
Curcumin in Cancer and Inflammation: An In-Depth Exploration of Molecular Interactions, Therapeutic Potentials, and the Role in Disease Management

Abstract
1. Introduction
2. Curcumin’s Molecular Interactions and Target Proteins
3. Curcumin Target Proteins in Cancer
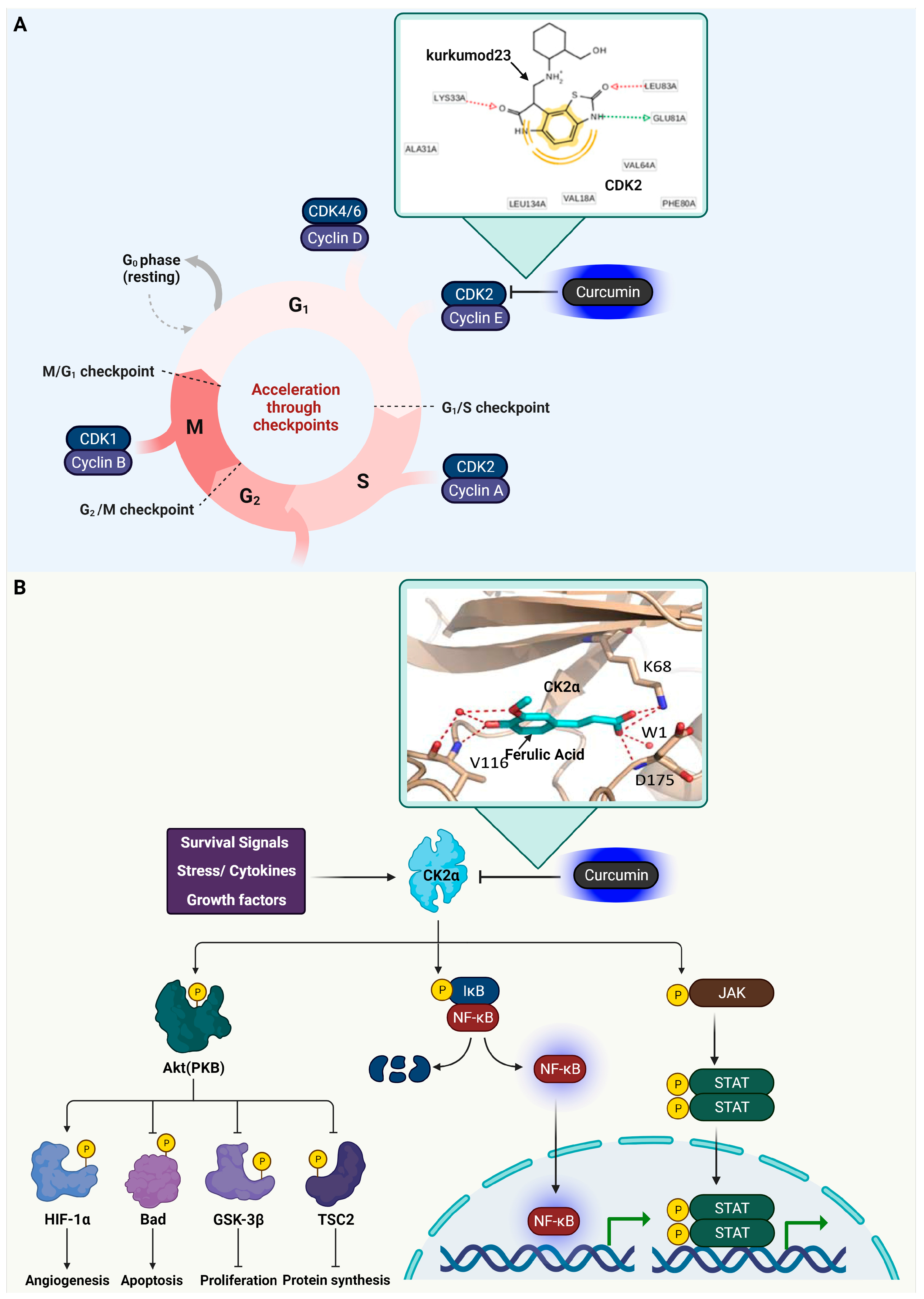
3.1. Cyclin-Dependent Kinase 2 (CDK2)
3.2. Casein Kinase 2 (CK2) Alpha
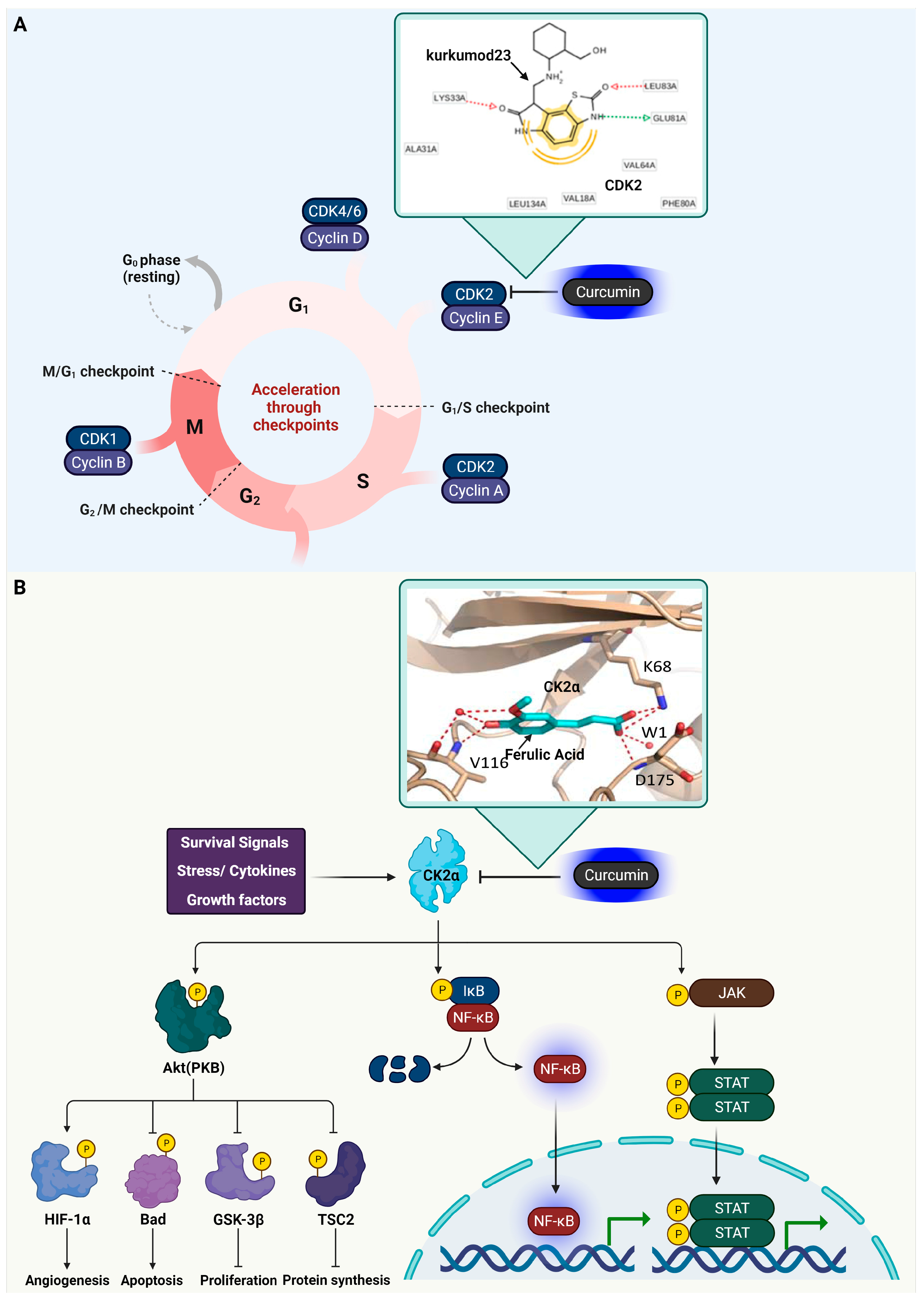
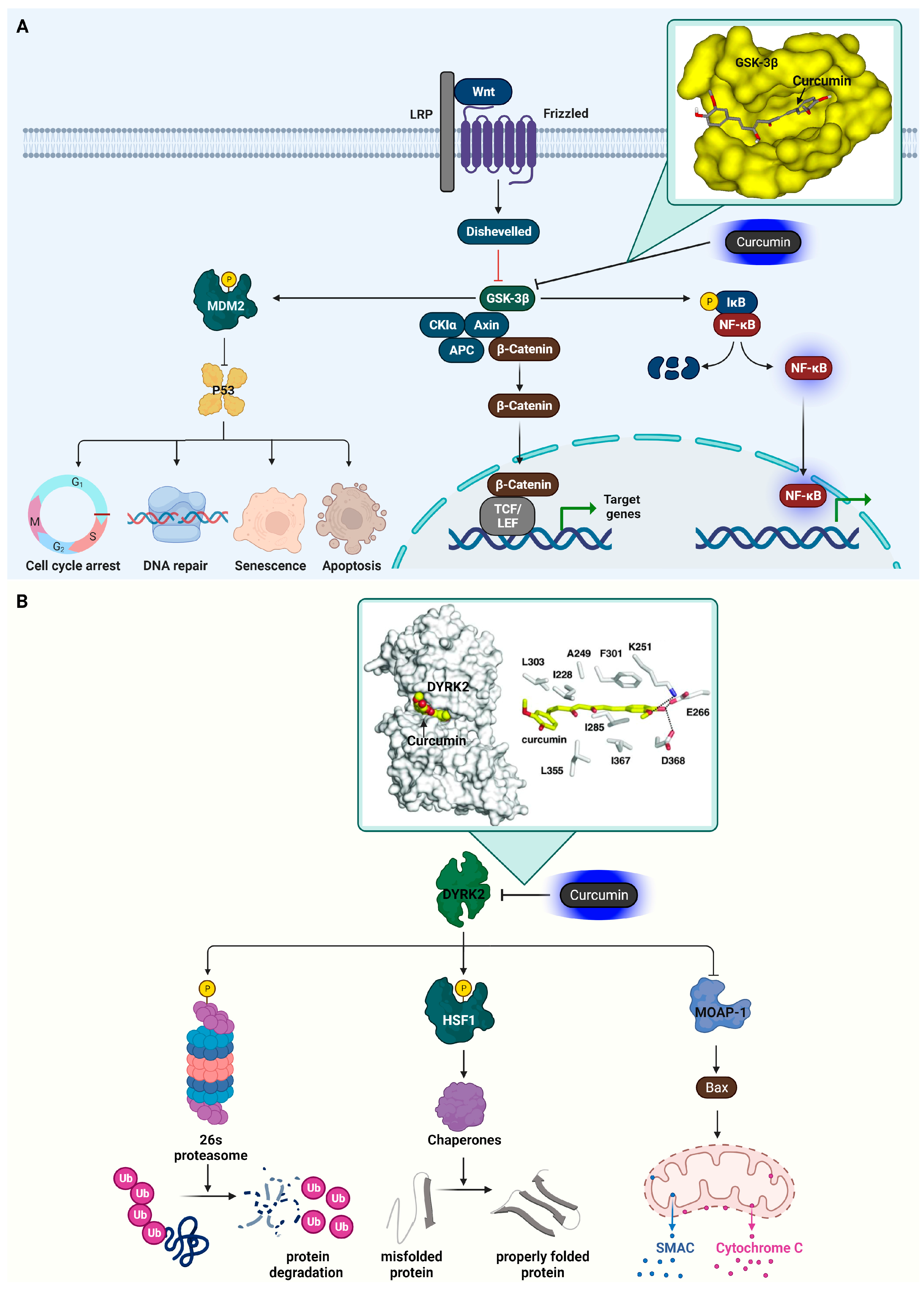
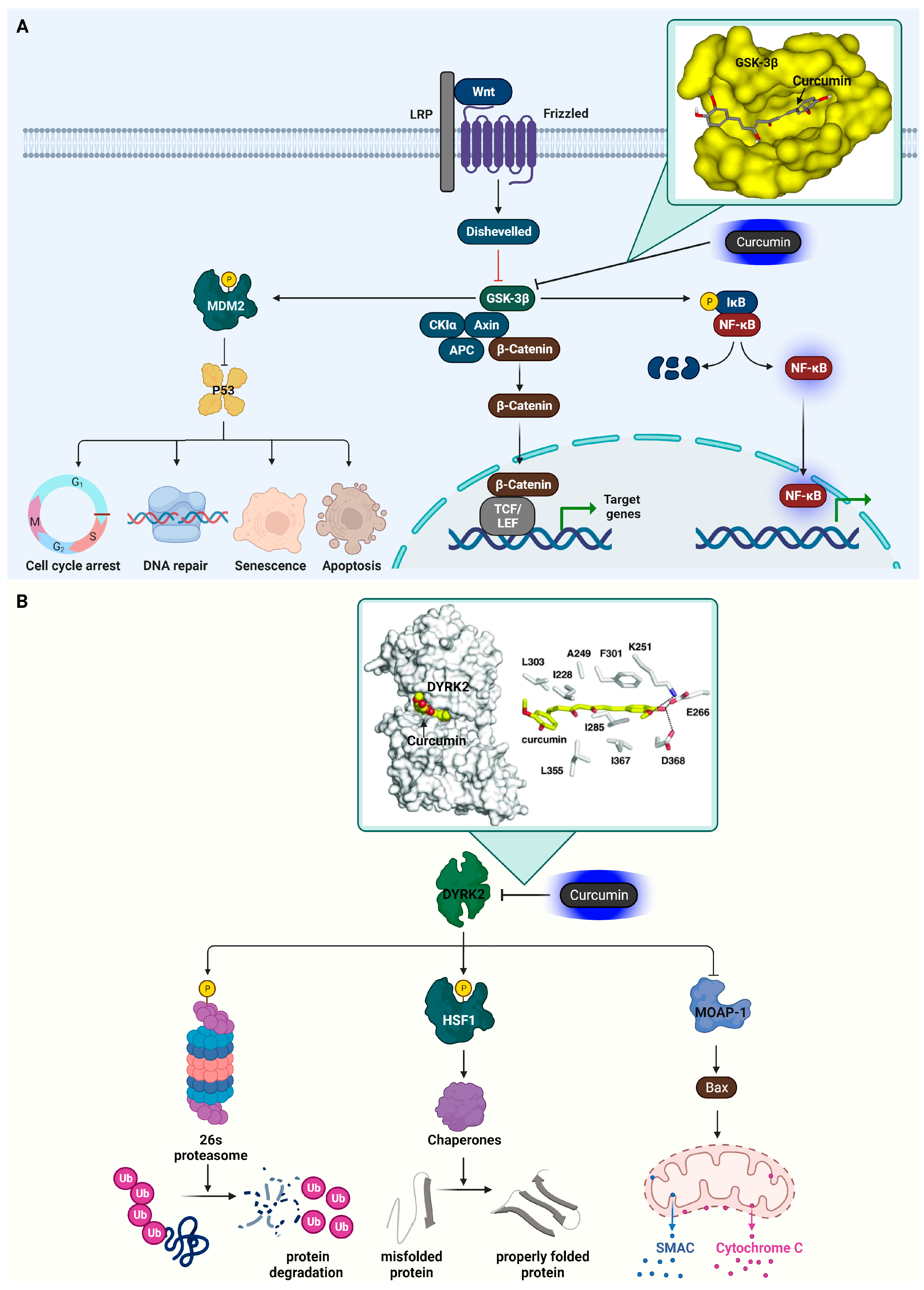
3.3. Glycogen Synthase Kinase-3 Beta (GSK3β)
3.4. Dual-Specificity Tyrosine-Regulated Kinase 2 (DYRK2)
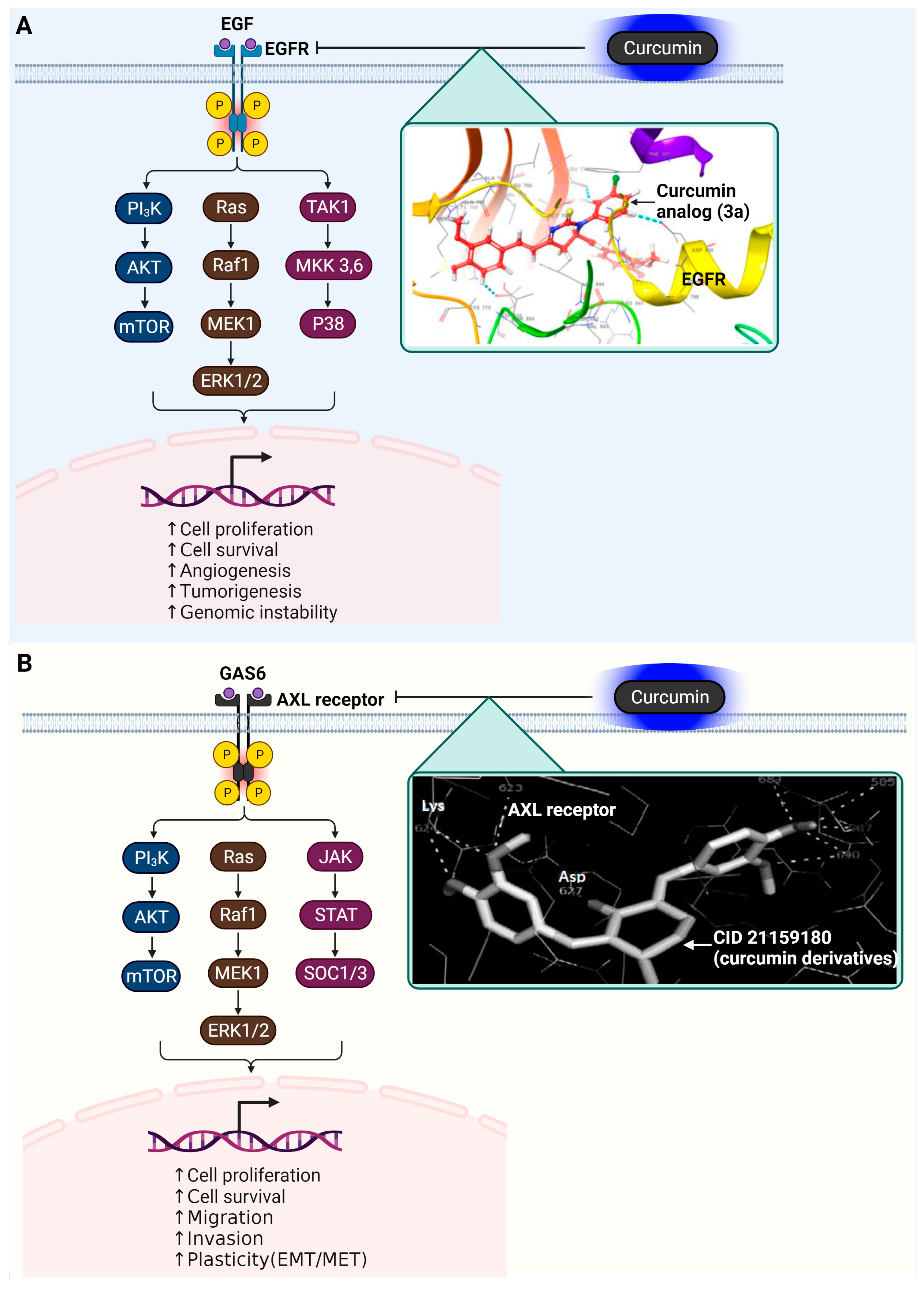
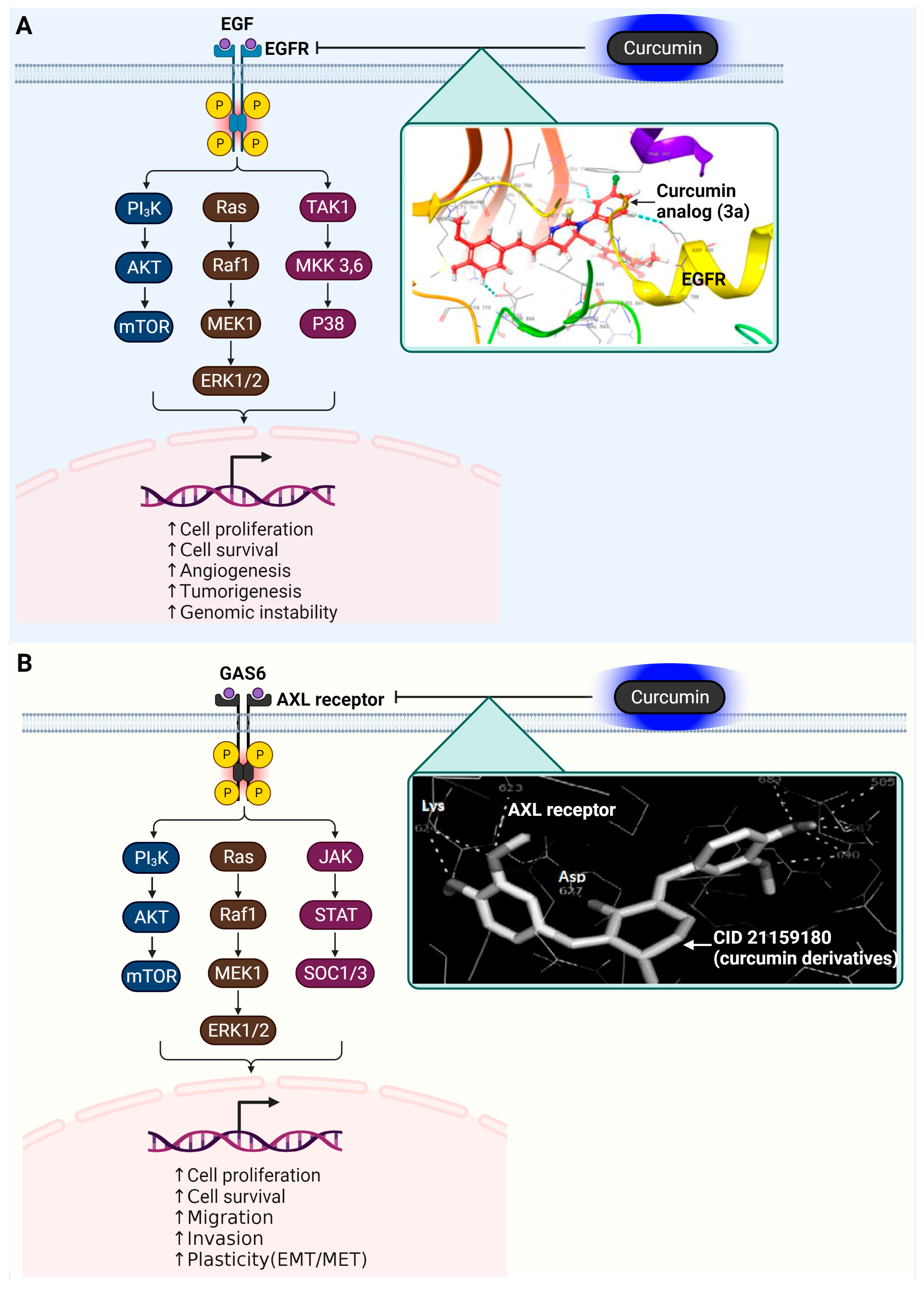
3.5. Epidermal Growth Factor Receptor (EGFR)
3.6. AXL Receptor Tyrosine Kinase
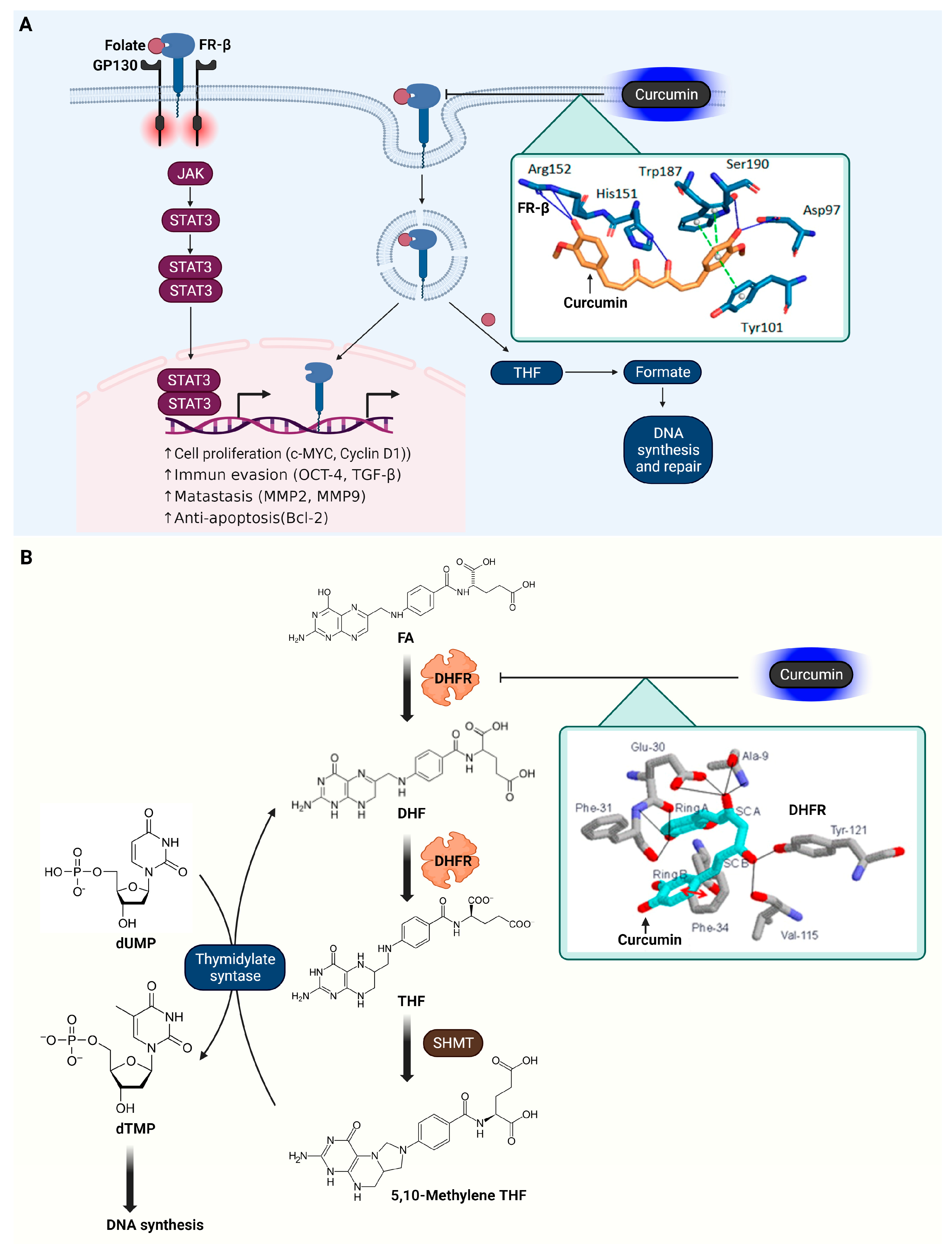
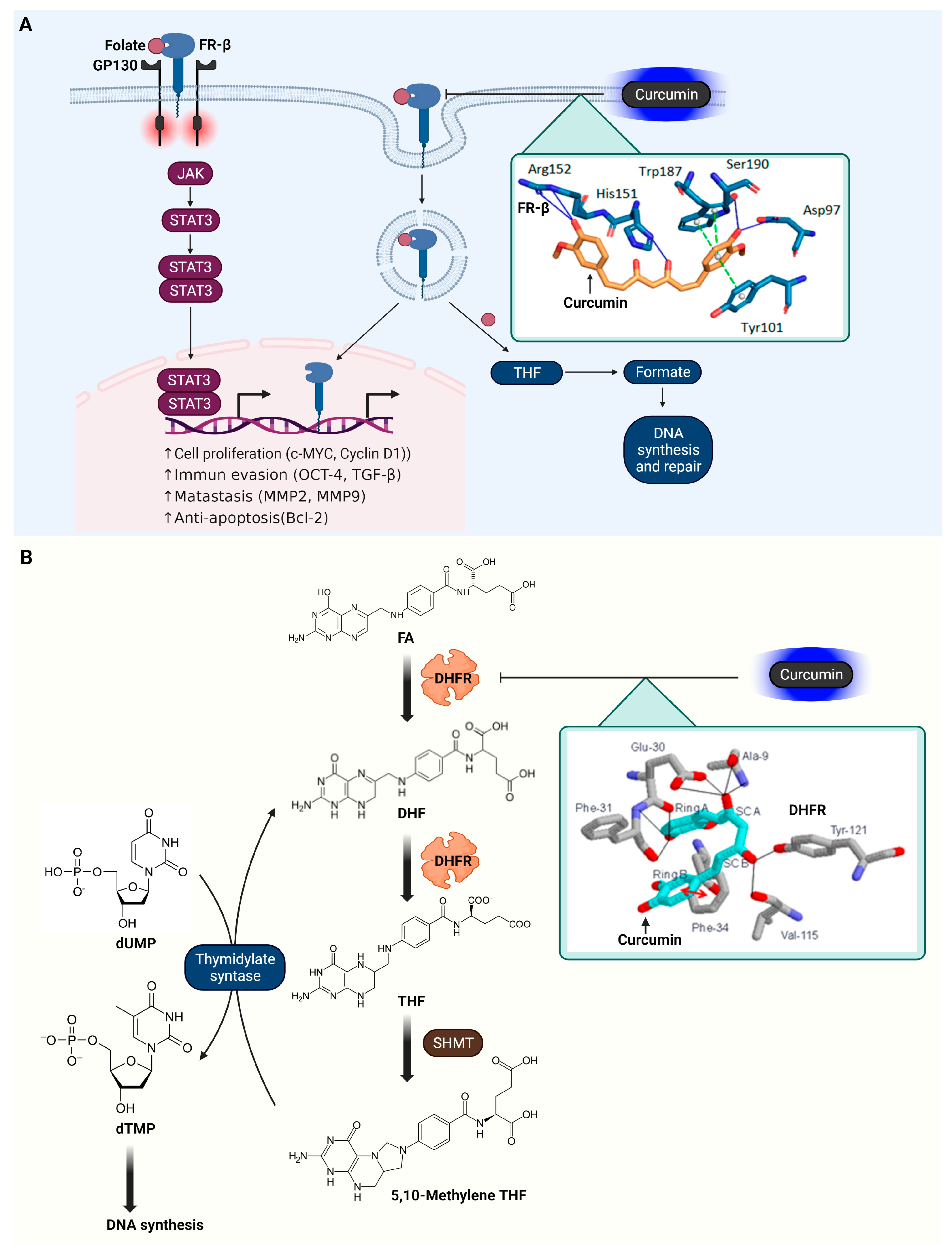
3.7. Folate Receptor β (FR-β)
3.8. Dihydrofolate Reductase (DHFR)
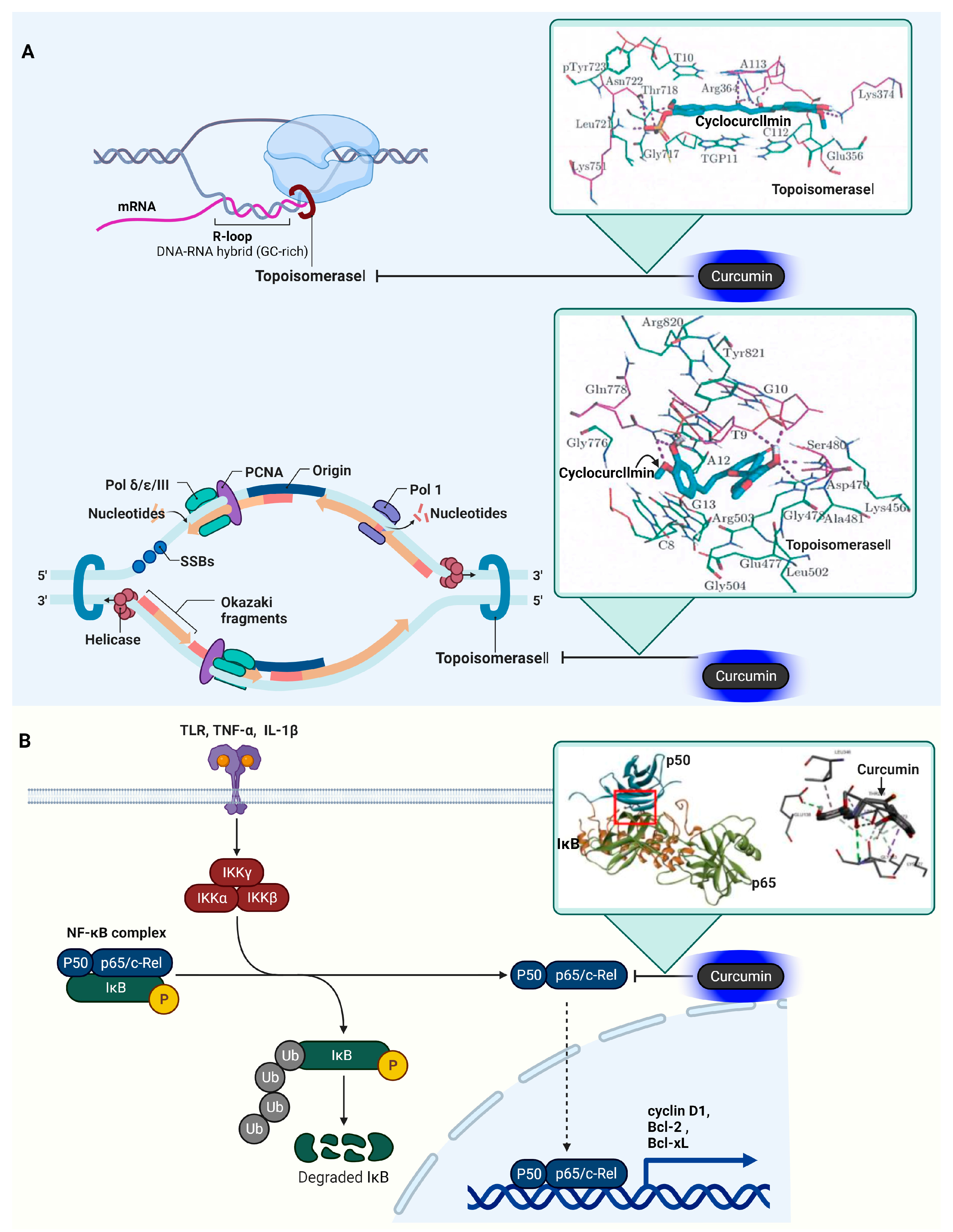
3.9. DNA Topoisomerase I (Topo I) and II (Topo II)
3.10. Nuclear Factor Kappa-Light-Chain-Enhancer of Activated B Cells (NF-κB)
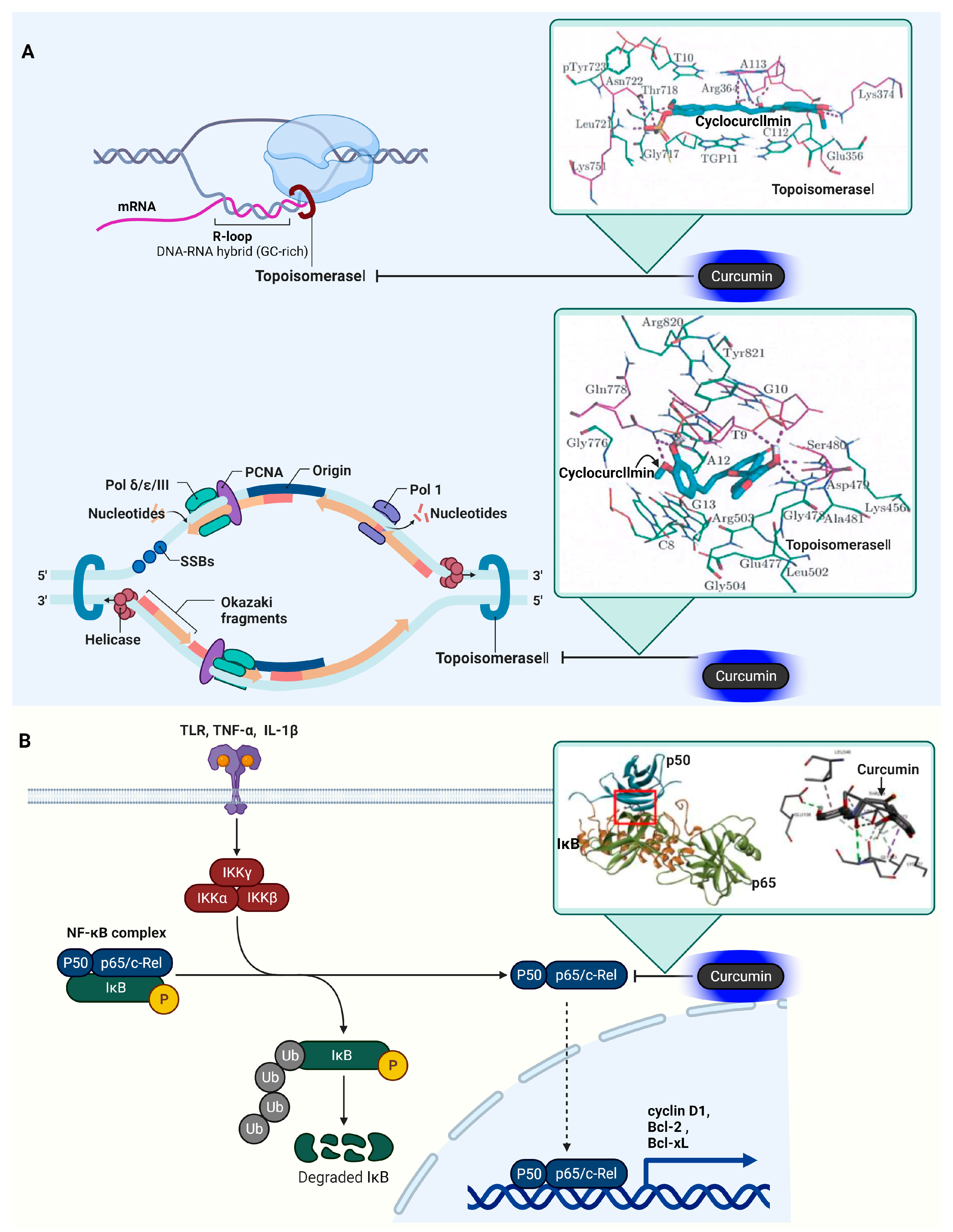
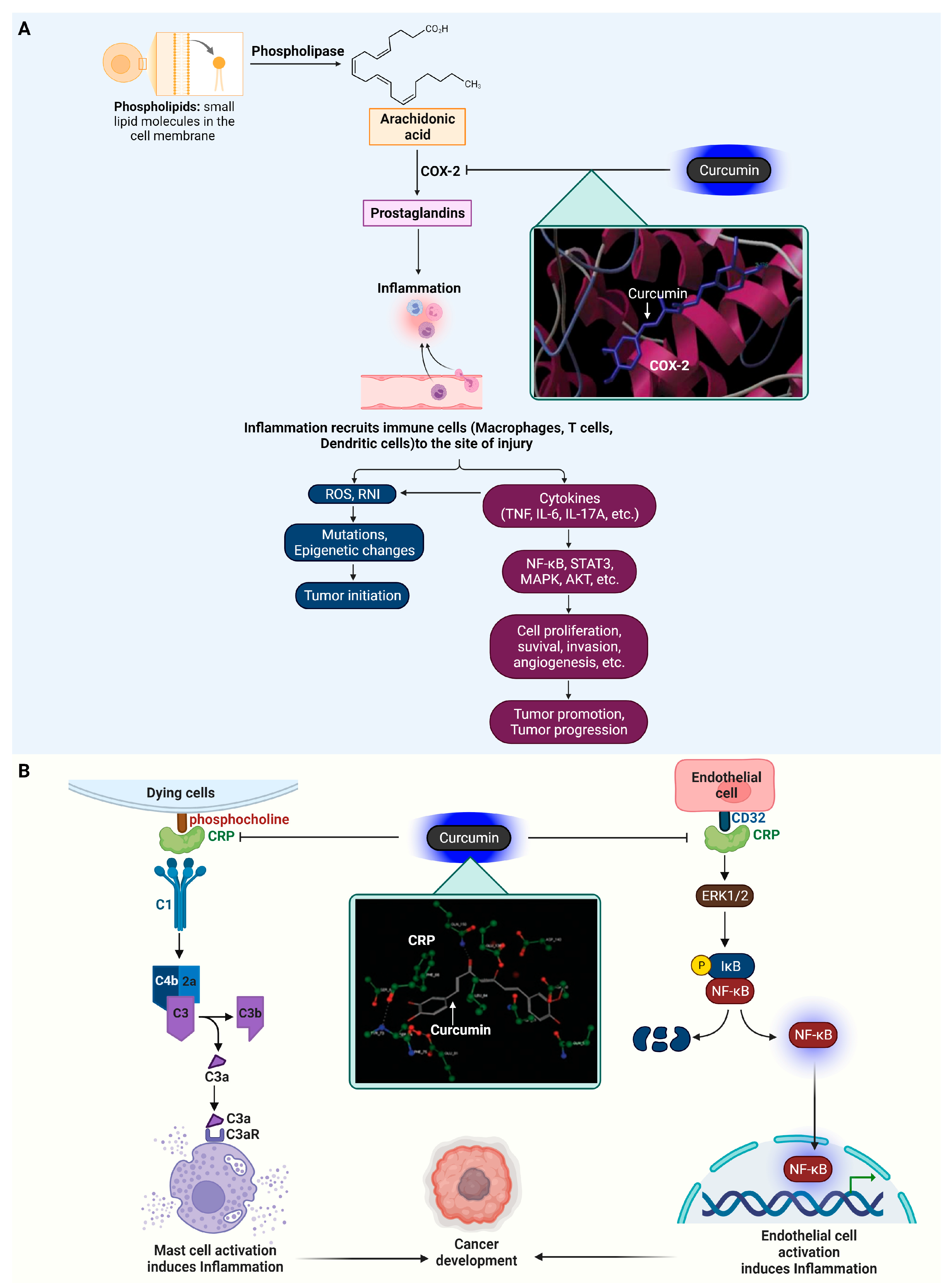
4. Curcumin Target Proteins in Inflammation
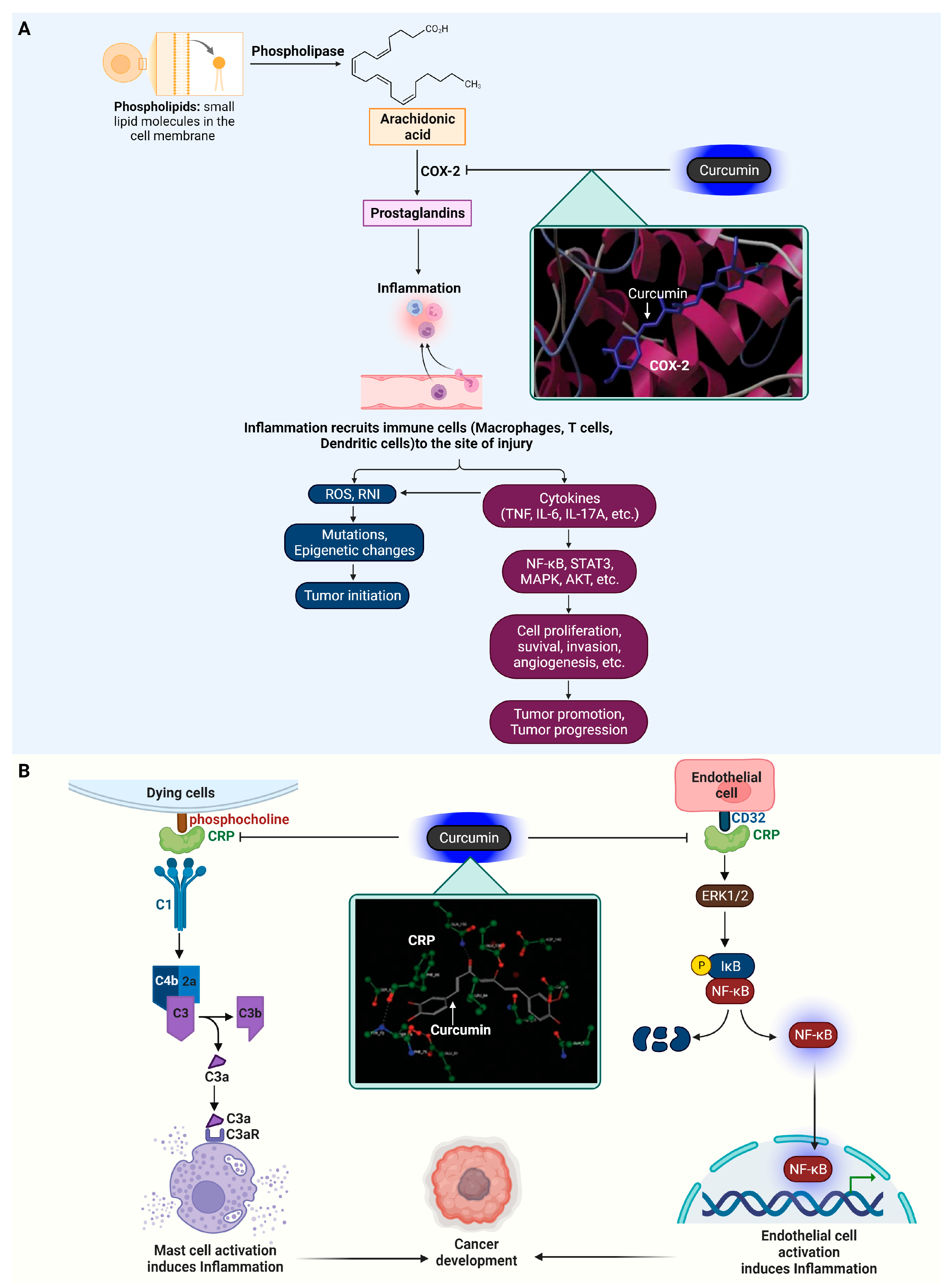
4.1. Cyclooxygenase-2 (COX-2)
4.2. C-Reactive Protein (CRP)
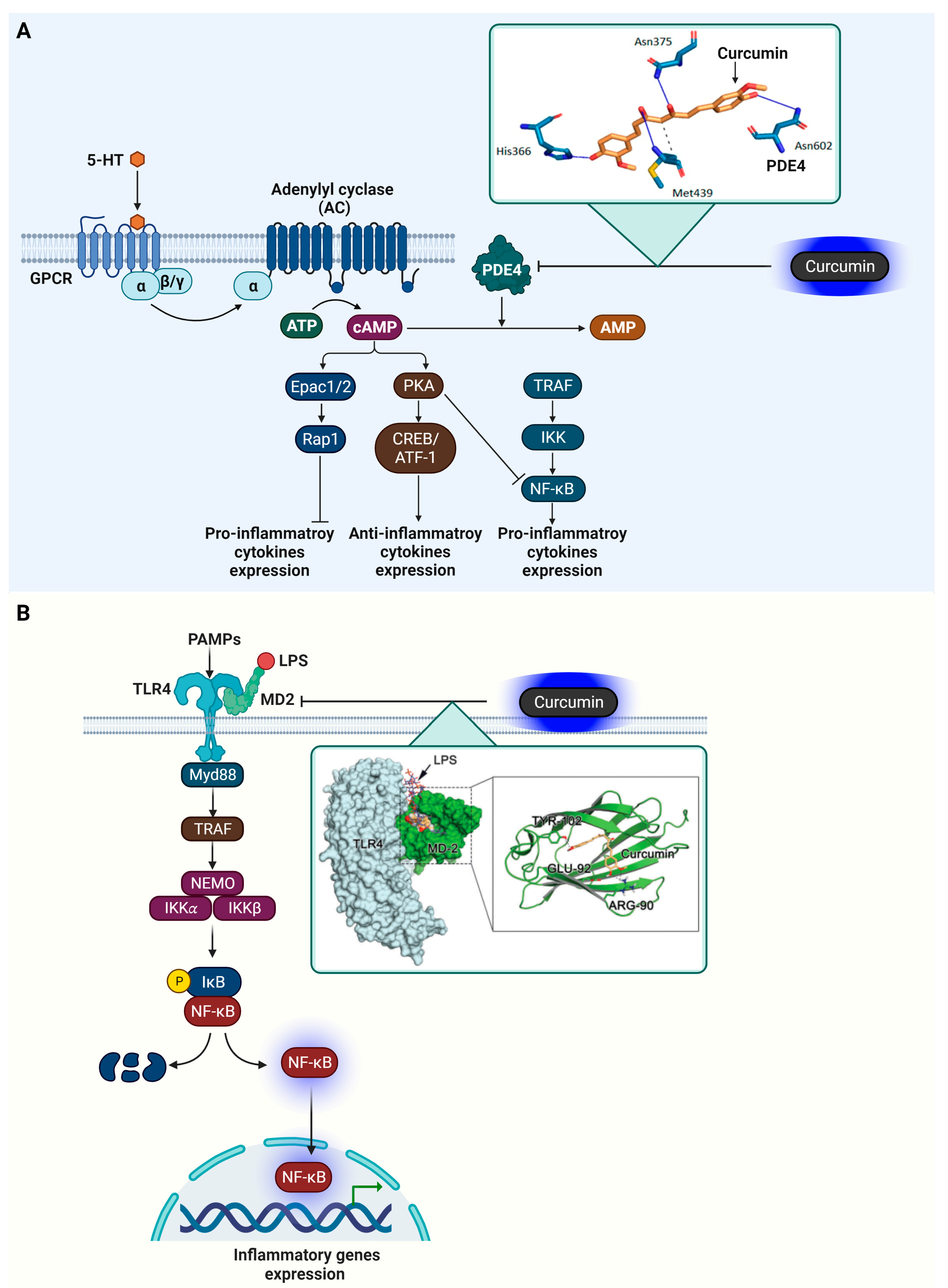
4.3. Phosphodiesterase 4 (PDE4)
4.4. Myeloid Differentiation Protein 2 (MD-2)
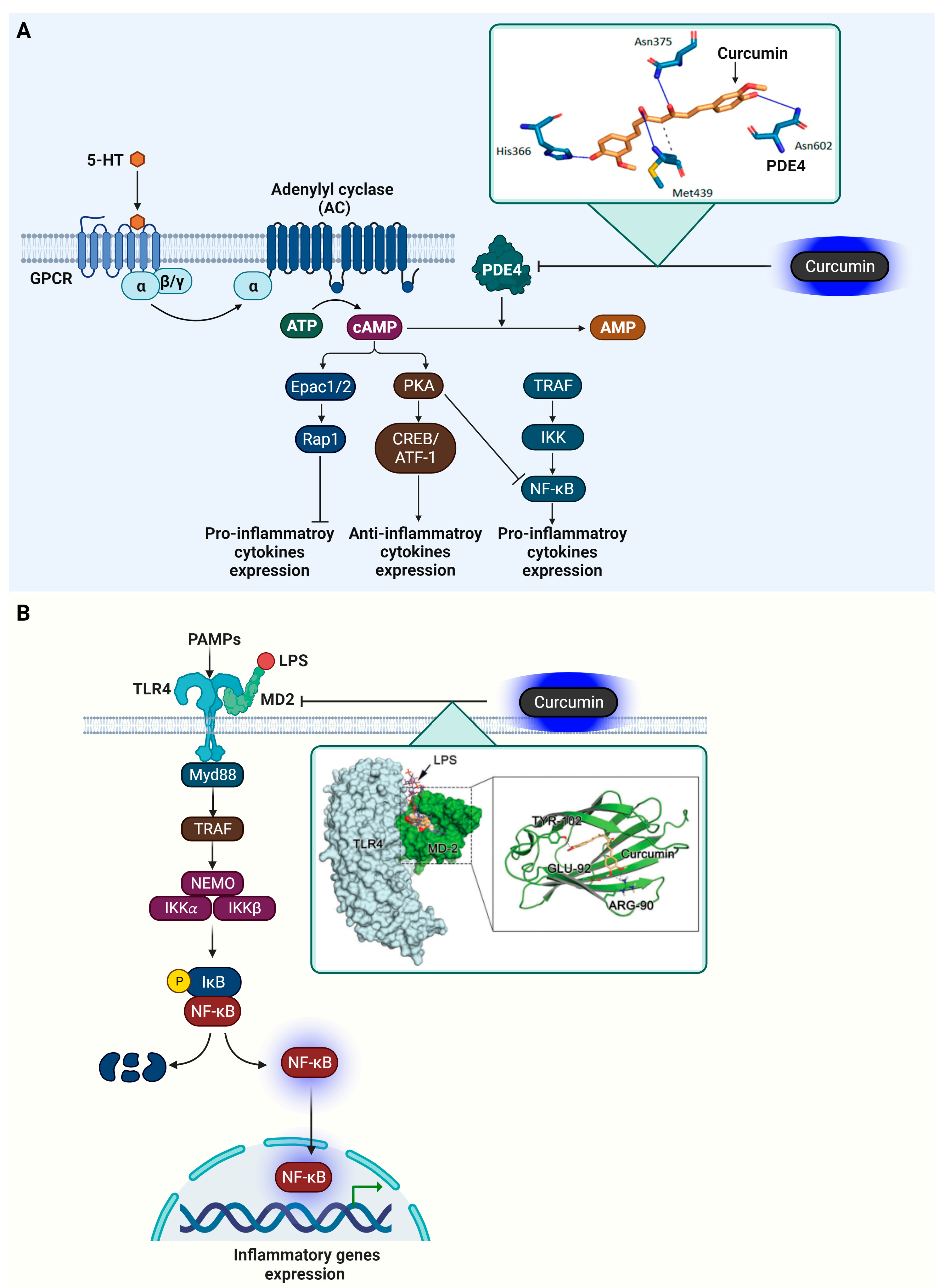
5. In Vitro Experiment Results of Curcumin Target Protein
6. Clinical Studies of Curcumin
6.1. Colorectal Cancer
6.2. Myeloid Leukemia (CML)
6.3. Prostate Cancer
6.4. Head and Neck Cancer
6.5. Multiple Myeloma
7. Conclusions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Joshi, P.; Verma, K.; Semwal, D.K.; Dwivedi, J.; Sharma, S. Mechanism insights of curcumin and its analogues in cancer: An update. Phytother. Res. 2023, 37, 5435–5463. [Google Scholar] [CrossRef] [PubMed]
- Zang, W.B.; Wei, H.L.; Zhang, W.W.; Ma, W.; Li, J.; Yao, Y. Curcumin hybrid molecules for the treatment of Alzheimer’s disease: Structure and pharmacological activities. Eur. J. Med. Chem. 2023, 265, 116070. [Google Scholar] [CrossRef] [PubMed]
- Garodia, P.; Hegde, M.; Kunnumakkara, A.B.; Aggarwal, B.B. Curcumin, inflammation, and neurological disorders: How are they linked? Integr. Med. Res. 2023, 12, 100968. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Li, Z.; Huang, Y.; Xu, Y.; Zou, B. Nanotechnology and curcumin: A novel and promising approach in digestive cancer therapy. Nanomedicine 2023, 18, 2081–2099. [Google Scholar] [CrossRef] [PubMed]
- Tang, F.; Liu, D.; Zhang, L.; Xu, L.Y.; Zhang, J.N.; Zhao, X.L.; Ao, H.; Peng, C. Targeting endothelial cells with golden spice curcumin: A promising therapy for cardiometabolic multimorbidity. Pharmacol. Res. 2023, 197, 106953. [Google Scholar] [CrossRef] [PubMed]
- Ratan, C.; Arian, A.M.; Rajendran, R.; Jayakumar, R.; Masson, M.; Mangalathillam, S. Nano-based formulations of curcumin: Elucidating the potential benefits and future prospects in skin cancer. Biomed. Mater. 2023, 18, 052008. [Google Scholar] [CrossRef] [PubMed]
- Ansari, L.; Mashayekhi-Sardoo, H.; Rahimi, V.B.; Yahyazadeh, R.; Ghayour-Mobarhan, M.; Askari, V.R. Curcumin-based nanoformulations alleviate wounds and related disorders: A comprehensive review. Biofactors 2023, 49, 736–781. [Google Scholar] [CrossRef]
- Grajales, D.B.; Kar, S. Exploring Monkeypox: Prospects for therapeutics through computational-aided drug discovery. Mol. Divers. 2023. [Google Scholar] [CrossRef]
- Kaur, T.; Madgulkar, A.; Bhalekar, M.; Asgaonkar, K. Molecular Docking in Formulation and Development. Curr. Drug Discov. Technol. 2019, 16, 30–39. [Google Scholar] [CrossRef]
- Parameswari, A.R.; Rajalakshmi, G.; Kumaradhas, P. A combined molecular docking and charge density analysis is a new approach for medicinal research to understand drug-receptor interaction: Curcumin-AChE model. Chem. Biol. Interact. 2015, 225, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Kumar, D.; Shri, R.; Kumar, S. Mechanistic Insights and Docking Studies of Phytomolecules as Potential Candidates in the Management of Cancer. Curr. Pharm. Des. 2022, 28, 2704–2724. [Google Scholar] [CrossRef]
- Li, J.; Fu, A.; Zhang, L. An Overview of Scoring Functions Used for Protein-Ligand Interactions in Molecular Docking. Interdiscip. Sci. 2019, 11, 320–328. [Google Scholar] [CrossRef]
- Anand, P.; Kunnumakkara, A.B.; Newman, R.A.; Aggarwal, B.B. Bioavailability of curcumin: Problems and promises. Mol. Pharm. 2007, 4, 807–818. [Google Scholar] [CrossRef] [PubMed]
- Ipar, V.S.; Dsouza, A.; Devarajan, P.V. Enhancing Curcumin Oral Bioavailability through Nanoformulations. Eur. J. Drug Metab. Pharmacokinet. 2019, 44, 459–480. [Google Scholar] [CrossRef] [PubMed]
- Tadesse, S.; Caldon, E.C.; Tilley, W.; Wang, S. Cyclin-Dependent Kinase 2 Inhibitors in Cancer Therapy: An Update. J. Med. Chem. 2019, 62, 4233–4251. [Google Scholar] [CrossRef] [PubMed]
- Chohan, T.A.; Qian, H.; Pan, Y.; Chen, J.Z. Cyclin-dependent kinase-2 as a target for cancer therapy: Progress in the development of CDK2 inhibitors as anti-cancer agents. Curr. Med. Chem. 2015, 22, 237–263. [Google Scholar] [CrossRef] [PubMed]
- Chu, C.; Geng, Y.; Zhou, Y.; Sicinski, P. Cyclin E in normal physiology and disease states. Trends Cell Biol. 2021, 31, 732–746. [Google Scholar] [CrossRef]
- Tadesse, S.; Anshabo, A.T.; Portman, N.; Lim, E.; Tilley, W.; Caldon, C.E.; Wang, S. Targeting CDK2 in cancer: Challenges and opportunities for therapy. Drug Discov. Today 2020, 25, 406–413. [Google Scholar] [CrossRef] [PubMed]
