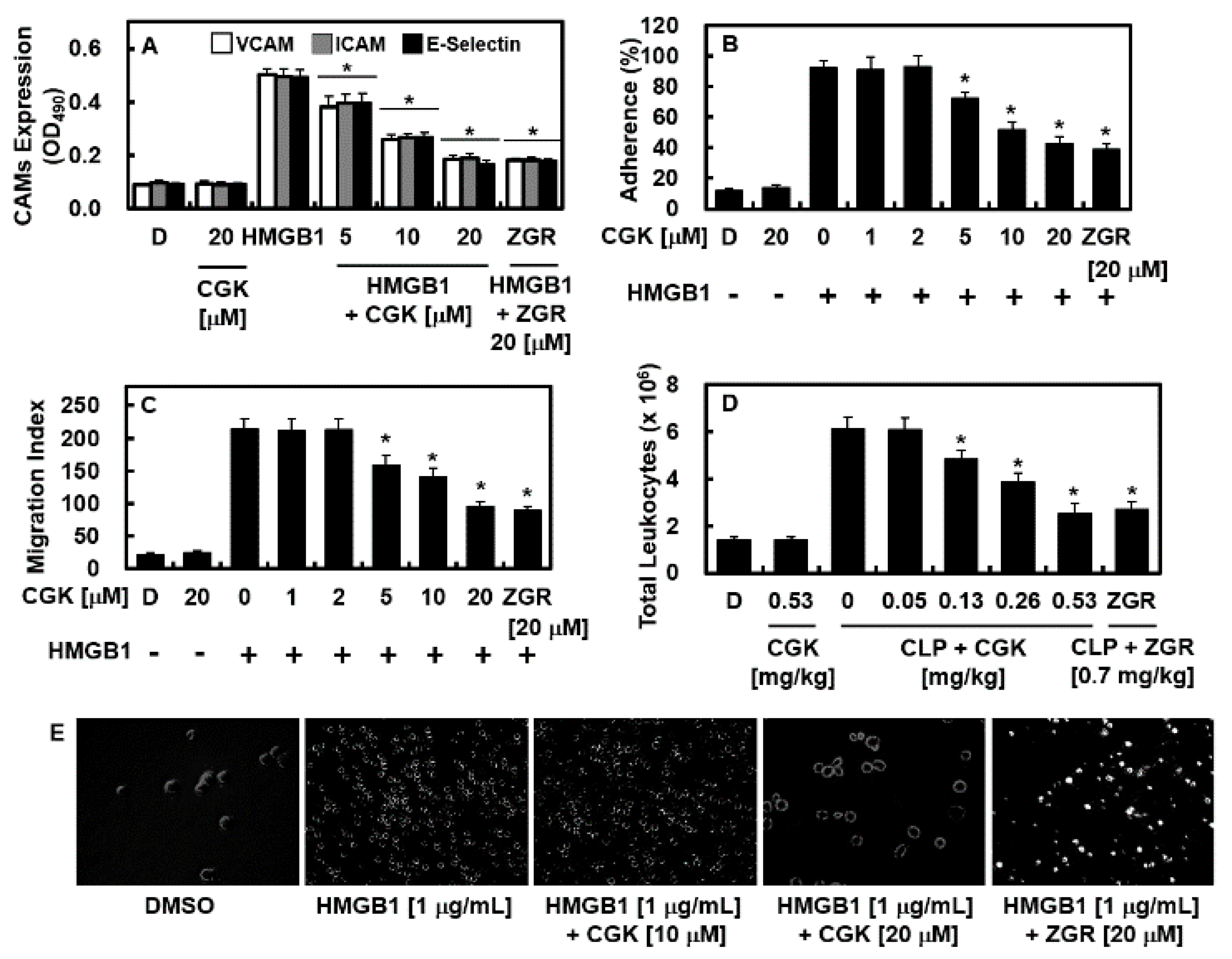
2.4. Effects of CGK012 on HMGB1-Mediated Expression of CAMs, Adhesion of Neutrophils, and Migration of Leukocytes
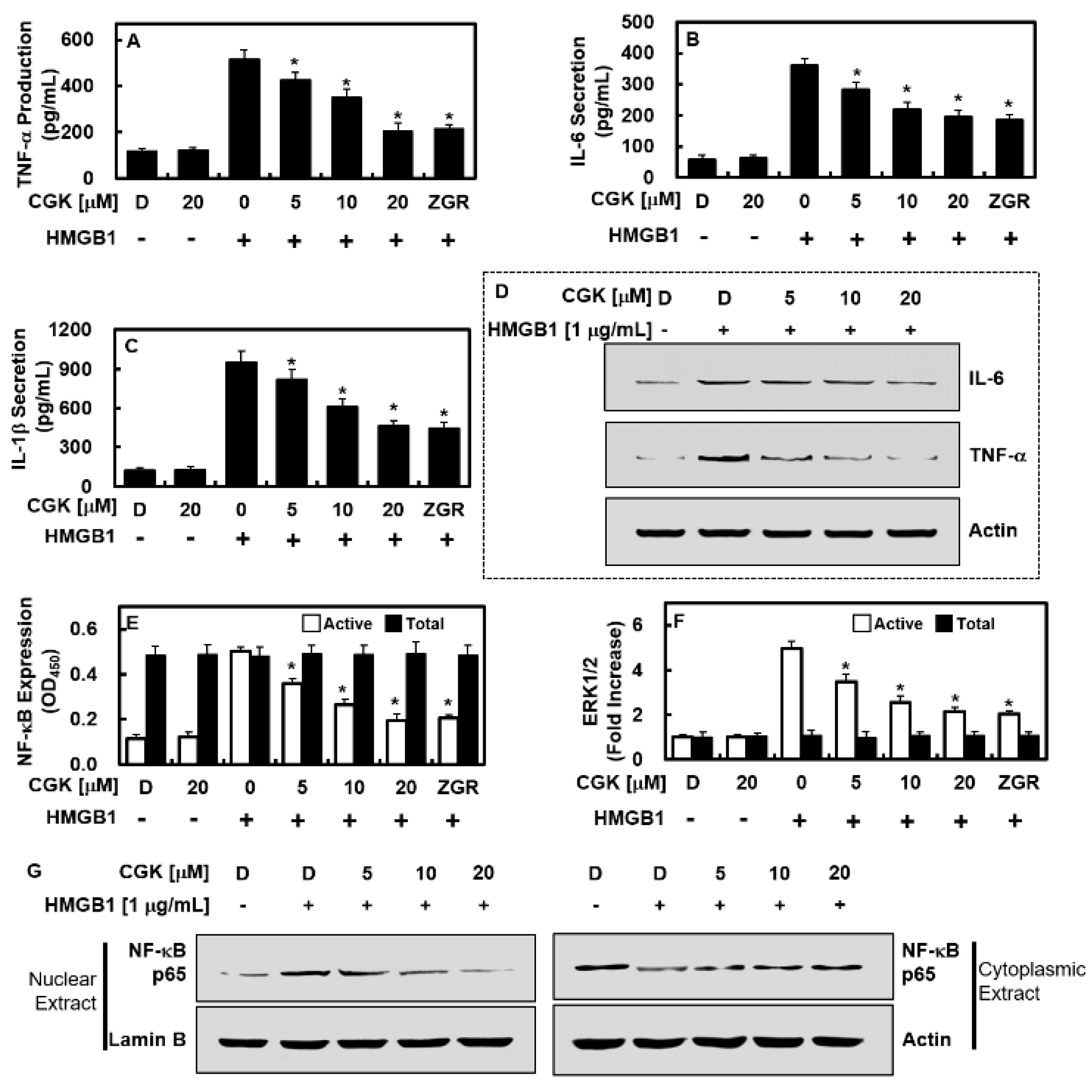
2.5. CGK012 Suppresses NF-κB/ERK Signaling and IL-1β, IL-6, and TNF-α Production
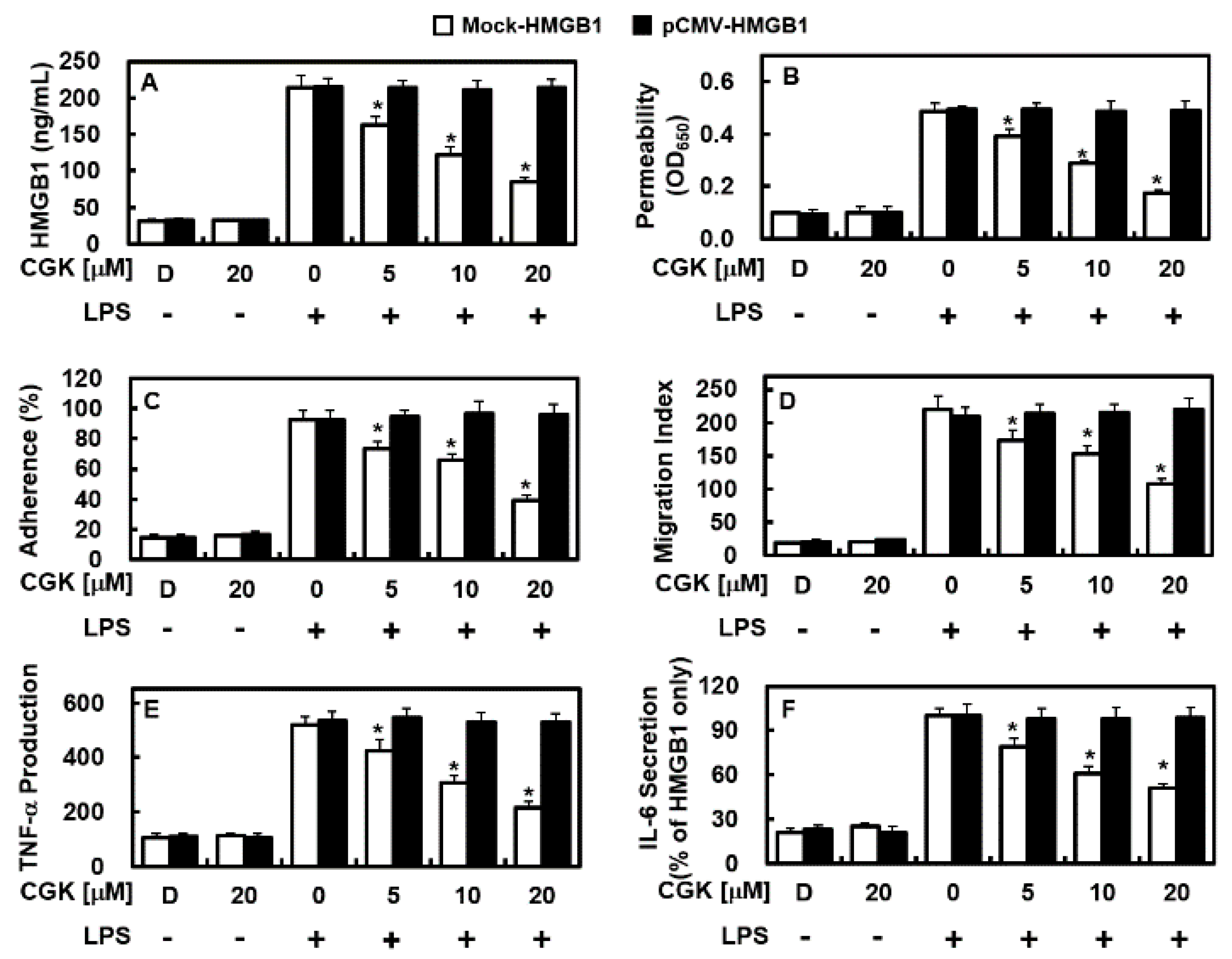
2.6. Overexpression of HMGB1 Prevents the Anti-Inflammatory Functions of CGK012
2.7. Effects of CGK012 Administration on Survival Rate and Tissue Injury Reduction in Mice with CLP-Induced Sepsis
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Reagents
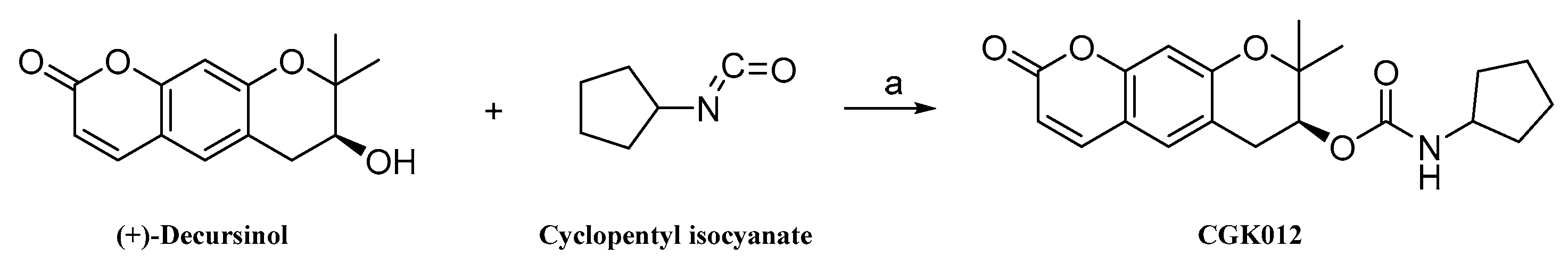
4.2. Method for the Synthesis of (7S)-(+)-Cyclopentyl Carbamic Acid 8,8-Dimethyl-2-oxo-6,7-dihydro-2H,8H-pyrano[3,2-g]chromen-7-yl-ester (CGK012)
4.3. Animals and CLP
4.4. Competitive ELISA for HMGB1
4.5. Expression Levels of CAMs and HMGB1 Receptors
4.6. Cell Viability Assay
4.7. Preparation of Cytoplasmic and Nuclear Extracts and Western Blotting Analyses
4.8. Cell–Cell Adhesion Assay
4.9. Permeability Assay
4.10. In Vitro Migration Assay
4.11. In Vivo Leukocyte Migration Assay
4.12. ELISAs for Phosphorylation of p38 MAPK/SAPK, NF-κB, TNF-α, ERK 1/2, IL-1β, and IL-6
4.13. Transfection for Stable Human HMGB1-Overexpressed HUVECs
4.14. H&E Staining and Histopathological Examinations
4.15. Measurement of Tissue Injury Markers
4.16. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hotchkiss, R.S.; Moldawer, L.L.; Opal, S.M.; Reinhart, K.; Turnbull, I.R.; Vincent, J.L. Sepsis and septic shock. Nat. Rev. Dis. Primers 2016, 2, 16045. [Google Scholar] [CrossRef]
