
Lack of the Histone Deacetylase SIRT1 Leads to Protection against Endoplasmic Reticulum Stress through the Upregulation of Heat Shock Proteins

 , ,
, ,  ,
,
Abstract
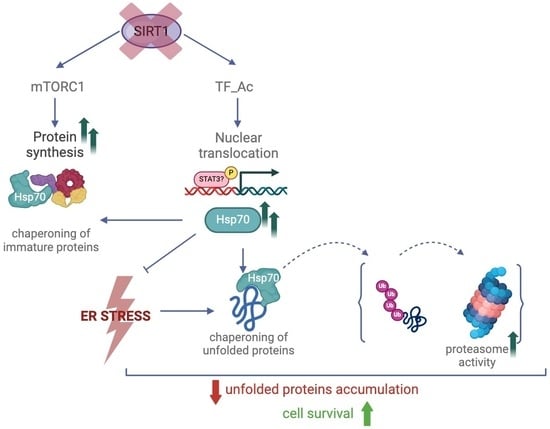
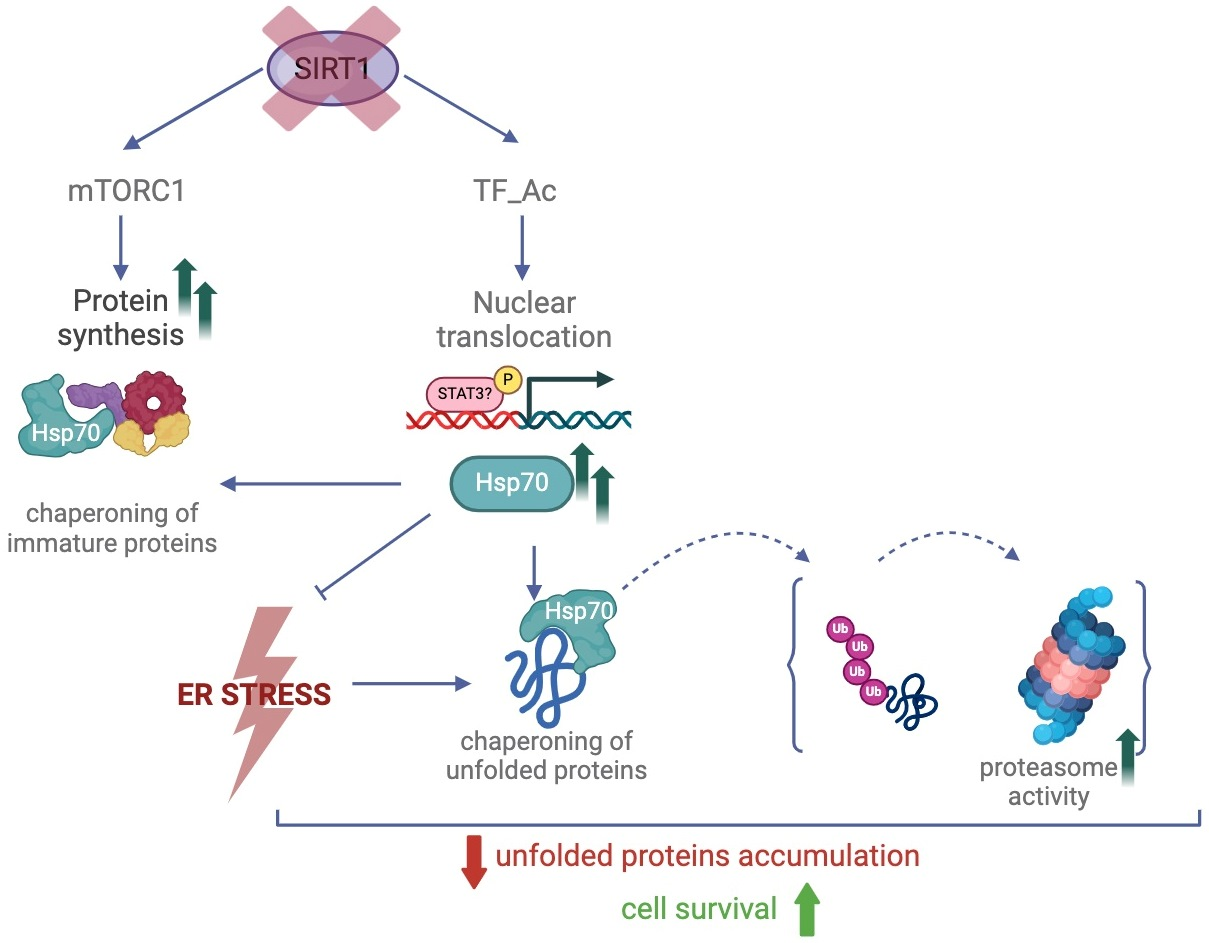{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
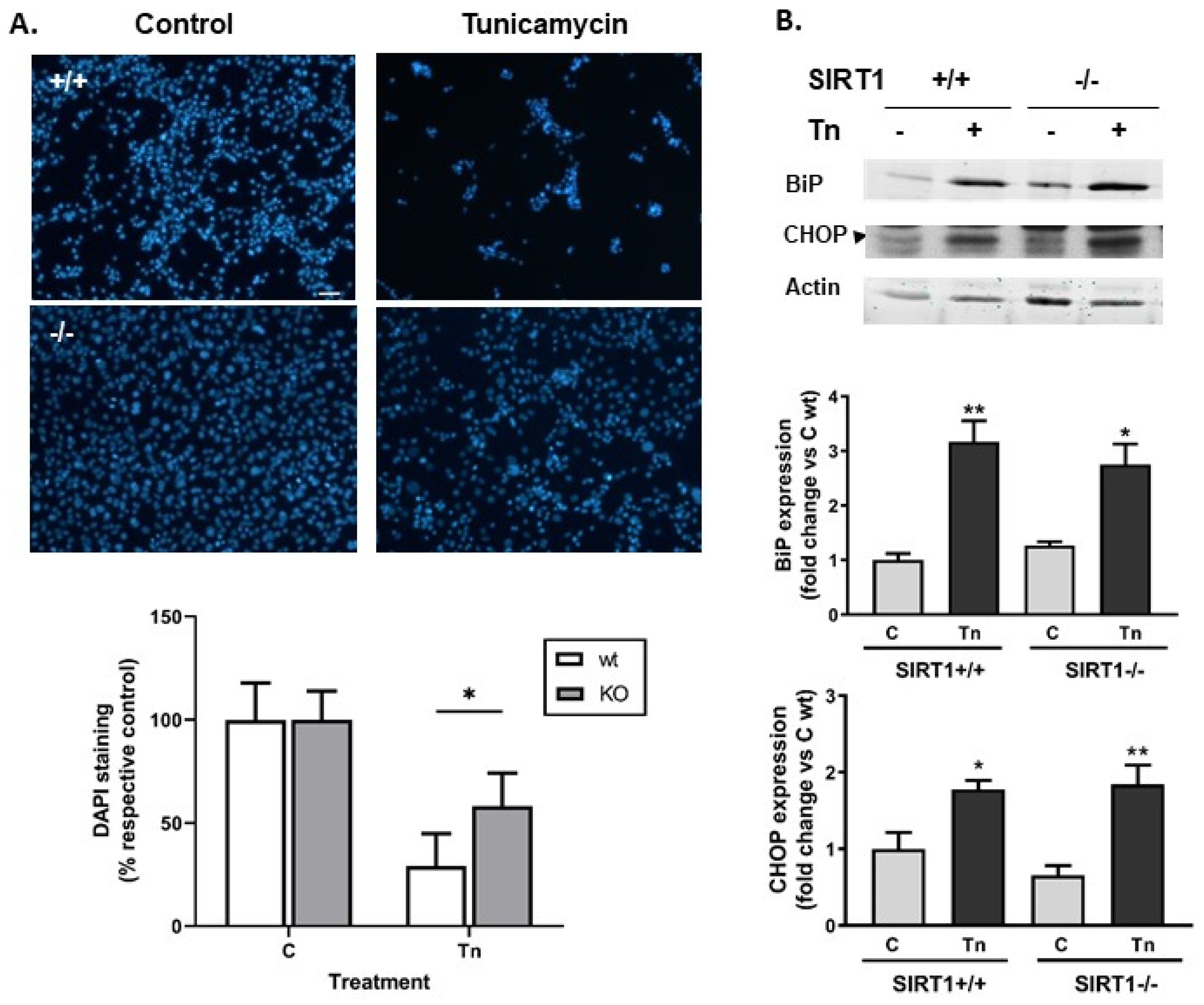
2.1. SIRT1-Deficient Cells Were Less Vulnerable to ER Stress Than wt Cells but Developed Similar UPR, and Resistance Was Independent of mTOR Hyperactivity
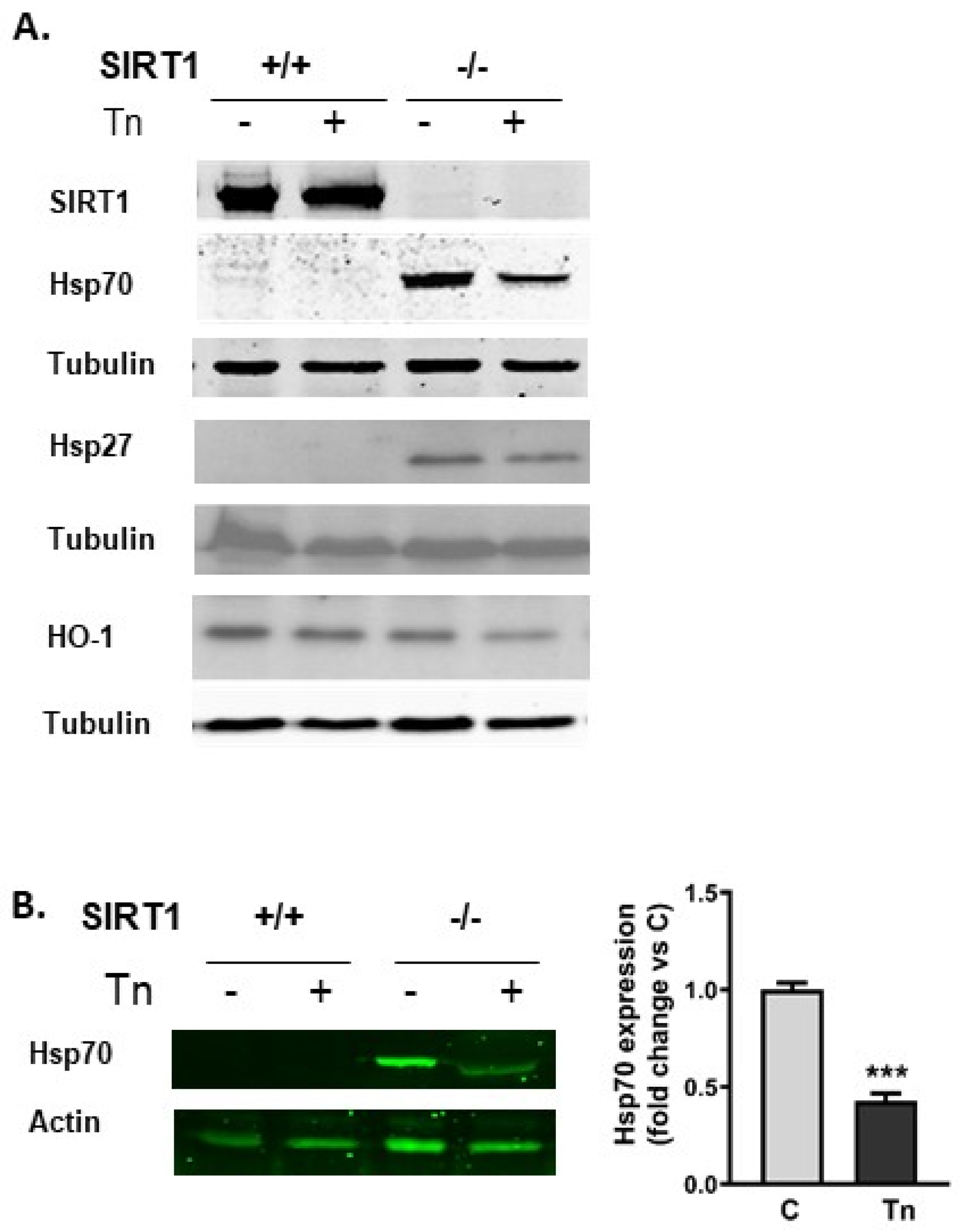
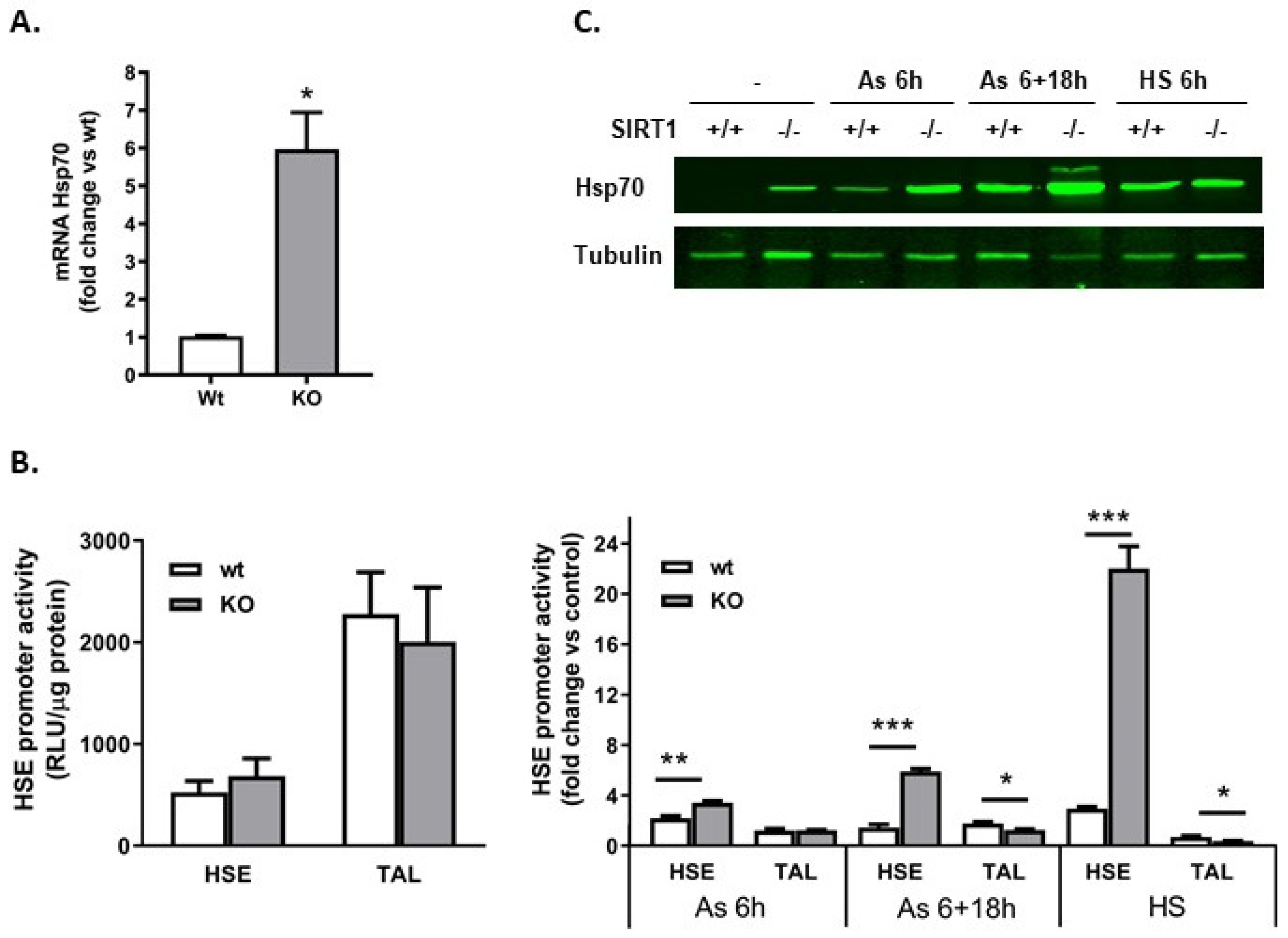
2.2. SIRT1−/− Cells Constitutively Express Heat Shock Proteins
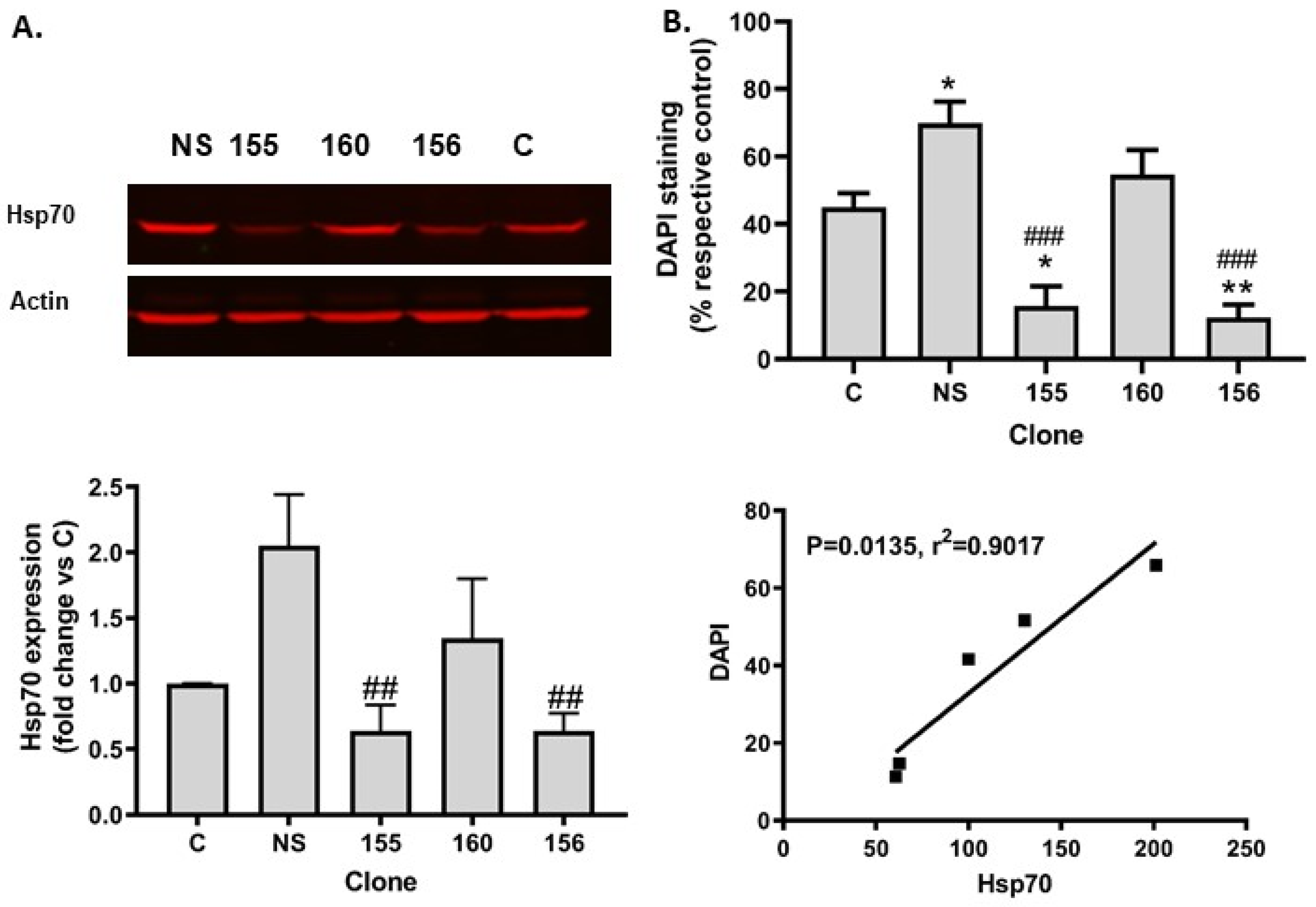
2.3. Hsp70 Silencing Abolished SIRT1 Resistance to Tunicamycin Toxicity
2.4. SIRT1 Absence Did Not Impair Proteasome Activity
2.5. Heat Shock Element (HSE) Was Not Involved in the Constitutive Production of Hsp70 in SIRT1−/− Cells
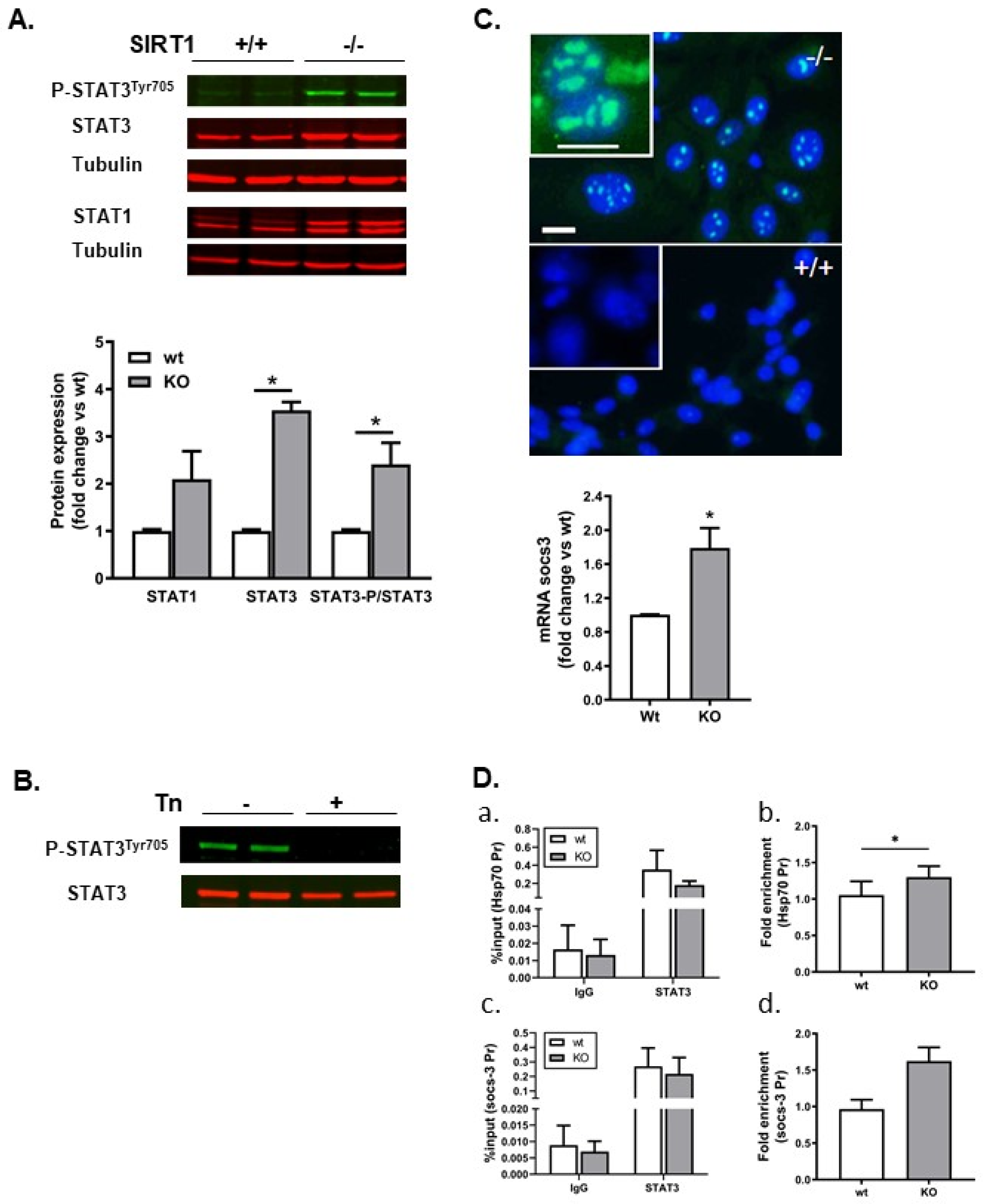
2.6. STAT3 and Its Target Gene Socs-3 Were Upregulated in SIRT1−/− Cells
3. Discussion
4. Materials and Methods
4.1. Cell Cultures and Treatments
4.2. Stable Cell Lines Expressing shRNA-Hsp70
4.3. Viability Assays
4.4. Immunofluorescence
4.5. Western Blot
4.6. Real-Time PCR
4.7. Proteasome Activity Assay
4.8. HSE-Luciferase Reporter Assay
4.9. Chromatin Immunoprecipitation (ChIP)
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Giblin, W.; Skinner, M.E.; Lombard, D.B. Sirtuins: Guardians of Mammalian Healthspan. Trends Genet. 2014, 30, 271–286. [Google Scholar] [CrossRef]
- Kwon, H.S.; Ott, M. The Ups and Downs of SIRT1. Trends Biochem. Sci. 2008, 33, 517–525. [Google Scholar] [CrossRef]
- Pantazi, E.; Zaouali, M.A.; Bejaoui, M.; Serafin, A.; Folch-Puy, E.; Petegnief, V.; De Vera, N.; Ben Abdennebi, H.; Rimola, A.; Rosello-Catafau, J. Silent Information Regulator 1 Protects the Liver against Ischemia-Reperfusion Injury: Implications in Steatotic Liver Ischemic Preconditioning. Mol. Neurobiol. 2014, 27, 493–503. [Google Scholar] [CrossRef]
