
ER Stress-Activated HSF1 Governs Cancer Cell Resistance to USP7 Inhibitor-Based Chemotherapy through the PERK Pathway
Abstract
1. Introduction
2. Results
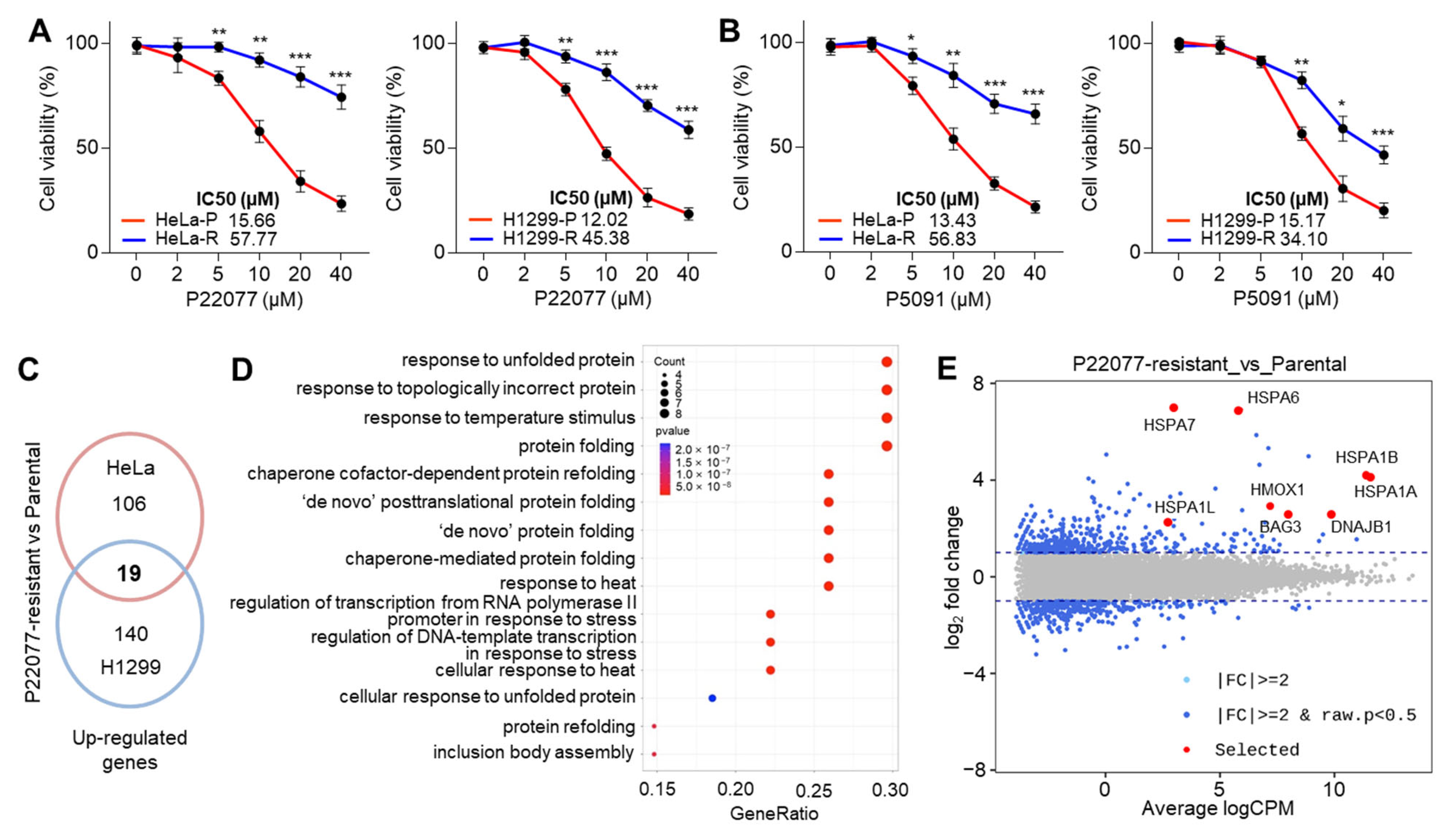
2.1. Heat Stress Response (HSR)- and Unfolded Protein