- Ziesmann, M.T.; Marshall, J.C. Multiple Organ Dysfunction: The Defining Syndrome of Sepsis. Surg. Infect. 2018, 19, 184–190. [Google Scholar] [CrossRef]
- Ologunde, R.; Zhao, H.; Lu, K.; Ma, D. Organ cross talk and remote organ damage following acute kidney injury. Int. Urol. Nephrol. 2014, 46, 2337–2345. [Google Scholar] [CrossRef]
- Riedemann, N.C.; Guo, R.F.; Ward, P.A. Novel strategies for the treatment of sepsis. Nat. Med. 2003, 9, 517–524. [Google Scholar] [CrossRef]
- Abraham, E.; Arcaroli, J.; Carmody, A.; Wang, H.; Tracey, K.J. HMG-1 as a mediator of acute lung inflammation. J. Immunol. 2000, 165, 2950–2954. [Google Scholar] [CrossRef]
- Dickson, K.; Lehmann, C. Inflammatory Response to Different Toxins in Experimental Sepsis Models. Int. J. Mol. Sci. 2019, 20, 4341. [Google Scholar] [CrossRef]
- Wang, H.; Bloom, O.; Zhang, M.; Vishnubhakat, J.M.; Ombrellino, M.; Che, J.; Frazier, A.; Yang, H.; Ivanova, S.; Borovikova, L.; et al. HMG-1 as a late mediator of endotoxin lethality in mice. Science 1999, 285, 248–251. [Google Scholar] [CrossRef]
- Bae, J.S. Role of high mobility group box 1 in inflammatory disease: Focus on sepsis. Arch. Pharm. Res. 2012, 35, 1511–1523. [Google Scholar] [CrossRef]
- Kumar, N.R.; Balraj, T.A.; Kempegowda, S.N.; Prashant, A. Multidrug-Resistant Sepsis: A Critical Healthcare Challenge. Antibiotics 2024, 13, 46. [Google Scholar] [CrossRef]