- Donmez, G. The Neurobiology of Sirtuins and Their Role in Neurodegeneration. Trends Pharm. Sci. 2012, 33, 494–501. [Google Scholar] [CrossRef]
- Zhang, W.; Xiao, D.; Li, X.; Zhang, Y.; Rasouli, J.; Casella, G.; Boehm, A.; Hwang, D.; Ishikawa, L.L.; Thome, R.; et al. SIRT1 Inactivation Switches Reactive Astrocytes to an Antiinflammatory Phenotype in CNS Autoimmunity. J. Clin. Investig. 2022, 132, e151803. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Gharavi, R.; Pitta, M.; Gleichmann, M.; Mattson, M.P. Nicotinamide Prevents NAD + Depletion and Protects Neurons against Excitotoxicity and Cerebral Ischemia: NAD+ Consumption by SIRT1 May Endanger Energetically Compromised Neurons. Neuromol. Med. 2009, 11, 28–42. [Google Scholar] [CrossRef]
- Kakefuda, K.; Fujita, Y.; Oyagi, A.; Hyakkoku, K.; Kojima, T.; Umemura, K.; Tsuruma, K.; Shimazawa, M.; Ito, M.; Nozawa, Y.; et al. Sirtuin 1 Overexpression Mice Show a Reference Memory Deficit, but Not Neuroprotection. Biochem. Biophys. Res. Commun. 2009, 387, 784–788. [Google Scholar] [CrossRef]
- Wang, F.M.; Chen, Y.J.; Ouyang, H.J. Regulation of Unfolded Protein Response Modulator XBP1s by Acetylation and Deacetylation. Biochem. J. 2011, 433, 245–252. [Google Scholar] [CrossRef]
- Sánchez-Hidalgo, A.C.; Muñoz, M.F.; Herrera, A.J.; Espinosa-Oliva, A.M.; Stowell, R.; Ayala, A.; Machado, A.; Venero, J.L.; de Pablos, R.M. Chronic Stress Alters the Expression Levels of Longevity-Related Genes in the Rat Hippocampus. Neurochem. Int. 2016, 97, 181–192. [Google Scholar] [CrossRef]