- Volkart, P.A.; Bitencourt-Ferreira, G.; Souto, A.A.; de Azevedo, W.F. Cyclin-Dependent Kinase 2 in Cellular Senescence and Cancer. A Structural and Functional Review. Curr. Drug Targets 2019, 20, 716–726. [Google Scholar] [CrossRef]
- Patel, D.A.; Patel, S.S.; Patel, H.D. Advances in synthesis and biological evaluation of CDK2 inhibitors for cancer therapy. Bioorg Chem. 2023, 143, 107045. [Google Scholar] [CrossRef]
- Sumirtanurdin, R.; Sungkar, S.; Hisprastin, Y.; Sidharta, K.D.; Nurhikmah, D.D. Molecular Docking Simulation Studies of Curcumin and Its Derivatives as Cyclin-Dependent Kinase 2 Inhibitors. Turk. J. Pharm. Sci. 2020, 17, 417–423. [Google Scholar] [CrossRef]
- Pinna, L.A.; Meggio, F. Protein kinase CK2 (“casein kinase-2”) and its implication in cell division and proliferation. Prog. Cell Cycle Res. 1997, 3, 77–97. [Google Scholar] [CrossRef] [PubMed]
- Guerra, B.; Issinger, O.G. Protein kinase CK2 and its role in cellular proliferation, development and pathology. Electrophoresis 1999, 20, 391–408. [Google Scholar] [CrossRef]
- Lee, S.W.; Song, Y.S.; Lee, S.Y.; Yoon, Y.G.; Lee, S.H.; Park, B.S.; Yun, I.; Choi, H.; Kim, K.; Chung, W.T.; et al. Downregulation of protein kinase CK2 activity facilitates tumor necrosis factor-α-mediated chondrocyte death through apoptosis and autophagy. PLoS ONE 2011, 6, e19163. [Google Scholar] [CrossRef] [PubMed]
- Ersahin, T.; Tuncbag, N.; Cetin-Atalay, R. The PI3K/AKT/mTOR interactive pathway. Mol. Biosyst. 2015, 11, 1946–1954. [Google Scholar] [CrossRef] [PubMed]
- Porta, C.; Paglino, C.; Mosca, A. Targeting PI3K/Akt/mTOR Signaling in Cancer. Front. Oncol. 2014, 4, 64. [Google Scholar] [CrossRef] [PubMed]
- LoRusso, P.M. Inhibition of the PI3K/AKT/mTOR Pathway in Solid Tumors. J. Clin. Oncol. 2016, 34, 3803–3815. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Chen, Y.; Liu, G.; Li, C.; Song, Y.; Cao, Z.; Li, W.; Hu, J.; Lu, C.; Liu, Y. PI3K/AKT pathway as a key link modulates the multidrug resistance of cancers. Cell Death Dis. 2020, 11, 797. [Google Scholar] [CrossRef] [PubMed]
- Guerra, B. Protein kinase CK2 subunits are positive regulators of AKT kinase. Int. J. Oncol. 2006, 28, 685–693. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-κB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef]
- Taniguchi, K.; Karin, M. NF-κB, inflammation, immunity and cancer: Coming of age. Nat. Rev. Immunol. 2018, 18, 309–324. [Google Scholar] [CrossRef]
- Wang, D.; Westerheide, S.D.; Hanson, J.L.; Baldwin, A.S., Jr. Tumor necrosis factor alpha-induced phosphorylation of RelA/p65 on Ser529 is controlled by casein kinase II. J. Biol. Chem. 2000, 275, 32592–32597. [Google Scholar] [CrossRef]
- Seif, F.; Khoshmirsafa, M.; Aazami, H.; Mohsenzadegan, M.; Sedighi, G.; Bahar, M. The role of JAK-STAT signaling pathway and its regulators in the fate of T helper cells. Cell Commun. Signal 2017, 15, 23. [Google Scholar] [CrossRef] [PubMed]
- Villarino, A.V.; Kanno, Y.; O’Shea, J.J. Mechanisms and consequences of Jak-STAT signaling in the immune system. Nat. Immunol. 2017, 18, 374–384. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Qin, H.; Frank, S.J.; Deng, L.; Litchfield, D.W.; Tefferi, A.; Pardanani, A.; Lin, F.T.; Li, J.; Sha, B.; et al. A CK2-dependent mechanism for activation of the JAK-STAT signaling pathway. Blood 2011, 118, 156–166. [Google Scholar] [CrossRef] [PubMed]
- Cozza, G.; Zonta, F.; Vedove, A.D.; Venerando, A.; Dall’Acqua, S.; Battistutta, R.; Ruzzene, M.; Lolli, G. Biochemical and cellular mechanism of protein kinase CK2 inhibition by deceptive curcumin. FEBS J. 2020, 287, 1850–1864. [Google Scholar] [CrossRef] [PubMed]
- Takahashi-Yanaga, F.; Sasaguri, T. GSK-3beta regulates cyclin D1 expression: A new target for chemotherapy. Cell Signal 2008, 20, 581–589. [Google Scholar] [CrossRef]
- Lin, J.; Song, T.; Li, C.; Mao, W. GSK-3β in DNA repair, apoptosis, and resistance of chemotherapy, radiotherapy of cancer. Biochim. Biophys. Acta Mol. Cell Res. 2020, 1867, 118659. [Google Scholar] [CrossRef]
- Song, M.; Bode, A.M.; Dong, Z.; Lee, M.H. AKT as a Therapeutic Target for Cancer. Cancer Res. 2019, 79, 1019–1031. [Google Scholar] [CrossRef] [PubMed]
- Vallée, A.; Lecarpentier, Y. Crosstalk between Peroxisome Proliferator-Activated Receptor Gamma and the Canonical WNT/β-Catenin Pathway in Chronic Inflammation and Oxidative Stress during Carcinogenesis. Front. Immunol. 2018, 9, 745. [Google Scholar] [CrossRef]
- Manoukian, A.S.; Woodgett, J.R. Role of glycogen synthase kinase-3 in cancer: Regulation by Wnts and other signaling pathways. Adv. Cancer Res. 2002, 84, 203–229. [Google Scholar] [CrossRef] [PubMed]
- Valvezan, A.J.; Zhang, F.; Diehl, J.A.; Klein, P.S. Adenomatous polyposis coli (APC) regulates multiple signaling pathways by enhancing glycogen synthase kinase-3 (GSK-3) activity. J. Biol. Chem. 2012, 287, 3823–3832. [Google Scholar] [CrossRef]
- Peifer, M.; Polakis, P. Wnt signaling in oncogenesis and embryogenesis—A look outside the nucleus. Science 2000, 287, 1606–1609. [Google Scholar] [CrossRef] [PubMed]
- Lustig, B.; Behrens, J. The Wnt signaling pathway and its role in tumor development. J. Cancer Res. Clin. Oncol. 2003, 129, 199–221. [Google Scholar] [CrossRef]
- Giles, R.H.; van Es, J.H.; Clevers, H. Caught up in a Wnt storm: Wnt signaling in cancer. Biochim. Biophys. Acta 2003, 1653, 1–24. [Google Scholar] [CrossRef]
- Hoeflich, K.P.; Luo, J.; Rubie, E.A.; Tsao, M.S.; Jin, O.; Woodgett, J.R. Requirement for glycogen synthase kinase-3beta in cell survival and NF-kappaB activation. Nature 2000, 406, 86–90. [Google Scholar] [CrossRef]