- Reed, K.R.; Song, F.; Young, M.A.; Hassan, N.; Antoine, D.J.; Gemici, N.B.; Clarke, A.R.; Jenkins, J.R. Secreted HMGB1 from Wnt activated intestinal cells is required to maintain a crypt progenitor phenotype. Oncotarget 2016, 7, 51665–51673. [Google Scholar] [CrossRef]
- Choi, P.J.; Yuseok, O.; Her, J.H.; Yun, E.; Song, G.Y.; Oh, S. Anti-proliferative activity of CGK012 against multiple myeloma cells via Wnt/β-catenin signaling attenuation. Leuk. Res. 2017, 60, 103–108. [Google Scholar] [CrossRef]
- Lee, W.; Ku, S.K.; Bae, J.S. Zingerone reduces HMGB1-mediated septic responses and improves survival in septic mice. Toxicol. Appl. Pharm. 2017, 329, 202–211. [Google Scholar] [CrossRef]
- Lee, B.S.; Lee, C.; Yang, S.; Ku, S.K.; Bae, J.S. Renal protective effects of zingerone in a mouse model of sepsis. BMB Rep. 2019, 52, 271–276. [Google Scholar] [CrossRef]
- Lee, W.; Hwang, M.H.; Lee, Y.; Bae, J.S. Protective effects of zingerone on lipopolysaccharide-induced hepatic failure through the modulation of inflammatory pathways. Chem. Biol. Interact. 2018, 281, 106–110. [Google Scholar] [CrossRef]
- Bonaldi, T.; Talamo, F.; Scaffidi, P.; Ferrera, D.; Porto, A.; Bachi, A.; Rubartelli, A.; Agresti, A.; Bianchi, M.E. Monocytic cells hyperacetylate chromatin protein HMGB1 to redirect it towards secretion. EMBO J. 2003, 22, 5551–5560. [Google Scholar] [CrossRef]
- Youn, J.H.; Shin, J.S. Nucleocytoplasmic shuttling of HMGB1 is regulated by phosphorylation that redirects it toward secretion. J. Immunol. 2006, 177, 7889–7897. [Google Scholar] [CrossRef]