- Marinova, Z.; Ren, M.; Wendland, J.R.; Leng, Y.; Liang, M.-H.; Yasuda, S.; Leeds, P.; Chuang, D.-M. Valproic Acid Induces Functional Heat shock Protein 70 via Class I Histone Deacetylase Inhibition in Cortical Neurons: A Potential Role of Sp1 Acetylation. J. Neurochem. 2009, 111, 976–987. [Google Scholar] [CrossRef]
- Ren, M.; Leng, Y.; Jeong, M.; Leeds, P.R.; Chuang, D.-M. Valproic Acid Reduces Brain Damage Induced by Transient Focal Cerebral Ischemia in Rats: Potential Roles of Histone Deacetylase Inhibition and Heat Shock Protein Induction. J. Neurochem. 2004, 89, 1358–1367. [Google Scholar] [CrossRef]
- Rao, R.; Fiskus, W.; Ganguly, S.; Kambhampati, S.; Bhalla, K.N. HDAC Inhibitors and Chaperone Function. Adv. Cancer Res. 2012, 116, 239–262. [Google Scholar] [CrossRef]
- Chuang, D.-M.; Leng, Y.; Marinova, Z.; Kim, H.-J.; Chiu, C.-T. Multiple Roles of HDAC Inhibition in Neurodegenerative Conditions. Trends Neurosci. 2009, 32, 591–601. [Google Scholar] [CrossRef]
- Planas, A.M.; Soriano, M.A.; Estrada, A.; Sanz, O.; Martin, F.; Ferrer, I. The Heat Shock Stress Response after Brain Lesions: Induction of 72 KDa Heat Shock Protein (Cell Types Involved, Axonal Transport, Transcriptional Regulation) and Protein Synthesis Inhibition. Prog. Neurobiol. 1997, 51, 607–636. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sasi, B.K.; Sonawane, P.J.; Gupta, V.; Sahu, B.S.; Mahapatra, N.R. Coordinated Transcriptional Regulation of Hspa1a Gene by Multiple Transcription Factors: Crucial Roles for HSF-1, NF-Y, NF-KappaB, and CREB. J. Mol. Biol. 2014, 426, 116–135. [Google Scholar] [CrossRef]
- Fauconneau, B.; Petegnief, V.; Sanfeliu, C.; Piriou, A.; Planas, A.M. Induction of Heat Shock Proteins (HSPs) by Sodium Arsenite in Cultured Astrocytes and Reduction of Hydrogen Peroxide-Induced Cell Death. J. Neurochem. 2002, 83, 1338–1348. [Google Scholar] [CrossRef]