- Schwabe, R.F.; Brenner, D.A. Role of glycogen synthase kinase-3 in TNF-alpha-induced NF-kappaB activation and apoptosis in hepatocytes. Am. J. Physiol. Gastrointest. Liver Physiol. 2002, 283, G204–G211. [Google Scholar] [CrossRef]
- Pan, J.; Seeger-Nukpezah, T.; Golemis, E.A. The role of the cilium in normal and abnormal cell cycles: Emphasis on renal cystic pathologies. Cell Mol. Life Sci. 2013, 70, 1849–1874. [Google Scholar] [CrossRef] [PubMed]
- Bustanji, Y.; Taha, M.O.; Almasri, I.M.; Al-Ghussein, M.A.; Mohammad, M.K.; Alkhatib, H.S. Inhibition of glycogen synthase kinase by curcumin: Investigation by simulated molecular docking and subsequent in vitro/in vivo evaluation. J. Enzyme Inhib. Med. Chem. 2009, 24, 771–778. [Google Scholar] [CrossRef] [PubMed]
- Correa-Sáez, A.; Jiménez-Izquierdo, R.; Garrido-Rodríguez, M.; Morrugares, R.; Muñoz, E.; Calzado, M.A. Updating dual-specificity tyrosine-phosphorylation-regulated kinase 2 (DYRK2): Molecular basis, functions and role in diseases. Cell Mol. Life Sci. 2020, 77, 4747–4763. [Google Scholar] [CrossRef] [PubMed]
- Ito, D.; Yogosawa, S.; Mimoto, R.; Hirooka, S.; Horiuchi, T.; Eto, K.; Yanaga, K.; Yoshida, K. Dual-specificity tyrosine-regulated kinase 2 is a suppressor and potential prognostic marker for liver metastasis of colorectal cancer. Cancer Sci. 2017, 108, 1565–1573. [Google Scholar] [CrossRef]
- Morrugares, R.; Correa-Sáez, A.; Moreno, R.; Garrido-Rodríguez, M.; Muñoz, E.; de la Vega, L.; Calzado, M.A. Phosphorylation-dependent regulation of the NOTCH1 intracellular domain by dual-specificity tyrosine-regulated kinase 2. Cell Mol. Life Sci. 2020, 77, 2621–2639. [Google Scholar] [CrossRef] [PubMed]
- Takano, Y.; Yogosawa, S.; Imaizumi, Y.; Kamioka, H.; Kanegae, Y.; Eto, K.; Yoshida, K. DYRK2 promotes chemosensitivity via p53-mediated apoptosis after DNA damage in colorectal cancer. Cancer Sci. 2023, 114, 4558–4570. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Wang, X.; Wang, Z.; Banerjee, S.; Yang, J.; Huang, L.; Dixon, J.E. Site-specific proteasome phosphorylation controls cell proliferation and tumorigenesis. Nat. Cell Biol. 2016, 18, 202–212. [Google Scholar] [CrossRef] [PubMed]
- Moreno, R.; Banerjee, S.; Jackson, A.W.; Quinn, J.; Baillie, G.; Dixon, J.E.; Dinkova-Kostova, A.T.; Edwards, J.; de la Vega, L. The stress-responsive kinase DYRK2 activates heat shock factor 1 promoting resistance to proteotoxic stress. Cell Death Differ. 2021, 28, 1563–1578. [Google Scholar] [CrossRef]
- Banerjee, S.; Wei, T.; Wang, J.; Lee, J.J.; Gutierrez, H.L.; Chapman, O.; Wiley, S.E.; Mayfield, J.E.; Tandon, V.; Juarez, E.F.; et al. Inhibition of dual-specificity tyrosine phosphorylation-regulated kinase 2 perturbs 26S proteasome-addicted neoplastic progression. Proc. Natl. Acad. Sci. USA 2019, 116, 24881–24891. [Google Scholar] [CrossRef] [PubMed]
- Matsuura, K.; Huang, N.J.; Cocce, K.; Zhang, L.; Kornbluth, S. Downregulation of the proapoptotic protein MOAP-1 by the UBR5 ubiquitin ligase and its role in ovarian cancer resistance to cisplatin. Oncogene 2017, 36, 1698–1706. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; Ji, C.; Mayfield, J.E.; Goel, A.; Xiao, J.; Dixon, J.E.; Guo, X. Ancient drug curcumin impedes 26S proteasome activity by direct inhibition of dual-specificity tyrosine-regulated kinase 2. Proc. Natl. Acad. Sci. USA 2018, 115, 8155–8160. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.; Carpenter, G.; Coffey, R.J. EGF receptor ligands: Recent advances. F1000Research 2016, 5, 2270. [Google Scholar] [CrossRef]
- Wang, Z. ErbB Receptors and Cancer. Methods Mol. Biol. 2017, 1652, 3–35. [Google Scholar] [CrossRef]
- Tomas, A.; Futter, C.E.; Eden, E.R. EGF receptor trafficking: Consequences for signaling and cancer. Trends Cell Biol. 2014, 24, 26–34. [Google Scholar] [CrossRef]
- Wang, C.; He, Q.; Yin, Y.; Wu, Y.; Li, X. Clonorchis sinensis Granulin Promotes Malignant Transformation of Hepatocyte Through EGFR-Mediated RAS/MAPK/ERK and PI3K/Akt Signaling Pathways. Front. Cell Infect. Microbiol. 2021, 11, 734750. [Google Scholar] [CrossRef]
- Al Moustafa, A.E.; Achkhar, A.; Yasmeen, A. EGF-receptor signaling and epithelial-mesenchymal transition in human carcinomas. Front. Biosci. (Schol. Ed.) 2012, 4, 671–684. [Google Scholar] [CrossRef]
- García-Hernández, L.; García-Ortega, M.B.; Ruiz-Alcalá, G.; Carrillo, E.; Marchal, J.A.; García, M. The p38 MAPK Components and Modulators as Biomarkers and Molecular Targets in Cancer. Int. J. Mol. Sci. 2021, 23, 370. [Google Scholar] [CrossRef] [PubMed]
- Singh, D.; Attri, B.K.; Gill, R.K.; Bariwal, J. Review on EGFR Inhibitors: Critical Updates. Mini Rev. Med. Chem. 2016, 16, 1134–1166. [Google Scholar] [CrossRef] [PubMed]
- Ali, A.; Ali, A.; Tahir, A.; Bakht, M.A.; Salahuddin; Ahsan, M.J. Molecular Engineering of Curcumin, an Active Constituent of Curcuma longa L. (Turmeric) of the Family Zingiberaceae with Improved Antiproliferative Activity. Plants 2021, 10, 1559. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Wei, Y.; Wei, X. AXL receptor tyrosine kinase as a promising anti-cancer approach: Functions, molecular mechanisms and clinical applications. Mol. Cancer 2019, 18, 153. [Google Scholar] [CrossRef] [PubMed]
- Paccez, J.D.; Vogelsang, M.; Parker, M.I.; Zerbini, L.F. The receptor tyrosine kinase Axl in cancer: Biological functions and therapeutic implications. Int. J. Cancer 2014, 134, 1024–1033. [Google Scholar] [CrossRef] [PubMed]
- Nagata, K.; Ohashi, K.; Nakano, T.; Arita, H.; Zong, C.; Hanafusa, H.; Mizuno, K. Identification of the product of growth arrest-specific gene 6 as a common ligand for Axl, Sky, and Mer receptor tyrosine kinases. J. Biol. Chem. 1996, 271, 30022–30027. [Google Scholar] [CrossRef]