- Rabadi, M.M.; Xavier, S.; Vasko, R.; Kaur, K.; Goligorksy, M.S.; Ratliff, B.B. High-mobility group box 1 is a novel deacetylation target of Sirtuin1. Kidney Int. 2015, 87, 95–108. [Google Scholar] [CrossRef]
- Astiz, M.E.; Rackow, E.C. Septic shock. Lancet 1998, 351, 1501–1505. [Google Scholar] [CrossRef]
- Bogatcheva, N.V.; Verin, A.D. The role of cytoskeleton in the regulation of vascular endothelial barrier function. Microvasc. Res. 2008, 76, 202–207. [Google Scholar] [CrossRef]
- Tsung, A.; Tohme, S.; Billiar, T.R. High-mobility group box-1 in sterile inflammation. J. Intern. Med. 2014, 276, 425–443. [Google Scholar] [CrossRef]
- Li, L.; Lu, Y.Q. The Regulatory Role of High-Mobility Group Protein 1 in Sepsis-Related Immunity. Front. Immunol. 2020, 11, 601815. [Google Scholar] [CrossRef]
- He, M.; Zhang, B.; Wei, X.; Wang, Z.; Fan, B.; Du, P.; Zhang, Y.; Jian, W.; Chen, L.; Wang, L.; et al. HDAC4/5-HMGB1 signalling mediated by NADPH oxidase activity contributes to cerebral ischaemia/reperfusion injury. J. Cell. Mol. Med. 2013, 17, 531–542. [Google Scholar] [CrossRef]
- Denk, A.; Goebeler, M.; Schmid, S.; Berberich, I.; Ritz, O.; Lindemann, D.; Ludwig, S.; Wirth, T. Activation of NF-kappa B via the Ikappa B kinase complex is both essential and sufficient for proinflammatory gene expression in primary endothelial cells. J. Biol. Chem. 2001, 276, 28451–28458. [Google Scholar] [CrossRef]
- Pober, J.S.; Sessa, W.C. Evolving functions of endothelial cells in inflammation. Nat. Rev. Immunol. 2007, 7, 803–815. [Google Scholar] [CrossRef]