- Boyault, C.; Zhang, Y.; Fritah, S.; Caron, C.; Gilquin, B.; Kwon, S.H.; Garrido, C.; Yao, T.P.; Vourc’h, C.; Matthias, P.; et al. HDAC6 Controls Major Cell Response Pathways to Cytotoxic Accumulation of Protein Aggregates. Genes Dev. 2007, 21, 2172–2181. [Google Scholar] [CrossRef]
- Stephanou, A.; Isenberg, D.A.; Nakajima, K.; Latchman, D.S. Signal Transducer and Activator of Transcription-1 and Heat Shock Factor-1 Interact and Activate the Transcription of the Hsp-70 and Hsp-90beta Gene Promoters. J. Biol. Chem. 1999, 274, 1723–1728. [Google Scholar] [CrossRef]
- Yamagishi, N.; Fujii, H.; Saito, Y.; Hatayama, T. Hsp105beta Upregulates Hsp70 Gene Expression through Signal Transducer and Activator of Transcription-3. FEBS J. 2009, 276, 5870–5880. [Google Scholar] [CrossRef]
- Nie, Y.; Erion, D.M.; Yuan, Z.; Dietrich, M.; Shulman, G.I.; Horvath, T.L.; Gao, Q. STAT3 Inhibition of Gluconeogenesis Is Downregulated by SirT1. Nat. Cell Biol. 2009, 11, 492–500. [Google Scholar] [CrossRef]
- Westerheide, S.D.; Anckar, J.; Stevens, S.M., Jr.; Sistonen, L.; Morimoto, R.I. Stress-Inducible Regulation of Heat Shock Factor 1 by the Deacetylase SIRT1. Science 2009, 323, 1063–1066. [Google Scholar] [CrossRef]
- Ghosh, H.S.; McBurney, M.; Robbins, P.D. SIRT1 Negatively Regulates the Mammalian Target of Rapamycin. PLoS ONE 2010, 5, e9199. [Google Scholar] [CrossRef]
- Appenzeller-Herzog, C.; Hall, M.N. Bidirectional Crosstalk between Endoplasmic Reticulum Stress and MTOR Signaling. Trends Cell Biol. 2012, 22, 274–282. [Google Scholar] [CrossRef]
- Laplante, M.; Sabatini, D.M. Regulation of MTORC1 and Its Impact on Gene Expression at a Glance. J. Cell Sci. 2013, 126, 1713–1719. [Google Scholar] [CrossRef]
- Petegnief, V.; Font-Nieves, M.; Martin, M.E.; Salinas, M.; Planas, A.M. Nitric Oxide Mediates NMDA-Induced Persistent Inhibition of Protein Synthesis through Dephosphorylation of Eukaryotic Initiation Factor 4E-Binding Protein 1 and Eukaryotic Initiation Factor 4G Proteolysis. Biochem. J. 2008, 411, 667–677. [Google Scholar] [CrossRef][Green Version]