- Tanaka, M.; Siemann, D.W. Gas6/Axl Signaling Pathway in the Tumor Immune Microenvironment. Cancers 2020, 12, 1850. [Google Scholar] [CrossRef]
- Stenhoff, J.; Dahlbäck, B.; Hafizi, S. Vitamin K-dependent Gas6 activates ERK kinase and stimulates growth of cardiac fibroblasts. Biochem. Biophys. Res. Commun. 2004, 319, 871–878. [Google Scholar] [CrossRef] [PubMed]
- Geng, K.; Kumar, S.; Kimani, S.G.; Kholodovych, V.; Kasikara, C.; Mizuno, K.; Sandiford, O.; Rameshwar, P.; Kotenko, S.V.; Birge, R.B. Requirement of Gamma-Carboxyglutamic Acid Modification and Phosphatidylserine Binding for the Activation of Tyro3, Axl, and Mertk Receptors by Growth Arrest-Specific 6. Front. Immunol. 2017, 8, 1521. [Google Scholar] [CrossRef]
- Axelrod, H.; Pienta, K.J. Axl as a mediator of cellular growth and survival. Oncotarget 2014, 5, 8818–8852. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Siemann, D.W. Therapeutic Targeting of the Gas6/Axl Signaling Pathway in Cancer. Int. J. Mol. Sci. 2021, 22, 9953. [Google Scholar] [CrossRef] [PubMed]
- Schoumacher, M.; Burbridge, M. Key Roles of AXL and MER Receptor Tyrosine Kinases in Resistance to Multiple Anticancer Therapies. Curr. Oncol. Rep. 2017, 19, 19. [Google Scholar] [CrossRef] [PubMed]
- Engelsen, A.S.T.; Lotsberg, M.L.; Khouzam, R.A.; Thiery, J.P.; Lorens, J.B.; Chouaib, S.; Terry, S. Dissecting the Role of AXL in Cancer Immune Escape and Resistance to Immune Checkpoint Inhibition. Front. Immunol. 2022, 13, 869676. [Google Scholar] [CrossRef] [PubMed]
- Tai, K.Y.; Shieh, Y.S.; Lee, C.S.; Shiah, S.G.; Wu, C.W. Axl promotes cell invasion by inducing MMP-9 activity through activation of NF-kappaB and Brg-1. Oncogene 2008, 27, 4044–4055. [Google Scholar] [CrossRef] [PubMed]
- Yeh, C.Y.; Shin, S.M.; Yeh, H.H.; Wu, T.J.; Shin, J.W.; Chang, T.Y.; Raghavaraju, G.; Lee, C.T.; Chiang, J.H.; Tseng, V.S.; et al. Transcriptional activation of the Axl and PDGFR-α by c-Met through a ras- and Src-independent mechanism in human bladder cancer. BMC Cancer 2011, 11, 139. [Google Scholar] [CrossRef]
- Antony, J.; Zanini, E.; Kelly, Z.; Tan, T.Z.; Karali, E.; Alomary, M.; Jung, Y.; Nixon, K.; Cunnea, P.; Fotopoulou, C.; et al. The tumour suppressor OPCML promotes AXL inactivation by the phosphatase PTPRG in ovarian cancer. EMBO Rep. 2018, 19, e45670. [Google Scholar] [CrossRef]
- Pei, J.P.; Wang, Y.; Ma, L.P.; Wang, X.; Liu, L.; Zhang, Y.; Jin, R.; Ren, Z.Q.; Deng, Y.; Shen, J.K.; et al. AXL antibody and AXL-ADC mediate antitumor efficacy via targeting AXL in tumor-intrinsic epithelial-mesenchymal transition and tumor-associated M2-like macrophage. Acta Pharmacol. Sin. 2023, 44, 1290–1303. [Google Scholar] [CrossRef]
- Asiedu, M.K.; Beauchamp-Perez, F.D.; Ingle, J.N.; Behrens, M.D.; Radisky, D.C.; Knutson, K.L. AXL induces epithelial-to-mesenchymal transition and regulates the function of breast cancer stem cells. Oncogene 2014, 33, 1316–1324. [Google Scholar] [CrossRef]
- Ghrifi, F.; Allam, L.; Wiame, L.; Ibrahimi, A. Curcumin-Synthetic Analogs Library Screening by Docking and Quantitative Structure-Activity Relationship Studies for AXL Tyrosine Kinase Inhibition in Cancers. J. Comput. Biol. 2019, 26, 1156–1167. [Google Scholar] [CrossRef] [PubMed]
- Yi, Y.S. Folate Receptor-Targeted Diagnostics and Therapeutics for Inflammatory Diseases. Immune Netw. 2016, 16, 337–343. [Google Scholar] [CrossRef] [PubMed]
- Minami, K.; Hiwatashi, K.; Ueno, S.; Sakoda, M.; Iino, S.; Okumura, H.; Hashiguchi, M.; Kawasaki, Y.; Kurahara, H.; Mataki, Y.; et al. Prognostic significance of CD68, CD163 and Folate receptor-β positive macrophages in hepatocellular carcinoma. Exp. Ther. Med. 2018, 15, 4465–4476. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Li, H.; Lee, R.J. Targeted drug delivery via folate receptors. Expert. Opin. Drug Deliv. 2008, 5, 309–319. [Google Scholar] [CrossRef]
- Nagai, T.; Tanaka, M.; Tsuneyoshi, Y.; Xu, B.; Michie, S.A.; Hasui, K.; Hirano, H.; Arita, K.; Matsuyama, T. Targeting tumor-associated macrophages in an experimental glioma model with a recombinant immunotoxin to folate receptor beta. Cancer Immunol. Immunother. 2009, 58, 1577–1586. [Google Scholar] [CrossRef]
- Frigerio, B.; Bizzoni, C.; Jansen, G.; Leamon, C.P.; Peters, G.J.; Low, P.S.; Matherly, L.H.; Figini, M. Folate receptors and transporters: Biological role and diagnostic/therapeutic targets in cancer and other diseases. J. Exp. Clin. Cancer Res. 2019, 38, 125. [Google Scholar] [CrossRef]
- Furlan, V.; Konc, J.; Bren, U. Inverse Molecular Docking as a Novel Approach to Study Anticarcinogenic and Anti-Neuroinflammatory Effects of Curcumin. Molecules 2018, 23, 3351. [Google Scholar] [CrossRef]
- Abali, E.E.; Skacel, N.E.; Celikkaya, H.; Hsieh, Y.C. Regulation of human dihydrofolate reductase activity and expression. Vitam. Horm. 2008, 79, 267–292. [Google Scholar] [CrossRef]
- Schweitzer, B.I.; Dicker, A.P.; Bertino, J.R. Dihydrofolate reductase as a therapeutic target. FASEB J. 1990, 4, 2441–2452. [Google Scholar] [CrossRef]
- Raimondi, M.V.; Randazzo, O.; La Franca, M.; Barone, G.; Vignoni, E.; Rossi, D.; Collina, S. DHFR Inhibitors: Reading the Past for Discovering Novel Anticancer Agents. Molecules 2019, 24, 1140. [Google Scholar] [CrossRef]
- Bertino, J.R. Cancer research: From folate antagonism to molecular targets. Best. Pract. Res. Clin. Haematol. 2009, 22, 577–582. [Google Scholar] [CrossRef]