- Sehnert, B.; Burkhardt, H.; Wessels, J.T.; Schroder, A.; May, M.J.; Vestweber, D.; Zwerina, J.; Warnatz, K.; Nimmerjahn, F.; Schett, G.; et al. NF-kappaB inhibitor targeted to activated endothelium demonstrates a critical role of endothelial NF-kappaB in immune-mediated diseases. Proc. Natl. Acad. Sci. USA 2013, 110, 16556–16561. [Google Scholar] [CrossRef]
- Ding, J.; Song, D.; Ye, X.; Liu, S.F. A pivotal role of endothelial-specific NF-kappaB signaling in the pathogenesis of septic shock and septic vascular dysfunction. J. Immunol. 2009, 183, 4031–4038. [Google Scholar] [CrossRef]
- Gareus, R.; Kotsaki, E.; Xanthoulea, S.; van der Made, I.; Gijbels, M.J.; Kardakaris, R.; Polykratis, A.; Kollias, G.; de Winther, M.P.; Pasparakis, M. Endothelial cell-specific NF-kappaB inhibition protects mice from atherosclerosis. Cell Metab. 2008, 8, 372–383. [Google Scholar] [CrossRef]
- Biala, G.; Kedzierska, E.; Kruk-Slomka, M.; Orzelska-Gorka, J.; Hmaidan, S.; Skrok, A.; Kaminski, J.; Havrankova, E.; Nadaska, D.; Malik, I. Research in the Field of Drug Design and Development. Pharmaceuticals 2023, 16, 1283. [Google Scholar] [CrossRef]
- Kim, J.E.; Lee, W.; Yang, S.; Cho, S.H.; Baek, M.C.; Song, G.Y.; Bae, J.S. Suppressive effects of rare ginsenosides, Rk1 and Rg5, on HMGB1-mediated septic responses. Food Chem. Toxicol. 2019, 124, 45–53. [Google Scholar] [CrossRef]
- Lee, I.C.; Bae, J.S. Pelargonidin Protects Against Renal Injury in a Mouse Model of Sepsis. J. Med. Food 2019, 22, 57–61. [Google Scholar] [CrossRef]