- Chiarini, F.; Evangelisti, C.; McCubrey, J.A.; Martelli, A.M. Current Treatment Strategies for Inhibiting MTOR in Cancer. Trends Pharmacol. Sci. 2015, 36, 124–135. [Google Scholar] [CrossRef]
- Kastle, M.; Woschee, E.; Grune, T. Histone Deacetylase 6 (HDAC6) Plays a Crucial Role in P38MAPK-Dependent Induction of Heme Oxygenase-1 (HO-1) in Response to Proteasome Inhibition. Free Radic. Biol. Med. 2012, 53, 2092–2101. [Google Scholar] [CrossRef]
- Kimura, K.; Yamada, T.; Matsumoto, M.; Kido, Y.; Hosooka, T.; Asahara, S.; Matsuda, T.; Ota, T.; Watanabe, H.; Sai, Y.; et al. Endoplasmic Reticulum Stress Inhibits STAT3-Dependent Suppression of Hepatic Gluconeogenesis via Dephosphorylation and Deacetylation. Diabetes 2012, 61, 61–73. [Google Scholar] [CrossRef]
- Hutchins, A.P.; Diez, D.; Takahashi, Y.; Ahmad, S.; Jauch, R.; Tremblay, M.L.; Miranda-Saavedra, D. Distinct Transcriptional Regulatory Modules Underlie STAT3’s Cell Type-Independent and Cell Type-Specific Functions. Nucleic Acids Res. 2013, 41, 2155–2170. [Google Scholar] [CrossRef]
- Hutchins, A.P.; Diez, D.; Miranda-Saavedra, D. Genomic and Computational Approaches to Dissect the Mechanisms of STAT3’s Universal and Cell Type-Specific Functions. JAKSTAT 2013, 2, e25097. [Google Scholar] [CrossRef]
- Bernier, M.; Paul, R.K.; Martin-Montalvo, A.; Scheibye-Knudsen, M.; Song, S.; He, H.J.; Armour, S.M.; Hubbard, B.P.; Bohr, V.A.; Wang, L.; et al. Negative Regulation of STAT3 Protein-Mediated Cellular Respiration by SIRT1 Protein. J. Biol. Chem. 2011, 286, 19270–19279. [Google Scholar] [CrossRef]
- Park, J.-M.; Jo, S.-H.; Kim, M.-Y.; Kim, T.-H.; Ahn, Y.-H. Role of Transcription Factor Acetylation in the Regulation of Metabolic Homeostasis. Protein Cell 2015, 6, 804–813. [Google Scholar] [CrossRef]