- Bhagat, K.; Kumar, N.; Gulati, H.K.; Sharma, A.; Kaur, A.; Singh, J.V.; Singh, H.; Bedi, P.M.S. Dihydrofolate reductase inhibitors: Patent landscape and phases of clinical development (2001–2021). Expert Opin. Ther. Pat. 2022, 32, 1079–1095. [Google Scholar] [CrossRef] [PubMed]
- Hobani, Y.; Jerah, A.; Bidwai, A. A comparative molecular docking study of curcumin and methotrexate to dihydrofolate reductase. Bioinformation 2017, 13, 63–66. [Google Scholar] [CrossRef] [PubMed]
- Gokduman, K. Strategies Targeting DNA Topoisomerase I in Cancer Chemotherapy: Camptothecins, Nanocarriers for Camptothecins, Organic Non-Camptothecin Compounds and Metal Complexes. Curr. Drug Targets 2016, 17, 1928–1939. [Google Scholar] [CrossRef]
- Arun, B.; Frenkel, E.P. Topoisomerase I inhibition with topotecan: Pharmacologic and clinical issues. Expert. Opin. Pharmacother. 2001, 2, 491–505. [Google Scholar] [CrossRef]
- Singh, S.; Pandey, V.P.; Yadav, K.; Yadav, A.; Dwivedi, U.N. Natural Products as Anti-Cancerous Therapeutic Molecules Targeted towards Topoisomerases. Curr. Protein Pept. Sci. 2020, 21, 1103–1142. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, E.L.; Osheroff, N. Etoposide, topoisomerase II and cancer. Curr. Med. Chem. Anticancer. Agents 2005, 5, 363–372. [Google Scholar] [CrossRef]
- Ma, Y.; North, B.J.; Shu, J. Regulation of topoisomerase II stability and activity by ubiquitination and SUMOylation: Clinical implications for cancer chemotherapy. Mol. Biol. Rep. 2021, 48, 6589–6601. [Google Scholar] [CrossRef]
- Elfadadny, A.; Ragab, R.F.; Hamada, R.; Al Jaouni, S.K.; Fu, J.; Mousa, S.A.; El-Far, A.H. Natural bioactive compounds-doxorubicin combinations targeting topoisomerase II-alpha: Anticancer efficacy and safety. Toxicol. Appl. Pharmacol. 2023, 461, 116405. [Google Scholar] [CrossRef]
- Kumar, A.; Bora, U. Molecular docking studies of curcumin natural derivatives with DNA topoisomerase I and II-DNA complexes. Interdiscip. Sci. 2014, 6, 285–291. [Google Scholar] [CrossRef]
- Witzel, I.I.; Koh, L.F.; Perkins, N.D. Regulation of cyclin D1 gene expression. Biochem. Soc. Trans. 2010, 38, 217–222. [Google Scholar] [CrossRef] [PubMed]
- Sevilla, L.; Zaldumbide, A.; Pognonec, P.; Boulukos, K.E. Transcriptional regulation of the bcl-x gene encoding the anti-apoptotic Bcl-xL protein by Ets, Rel/NFkappaB, STAT and AP1 transcription factor families. Histol. Histopathol. 2001, 16, 595–601. [Google Scholar] [CrossRef] [PubMed]
- Al-Khayri, J.M.; Sahana, G.R.; Nagella, P.; Joseph, B.V.; Alessa, F.M.; Al-Mssallem, M.Q. Flavonoids as Potential Anti-Inflammatory Molecules: A Review. Molecules 2022, 27, 2901. [Google Scholar] [CrossRef] [PubMed]
- Karin, M. NF-kappaB and cancer: Mechanisms and targets. Mol. Carcinog. 2006, 45, 355–361. [Google Scholar] [CrossRef] [PubMed]
- Huerta, S.; Heinzerling, J.H.; Anguiano-Hernandez, Y.M.; Huerta-Yepez, S.; Lin, J.; Chen, D.; Bonavida, B.; Livingston, E.H. Modification of gene products involved in resistance to apoptosis in metastatic colon cancer cells: Roles of Fas, Apaf-1, NFkappaB, IAPs, Smac/DIABLO, and AIF. J. Surg. Res. 2007, 142, 184–194. [Google Scholar] [CrossRef] [PubMed]
- Saeed, M.E.M.; Yücer, R.; Dawood, M.; Hegazy, M.F.; Drif, A.; Ooko, E.; Kadioglu, O.; Seo, E.J.; Kamounah, F.S.; Titinchi, S.J.; et al. In Silico and In Vitro Screening of 50 Curcumin Compounds as EGFR and NF-κB Inhibitors. Int. J. Mol. Sci. 2022, 23, 3966. [Google Scholar] [CrossRef] [PubMed]
- Jantarawong, S.; Swangphon, P.; Lauterbach, N.; Panichayupakaranant, P.; Pengjam, Y. Modified Curcuminoid-Rich Extract Liposomal CRE-SDInhibits Osteoclastogenesis via the Canonical NF-κB Signaling Pathway. Pharmaceutics 2023, 15, 2248. [Google Scholar] [CrossRef] [PubMed]
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef]
- Khandia, R.; Munjal, A. Interplay between inflammation and cancer. Adv. Protein Chem. Struct. Biol. 2020, 119, 199–245. [Google Scholar] [CrossRef]
- Guadagni, F.; Ferroni, P.; Palmirotta, R.; Portarena, I.; Formica, V.; Roselli, M. Review. TNF/VEGF cross-talk in chronic inflammation-related cancer initiation and progression: An early target in anticancer therapeutic strategy. In Vivo 2007, 21, 147–161. [Google Scholar] [PubMed]
- Liu, Q.; Li, A.; Tian, Y.; Wu, J.D.; Liu, Y.; Li, T.; Chen, Y.; Han, X.; Wu, K. The CXCL8-CXCR1/2 pathways in cancer. Cytokine Growth Factor. Rev. 2016, 31, 61–71. [Google Scholar] [CrossRef] [PubMed]
- Landskron, G.; De la Fuente, M.; Thuwajit, P.; Thuwajit, C.; Hermoso, M.A. Chronic inflammation and cytokines in the tumor microenvironment. J. Immunol. Res. 2014, 2014, 149185. [Google Scholar] [CrossRef] [PubMed]
- Goswami, K.K.; Bose, A.; Baral, R. Macrophages in tumor: An inflammatory perspective. Clin. Immunol. 2021, 232, 108875. [Google Scholar] [CrossRef] [PubMed]
- Padoan, A.; Plebani, M.; Basso, D. Inflammation and Pancreatic Cancer: Focus on Metabolism, Cytokines, and Immunity. Int. J. Mol. Sci. 2019, 20, 676. [Google Scholar] [CrossRef] [PubMed]
- Műzes, G.; Molnár, B.; Sipos, F. Regulatory T cells in inflammatory bowel diseases and colorectal cancer. World J. Gastroenterol. 2012, 18, 5688–5694. [Google Scholar] [CrossRef]
- Maru, G.B.; Gandhi, K.; Ramchandani, A.; Kumar, G. The role of inflammation in skin cancer. Adv. Exp. Med. Biol. 2014, 816, 437–469. [Google Scholar] [CrossRef]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, inflammation, and cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef]
- Simon, L.S. Role and regulation of cyclooxygenase-2 during inflammation. Am. J. Med. 1999, 106, 37s–42s. [Google Scholar] [CrossRef]
- Yu, T.; Lao, X.; Zheng, H. Influencing COX-2 Activity by COX Related Pathways in Inflammation and Cancer. Mini Rev. Med. Chem. 2016, 16, 1230–1243. [Google Scholar] [CrossRef]