- Hofbauer, R.; Moser, D.; Salfinger, H.; Frass, M.; Kapiotis, S. Sufentanil inhibits migration of human leukocytes through human endothelial cell monolayers. Anesth. Analg. 1998, 87, 1181–1185. [Google Scholar] [CrossRef]
- Bae, J.S.; Rezaie, A.R. Thrombin inhibits HMGB1-mediated proinflammatory signaling responses when endothelial protein C receptor is occupied by its natural ligand. BMB Rep. 2013, 46, 544–549. [Google Scholar] [CrossRef]
- Baek, D.H.; Kim, G.O.; Choi, H.J.; Yun, M.Y.; Park, D.H.; Song, G.Y.; Bae, J.S. Inhibitory Activities of GDX-365 on HMGB1-mediated Septic Responses. Biotechnol. Bioproc. Eng. 2023, 28, 623–631. [Google Scholar] [CrossRef]
- Kim, C.; Ryu, S.H.; Choi, H.; Park, D.H.; Bae, J.S. The Inhibitory Functions of Sparstolonin B against Ambient Fine Particulate Matter Induced Lung Injury. Biotechnol. Bioproc. Eng. 2022, 27, 949–960. [Google Scholar] [CrossRef]
- Lee, I.-C.; Bae, J.-S. Hepatic Protective Effects of Jujuboside B through the Modulation of Inflammatory Pathways. Biotechnol. Bioproc. Eng. 2022, 27, 336–343. [Google Scholar] [CrossRef]
- Kim, C.; Ryu, S.H.; Kim, N.; Lee, W.; Bae, J.-S. Renal Protective Effects of Sparstolonin B in a Mouse Model of Sepsis. Biotechnol. Bioproc. Eng. 2022, 27, 157–162. [Google Scholar] [CrossRef]
- Sim, H.; Noh, Y.; Choo, S.; Kim, N.; Lee, T.; Bae, J.S. Suppressive Activities of Fisetin on Particulate Matter-induced Oxidative Stress. Biotechnol. Bioproc. Eng. 2021, 26, 568–574. [Google Scholar] [CrossRef]
- Lee, W.H.; Choo, S.; Sim, H.; Bae, J.S. Inhibitory Activities of Ononin on Particulate Matter-induced Oxidative Stress. Biotechnol. Bioproc. Eng. 2021, 26, 208–215. [Google Scholar] [CrossRef]