- Filtz, T.M.; Vogel, W.K.; Leid, M. Regulation of Transcription Factor Activity by Interconnected, Post-Translational Modifications. Trends Pharmacol. Sci. 2014, 35, 76–85. [Google Scholar] [CrossRef]
- Leal, H.; Cardoso, J.; Valério, P.; Quatorze, M.; Carmona, V.; Cunha-Santos, J.; Pereira de Almeida, L.; Pereira, C.; Cavadas, C.; Gomes, P. SIRT2 Deficiency Exacerbates Hepatic Steatosis via a Putative Role of the ER Stress Pathway. Int. J. Mol. Sci. 2022, 23, 6790. [Google Scholar] [CrossRef]
- Kim, A.; Koo, J.H.; Lee, J.M.; Joo, M.S.; Kim, T.H.; Kim, H.; Jun, D.W.; Kim, S.G. NRF2-mediated SIRT3 Induction Protects Hepatocytes from ER Stress-induced Liver Injury. FASEB J. 2022, 36, e22170. [Google Scholar] [CrossRef]
- Cho, W.; Choi, S.W.; Oh, H.; Baygutalp, F.; Abd El-Aty, A.M.; Jeong, J.H.; Song, J.-H.; Shin, Y.K.; Jung, T.W. Musclin Attenuates Lipid Deposition in Hepatocytes through SIRT7/Autophagy-Mediated Suppression of ER Stress. Biochem. Biophys. Res. Commun. 2023, 658, 62–68. [Google Scholar] [CrossRef]
- Akkulak, A.; Yalcin, G.D. The Interaction of SIRT4 and Calreticulin during ER Stress in Glia Cells. Gene 2022, 814, 146135. [Google Scholar] [CrossRef]
- Wang, C.; Zhu, Y.; Chen, R.; Zhu, X.; Zhang, X. MicroRNA-143 Targets SIRT2 to Mediate the Histone Acetylation of PLAUR and Modulates Functions of Astrocytes in Spinal Cord Injury. Chem. Biol. Interact. 2024, 390, 110854. [Google Scholar] [CrossRef]
- Faraco, G.; Pancani, T.; Formentini, L.; Mascagni, P.; Fossati, G.; Leoni, F.; Moroni, F.; Chiarugi, A. Pharmacological Inhibition of Histone Deacetylases by Suberoylanilide Hydroxamic Acid Specifically Alters Gene Expression and Reduces Ischemic Injury in the Mouse Brain. Mol. Pharm. 2006, 70, 1876–1884. [Google Scholar] [CrossRef]
- Radons, J. The Human HSP70 Family of Chaperones: Where Do We Stand? Cell Stress Chaperones 2016, 21, 379–404. [Google Scholar] [CrossRef]