- Becker, R.C. COX-2 inhibitors. Tex. Heart Inst. J. 2005, 32, 380–383. [Google Scholar]
- Rossi, J.F.; Lu, Z.Y.; Massart, C.; Levon, K. Dynamic Immune/Inflammation Precision Medicine: The Good and the Bad Inflammation in Infection and Cancer. Front. Immunol. 2021, 12, 595722. [Google Scholar] [CrossRef] [PubMed]
- Howren, M.B.; Lamkin, D.M.; Suls, J. Associations of depression with C-reactive protein, IL-1, and IL-6: A meta-analysis. Psychosom. Med. 2009, 71, 171–186. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Zhou, L.; Wu, X.; Li, R.; Wen, J.; Sha, J.; Wen, X. The PI3K/AKT pathway in the pathogenesis of prostate cancer. Front. Biosci. (Landmark Ed.) 2016, 21, 1084–1091. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.E.; O’Keefe, R.A.; Grandis, J.R. Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat. Rev. Clin. Oncol. 2018, 15, 234–248. [Google Scholar] [CrossRef]
- Padhye, S.; Banerjee, S.; Chavan, D.; Pandye, S.; Swamy, K.V.; Ali, S.; Li, J.; Dou, Q.P.; Sarkar, F.H. Fluorocurcumins as cyclooxygenase-2 inhibitor: Molecular docking, pharmacokinetics and tissue distribution in mice. Pharm. Res. 2009, 26, 2438–2445. [Google Scholar] [CrossRef] [PubMed]
- Potempa, L.A.; Rajab, I.M.; Olson, M.E.; Hart, P.C. C-Reactive Protein and Cancer: Interpreting the Differential Bioactivities of Its Pentameric and Monomeric, Modified Isoforms. Front. Immunol. 2021, 12, 744129. [Google Scholar] [CrossRef] [PubMed]
- You, Y.K.; Wu, W.F.; Huang, X.R.; Li, H.D.; Ren, Y.P.; Zeng, J.C.; Chen, H.; Lan, H.Y. Deletion of Smad3 protects against C-reactive protein-induced renal fibrosis and inflammation in obstructive nephropathy. Int. J. Biol. Sci. 2021, 17, 3911–3922. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.S.; Sun, C.F. C-reactive protein and malignancy: Clinico-pathological association and therapeutic implication. Chang. Gung Med. J. 2009, 32, 471–482. [Google Scholar]
- Shakour, N.; Cabezas, R.; Santos, J.G.; Barreto, G.E.; Jamialahmadi, T.; Hadizadeh, F.; Sahebkar, A. Curcumin Can Bind and Interact with CRP: An in silico Study. Adv. Exp. Med. Biol. 2021, 1308, 91–100. [Google Scholar] [CrossRef]
- Crocetti, L.; Floresta, G.; Cilibrizzi, A.; Giovannoni, M.P. An Overview of PDE4 Inhibitors in Clinical Trials: 2010 to Early 2022. Molecules 2022, 27, 4964. [Google Scholar] [CrossRef]
- Xu, M.; Yu, X.; Meng, X.; Huang, S.; Zhang, Y.; Zhang, A.; Jia, Z. Inhibition of PDE4/PDE4B improves renal function and ameliorates inflammation in cisplatin-induced acute kidney injury. Am. J. Physiol. Renal Physiol. 2020, 318, F576–F588. [Google Scholar] [CrossRef]
- O’Donnell, J.M.; Zhang, H.T. Antidepressant effects of inhibitors of cAMP phosphodiesterase (PDE4). Trends Pharmacol. Sci. 2004, 25, 158–163. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zuo, J.; Tang, W. Phosphodiesterase-4 Inhibitors for the Treatment of Inflammatory Diseases. Front. Pharmacol. 2018, 9, 1048. [Google Scholar] [CrossRef]
- Christian, F.; Smith, E.L.; Carmody, R.J. The Regulation of NF-κB Subunits by Phosphorylation. Cells 2016, 5, 12. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Flórez, D.; Valor, L. Selective Phosphodiesterase Inhibitors: A New Therapeutic Option in Inflammation and Autoimmunity. Reumatol. Clin. 2016, 12, 303–306. [Google Scholar] [CrossRef] [PubMed]
- Hsien Lai, S.; Zervoudakis, G.; Chou, J.; Gurney, M.E.; Quesnelle, K.M. PDE4 subtypes in cancer. Oncogene 2020, 39, 3791–3802. [Google Scholar] [CrossRef] [PubMed]
- Park, S.H.; Kim, N.D.; Jung, J.K.; Lee, C.K.; Han, S.B.; Kim, Y. Myeloid differentiation 2 as a therapeutic target of inflammatory disorders. Pharmacol. Ther. 2012, 133, 291–298. [Google Scholar] [CrossRef] [PubMed]
- Billod, J.M.; Lacetera, A.; Guzmán-Caldentey, J.; Martín-Santamaría, S. Computational Approaches to Toll-Like Receptor 4 Modulation. Molecules 2016, 21, 994. [Google Scholar] [CrossRef]
- Cochet, F.; Peri, F. The Role of Carbohydrates in the Lipopolysaccharide (LPS)/Toll-Like Receptor 4 (TLR4) Signalling. Int. J. Mol. Sci. 2017, 18, 2318. [Google Scholar] [CrossRef]
- Wang, Z.; Chen, G.; Chen, L.; Liu, X.; Fu, W.; Zhang, Y.; Li, C.; Liang, G.; Cai, Y. Insights into the binding mode of curcumin to MD-2: Studies from molecular docking, molecular dynamics simulations and experimental assessments. Mol. Biosyst. 2015, 11, 1933–1938. [Google Scholar] [CrossRef]
- Lim, T.G.; Lee, S.Y.; Huang, Z.; Lim, D.Y.; Chen, H.; Jung, S.K.; Bode, A.M.; Lee, K.W.; Dong, Z. Curcumin suppresses proliferation of colon cancer cells by targeting CDK2. Cancer Prev. Res. 2014, 7, 466–474. [Google Scholar] [CrossRef]
- Sufi, S.A.; Adigopula, L.N.; Syed, S.B.; Mukherjee, V.; Coumar, M.S.; Rao, H.S.; Rajagopalan, R. In-silico and in-vitro anti-cancer potential of a curcumin analogue (1E,6E)-1, 7-di (1H-indol-3-yl) hepta-1,6-diene-3, 5-dione. Biomed. Pharmacother. 2017, 85, 389–398. [Google Scholar] [CrossRef]
- Li, W.; Wang, Z.; Xiao, X.; Han, L.; Wu, Z.; Ma, Q.; Cao, L. Curcumin attenuates hyperglycemia-driven EGF-induced invasive and migratory abilities of pancreatic cancer via suppression of the ERK and AKT pathways. Oncol. Rep. 2019, 41, 650–658. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.C.; Baek, S.H.; Lee, C. Curcumin-induced downregulation of Axl receptor tyrosine kinase inhibits cell proliferation and circumvents chemoresistance in non-small lung cancer cells. Int. J. Oncol. 2015, 47, 2296–2303. [Google Scholar] [CrossRef] [PubMed]
- Hong, W.; Guo, F.; Yu, N.; Ying, S.; Lou, B.; Wu, J.; Gao, Y.; Ji, X.; Wang, H.; Li, A.; et al. A Novel Folic Acid Receptor-Targeted Drug Delivery System Based on Curcumin-Loaded β-Cyclodextrin Nanoparticles for Cancer Treatment. Drug Des. Devel Ther. 2021, 15, 2843–2855. [Google Scholar] [CrossRef] [PubMed]