- Lee, W.; Lee, H.; Lee, T.; Park, E.K.; Bae, J.S. Inhibitory functions of maslinic acid, a natural triterpene, on HMGB1-mediated septic responses. Phytomedicine 2020, 69, 153200. [Google Scholar] [CrossRef]
- Lee, I.C.; Kim, D.Y.; Bae, J.S. Sulforaphane Reduces HMGB1-Mediated Septic Responses and Improves Survival Rate in Septic Mice. Am. J. Chin. Med. 2017, 45, 1253–1271. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, Y.J.; Heo, J.B.; Choi, Y.-J.; Cho, S.; Lee, T.; Song, G.Y.; Bae, J.-S. Antiseptic Functions of CGK012 against HMGB1-Mediated Septic Responses. Int. J. Mol. Sci. 2024, 25, 2976. https://doi.org/10.3390/ijms25052976
Park YJ, Heo JB, Choi Y-J, Cho S, Lee T, Song GY, Bae J-S. Antiseptic Functions of CGK012 against HMGB1-Mediated Septic Responses. International Journal of Molecular Sciences. 2024; 25(5):2976. https://doi.org/10.3390/ijms25052976
Chicago/Turabian StylePark, Yun Jin, Jong Beom Heo, Yoon-Jung Choi, Sanghee Cho, Taeho Lee, Gyu Yong Song, and Jong-Sup Bae. 2024. "Antiseptic Functions of CGK012 against HMGB1-Mediated Septic Responses" International Journal of Molecular Sciences 25, no. 5: 2976. https://doi.org/10.3390/ijms25052976
APA StylePark, Y. J., Heo, J. B., Choi, Y.-J., Cho, S., Lee, T., Song, G. Y., & Bae, J.-S. (2024). Antiseptic Functions of CGK012 against HMGB1-Mediated Septic Responses. International Journal of Molecular Sciences, 25(5), 2976. https://doi.org/10.3390/ijms25052976