- Kumar, V.; Kundu, S.; Singh, A.; Singh, S. Understanding the Role of Histone Deacetylase and Their Inhibitors in Neurodegenerative Disorders: Current Targets and Future Perspective. Curr. Neuropharmacol. 2022, 20, 158–178. [Google Scholar] [CrossRef]
- Han, Y.; Kang, Y.; Yu, J.; Yu, S.-L.; Park, H.-W.; Shin, J.; Park, S.-R.; Kang, J. Increase of Hspa1a and Hspa1b Genes in the Resting B Cells of Sirt1 Knockout Mice. Mol. Biol. Rep. 2019, 46, 4225–4234. [Google Scholar] [CrossRef]
- Tomita, T.; Hamazaki, J.; Hirayama, S.; McBurney, M.W.; Yashiroda, H.; Murata, S. Sirt1-Deficiency Causes Defective Protein Quality Control. Sci. Rep. 2015, 5, 12613. [Google Scholar] [CrossRef]
- Bocchini, C.E.; Kasembeli, M.M.; Roh, S.-H.; Tweardy, D.J. Contribution of Chaperones to STAT Pathway Signaling. JAK-STAT 2014, 3, e970459. [Google Scholar] [CrossRef]
- Jego, G.; Hermetet, F. Chaperoning STAT3/5 by Heat Shock Proteins: Interest of Their Targeting in Cancer Therapy. Cancers 2019, 12, 21. [Google Scholar] [CrossRef]
- Smith, M.R.; Syed, A.; Lukacsovich, T.; Purcell, J.; Barbaro, B.A.; Worthge, S.A.; Wei, S.R.; Pollio, G.; Magnoni, L.; Scali, C.; et al. A Potent and Selective Sirtuin 1 Inhibitor Alleviates Pathology in Multiple Animal and Cell Models of Huntington’s Disease. Hum. Mol. Genet. 2014, 23, 2995–3007. [Google Scholar] [CrossRef]
- Bosch-Presegue, L.; Raurell-Vila, H.; Marazuela-Duque, A.; Kane-Goldsmith, N.; Valle, A.; Oliver, J.; Serrano, L.; Vaquero, A. Stabilization of Suv39H1 by SirT1 Is Part of Oxidative Stress Response and Ensures Genome Protection. Mol. Cell 2011, 42, 210–223. [Google Scholar] [CrossRef]
- Martinez, A.; Hériché, J.-K.; Calvo, M.; Tischer, C.; Otxoa-de-Amezaga, A.; Pedragosa, J.; Bosch, A.; Planas, A.M.; Petegnief, V. Characterization of Microglia Behaviour in Healthy and Pathological Conditions with Image Analysis Tools. Open Biol. 2023, 13, 220200. [Google Scholar] [CrossRef]