- López-Lázaro, M.; Willmore, E.; Jobson, A.; Gilroy, K.L.; Curtis, H.; Padget, K.; Austin, C.A. Curcumin induces high levels of topoisomerase I- and II-DNA complexes in K562 leukemia cells. J. Nat. Prod. 2007, 70, 1884–1888. [Google Scholar] [CrossRef] [PubMed]
- Olivera, A.; Moore, T.W.; Hu, F.; Brown, A.P.; Sun, A.; Liotta, D.C.; Snyder, J.P.; Yoon, Y.; Shim, H.; Marcus, A.I.; et al. Inhibition of the NF-κB signaling pathway by the curcumin analog, 3,5-Bis(2-pyridinylmethylidene)-4-piperidone (EF31): Anti-inflammatory and anti-cancer properties. Int. Immunopharmacol. 2012, 12, 368–377. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.T.; Lysz, T.; Ferraro, T.; Abidi, T.F.; Laskin, J.D.; Conney, A.H. Inhibitory effects of curcumin on in vitro lipoxygenase and cyclooxygenase activities in mouse epidermis. Cancer Res. 1991, 51, 813–819. [Google Scholar]
- Cruz-Correa, M.; Shoskes, D.A.; Sanchez, P.; Zhao, R.; Hylind, L.M.; Wexner, S.D.; Giardiello, F.M. Combination treatment with curcumin and quercetin of adenomas in familial adenomatous polyposis. Clin. Gastroenterol. Hepatol. 2006, 4, 1035–1038. [Google Scholar] [CrossRef]
- Cruz-Correa, M.; Hylind, L.M.; Marrero, J.H.; Zahurak, M.L.; Murray-Stewart, T.; Casero, R.A., Jr.; Montgomery, E.A.; Iacobuzio-Donahue, C.; Brosens, L.A.; Offerhaus, G.J.; et al. Efficacy and Safety of Curcumin in Treatment of Intestinal Adenomas in Patients With Familial Adenomatous Polyposis. Gastroenterology 2018, 155, 668–673. [Google Scholar] [CrossRef] [PubMed]
- Carroll, R.E.; Benya, R.V.; Turgeon, D.K.; Vareed, S.; Neuman, M.; Rodriguez, L.; Kakarala, M.; Carpenter, P.M.; McLaren, C.; Meyskens, F.L., Jr.; et al. Phase IIa clinical trial of curcumin for the prevention of colorectal neoplasia. Cancer Prev. Res. 2011, 4, 354–364. [Google Scholar] [CrossRef] [PubMed]
- Ghalaut, V.S.; Sangwan, L.; Dahiya, K.; Ghalaut, P.S.; Dhankhar, R.; Saharan, R. Effect of imatinib therapy with and without turmeric powder on nitric oxide levels in chronic myeloid leukemia. J. Oncol. Pharm. Pract. 2012, 18, 186–190. [Google Scholar] [CrossRef]
- Mahammedi, H.; Planchat, E.; Pouget, M.; Durando, X.; Curé, H.; Guy, L.; Van-Praagh, I.; Savareux, L.; Atger, M.; Bayet-Robert, M.; et al. The New Combination Docetaxel, Prednisone and Curcumin in Patients with Castration-Resistant Prostate Cancer: A Pilot Phase II Study. Oncology 2016, 90, 69–78. [Google Scholar] [CrossRef]
- Kuriakose, M.A.; Ramdas, K.; Dey, B.; Iyer, S.; Rajan, G.; Elango, K.K.; Suresh, A.; Ravindran, D.; Kumar, R.R.; Prathiba, R.; et al. A Randomized Double-Blind Placebo-Controlled Phase IIB Trial of Curcumin in Oral Leukoplakia. Cancer Prev. Res. 2016, 9, 683–691. [Google Scholar] [CrossRef]
- Golombick, T.; Diamond, T.H.; Manoharan, A.; Ramakrishna, R. Monoclonal gammopathy of undetermined significance, smoldering multiple myeloma, and curcumin: A randomized, double-blind placebo-controlled cross-over 4g study and an open-label 8g extension study. Am. J. Hematol. 2012, 87, 455–460. [Google Scholar] [CrossRef]
- Priyadarsini, K.I. The chemistry of curcumin: From extraction to therapeutic agent. Molecules 2014, 19, 20091–20112. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Functional Classification | Target Protein | Protein Function | Compound | Binding Strength | Interactions Residues | Ref. |
|---|---|---|---|---|---|---|
| Kinase | CDK2 | G1-S phage transition | Curcumin | Binding affinity a = −7.8 kcal/mol | Hydogen bond (Glu12, Leu83), Van der Waals (Val18, Ile10) | [21] |
| Kurkumod23 (curcumin derivative)  | Binding affinity = −9.15 kcal/mol | Hydogen bond (Lys33, LEU83, Glu81), Van der Waals (Ala31, Ala44, Leu134, Val18, Phe80, Val64) | ||||
| Kinase | CK2α | Cancer cell proliferation | Ferulic Acid (curcumin derivative) 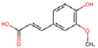 | LE score b = 0.61 kcal/mol | Hydogen bond (Lys68, Val116, Asp175) | [36] |
| Kinase | GSK3β | Involved Wnt and insulin signaling. | Curcumin | IC50 c = 66.3 nM | Hydogen bond (Val135, Ile62) | [49] |
| Kinase | DYRK2 | Cancer development and progression | Curcumin | IC50 = 5 nM | Hydogen bond (Lys251, Glu266, Asp368) Hydrophobic interaction (Ile228, Ala249, Ile285, Phe301, Leu303, Leu355, Ile367) | [58] |
| Receptor | EGFR | Cancer cell proliferation | 3a (curcumin analog)  | Docking d Score = −6.593 kcal/mol | π–π stacking (Asp855, Asp800, Leu718) | [66] |
| Receptor | AXL Tyrosine Kinase receptor | Promotes cancer development | CID 21159180 (curcumin analog) 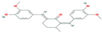 | Binding affinity = −9.0 kcal/mol | Hydogen bond (Met623, Glu585, Asp627) | [82] |
| Receptor | FR-β | Folic acid uptake | Curcumin | Docking Score = −63.30 kcal/mol | Hydogen bond (Arg152, His151, Ser190, Asp97) π–π stacking f (Trp187, Tyr101) | [88] |
| Enzyme | DHFR | DNA and RNA synthesis | Curcumin | ΔG e = −9.02 kcal/mol Ki = 243 nM | π–π stacking (Phe34) Hydogen bond (Glu30, Phe31, Val115, Tyr121) | [94] |
| Enzyme | Topo I and II | Relaxing DNA supercoils | Cyclo- Curcumin (curcumin derivative)  | Topo I ΔG = −10.33 kcal/mol Topo II ΔG = −11.16 kcal/mol | Topo I (Asp479, Ser480, and Gln778) Topo II (Asp479, Ser480, Gln778) | [101] |
| Transcription factor | NF-κB | Immune response, inflammation, and cell survival | Curcumin | Vina Score = −8.0 kcal/mol | Thr256, Ala257, Pro324, Pro344, Phe345, and Leu346 of p50 | [108] |
| Functional Classification | Target Protein | Protein Function | Compound | Binding Strength | Interactions Residues | Ref. |