- Snider, B.J.; Tee, L.Y.; Canzoniero, L.M.T.; Babcock, D.J.; Choi, D.W. NMDA Antagonists Exacerbate Neuronal Death Caused by Proteasome Inhibition in Cultured Cortical and Striatal Neurons. Eur. J. Neurosci. 2002, 15, 419–428. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Latorre, J.; de Vera, N.; Santalucía, T.; Balada, R.; Marazuela-Duque, A.; Vaquero, A.; Planas, A.M.; Petegnief, V. Lack of the Histone Deacetylase SIRT1 Leads to Protection against Endoplasmic Reticulum Stress through the Upregulation of Heat Shock Proteins. Int. J. Mol. Sci. 2024, 25, 2856. https://doi.org/10.3390/ijms25052856
Latorre J, de Vera N, Santalucía T, Balada R, Marazuela-Duque A, Vaquero A, Planas AM, Petegnief V. Lack of the Histone Deacetylase SIRT1 Leads to Protection against Endoplasmic Reticulum Stress through the Upregulation of Heat Shock Proteins. International Journal of Molecular Sciences. 2024; 25(5):2856. https://doi.org/10.3390/ijms25052856
Chicago/Turabian StyleLatorre, Jessica, Nuria de Vera, Tomàs Santalucía, Rafel Balada, Anna Marazuela-Duque, Alejandro Vaquero, Anna M. Planas, and Valérie Petegnief. 2024. "Lack of the Histone Deacetylase SIRT1 Leads to Protection against Endoplasmic Reticulum Stress through the Upregulation of Heat Shock Proteins" International Journal of Molecular Sciences 25, no. 5: 2856. https://doi.org/10.3390/ijms25052856
APA StyleLatorre, J., de Vera, N., Santalucía, T., Balada, R., Marazuela-Duque, A., Vaquero, A., Planas, A. M., & Petegnief, V. (2024). Lack of the Histone Deacetylase SIRT1 Leads to Protection against Endoplasmic Reticulum Stress through the Upregulation of Heat Shock Proteins. International Journal of Molecular Sciences, 25(5), 2856. https://doi.org/10.3390/ijms25052856