|---|---|---|---|---|---|---|
| Enzyme | COX-2 | Prostaglandin endoperoxide synthase | Curcumin | Binding affinity = −5.71 kcal/mol | Hydogen bond (Ala562) | [126] |
CDF (curcumin analog)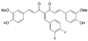 | Binding affinity = −7.91 kcal/mol | Hydogen bond (Glu346, Phe580, Asn101, Gln350) | ||||
| Enzyme | CRP | Activate the complement system | Curcumin | Docking Score = −18.0033 kcal/mol | Hydogen bond (Asp140, Gln150) | [130] |
| Enzyme | PDE4 | Modulation of cAMP signaling | Curcumin | Docking Score = −62.24 kcal/mol | Hydogen bond (His336, Asn375, Met439, Asn602) Hydrophobic interaction (Met439) | [88] |
| Co-factor protein | MD2 | Component of the TLR4 | Curcumin | KD value = 0.000379 M | Hydogen bond (Arg90, Glu92, Tyr102) | [141] |
| Cell Line | Dose | Target Protein | Findings | Ref. |
|---|---|---|---|---|
| HCT116 | 5, 10, 20, 40 µM curcumin | CDK2 | Curcumin suppresses CDK2 kinase activity dose-dependently and substantially reduces proliferation of these colon cancer cell lines. Induces G1 cell cycle arrest. | [142] |
| Jurkat Cells | 10 µM curcumin | CK2 alpha | Curcumin induces significant cell death and inhibits CK2. Activity in Jurkat cells; comparable effects to the CK2 inhibitor CX-4945. | [36] |
| SW480 | 10 µM ICA (Curcumin analogue) | GSK3β | Inhibits GSK-3β kinase. Cell cycle arrest at G0/G1 and G2/M phase. Induces apoptosis. | [143] |
| MDA-MB-231 and HaCaT cells | 10 μM curcumin | DYRK2 | Curcumin decreases 26S proteasome activity by 25–40%. | [58] |
| Bortezomib-Resistant RPMI8226.BR and MM.1S.BR | 10 μM curcumin | DYRK2 | Comparable cytotoxicity to curcumin despite resistance to bortezomib. | |
| BxPC-3 (Pancreatic Cancer Cells) | 20 μM curcumin | EGFR | Curcumin inhibited proliferation under high-glucose conditions and EGF stimulation. The invasive ability was suppressed by curcumin. Curcumin downregulated the activation of EGF/ERK and EGF/Akt pathways in high-glucose conditions and EGF treatment. | [144] |
| A549, H460 | curcumin doses (up to 20 μM) | AXL Tyrosine Kinase receptor | Curcumin inhibited Gas6-induced Axl phosphorylation, suggesting an inhibitory effect on Axl activation. Treatment with curcumin reduced cell viability in a dose-dependent manner. At 20 μM, only 30% (A549) and 22% (H460) of cells survived, indicating significant anti-proliferative effects. | [145] |
| HeLa | 2.5–40 μg/mL | FR-β | Curcumin-loaded nanoparticles showed the strongest anticancer activity, with an IC50 of 13.88 μg/mL. Enhanced cytotoxicity due to FR-mediated endocytosis. | [146] |
| K562 Leukemia | 10 µM curcumin | Topo I and Topo II | Curcumin induced higher levels of Topo I and Topo II–DNA complexes than standard inhibitors. | [147] |
| RAW264.7 Macrophages | 1–100 µM EF31 (Curcumin analog) | NF-κB | EF31 showed significantly more potent inhibition of LPS-induced NF-κB DNA binding than EF24 and curcumin, with an IC50 of ~5µM for EF31. | [148] |
| Mouse Epidermis | 3, 10, 30, 100 µM | COX-2 | Curcumin inhibited the metabolism of arachidonic acid to 5-HETE (40–83% inhibition) and 8-HETE (40–85% inhibition). IC50 = 5–10 µM. Curcumin markedly inhibited TPA- and arachidonic acid-induced epidermal inflammation. | [149] |
| Cancer Type | Description | Ref. |
|---|---|---|
| Colorectal cancer | Trials on familial adenomatous polyposis (FAP) patients with doses of 480 mg curcumin and 20 mg quercetin, showing a reduction in polyp size and number in most patients. Another trial with 1500 mg curcumin twice daily showed no significant differences. | [151,152] |
| Myeloid leukemia (CML) | Combined treatment of turmeric powder and imatinib more effective in reducing NO levels, suggesting turmeric’s potential as an adjunct therapy in CML | [153] |
| Prostate cancer | The Phase II clinical trial concluded that the combination of docetaxel and curcumin in treating metastatic castration-resistant prostate cancer demonstrated a high response rate, good tolerability, and patient acceptability. | [154] |
| Head and neck cancer | Randomized trial on oral leukoplakia with 3600 mg curcumin-containing product twice daily for six months. A higher rate of complete or partial responses was seen compared to placebo, with no significant differences in extended treatment or histological responses. | [155] |
| Multiple myeloma | Double-blind, placebo-controlled crossover study on MGUS or SMM patients with 4000 mg curcumin-containing product. Decreases noted in free light-chain ratio and urinary markers of bone resorption. | [156] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moon, D.-O. Curcumin in Cancer and Inflammation: An In-Depth Exploration of Molecular Interactions, Therapeutic Potentials, and the Role in Disease Management. Int. J. Mol. Sci. 2024, 25, 2911. https://doi.org/10.3390/ijms25052911
Moon D-O. Curcumin in Cancer and Inflammation: An In-Depth Exploration of Molecular Interactions, Therapeutic Potentials, and the Role in Disease Management. International Journal of Molecular Sciences. 2024; 25(5):2911. https://doi.org/10.3390/ijms25052911
Chicago/Turabian StyleMoon, Dong-Oh. 2024. "Curcumin in Cancer and Inflammation: An In-Depth Exploration of Molecular Interactions, Therapeutic Potentials, and the Role in Disease Management" International Journal of Molecular Sciences 25, no. 5: 2911. https://doi.org/10.3390/ijms25052911
APA StyleMoon, D.-O. (2024). Curcumin in Cancer and Inflammation: An In-Depth Exploration of Molecular Interactions, Therapeutic Potentials, and the Role in Disease Management. International Journal of Molecular Sciences, 25(5), 2911. https://doi.org/10.3390/ijms25052911