Response (UPR)-Associated Genes Are Increased in Ubiquitin-Specific Protease 7 Inhibitor (USP7i)-Resistant Cancer Cells
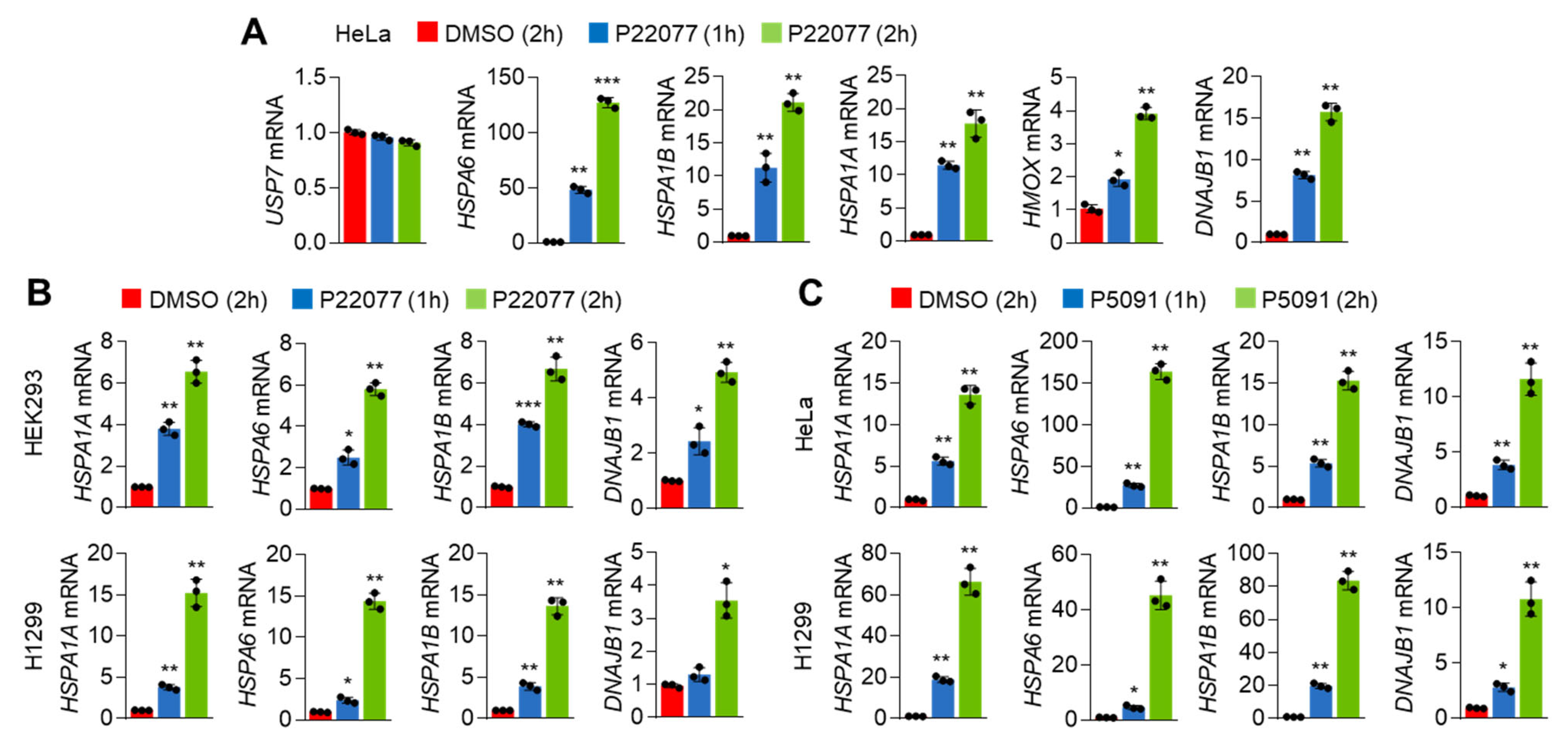
2.2. USP7i Induces the Expression of HSR- and UPR-Associated Genes in Cancer Cells
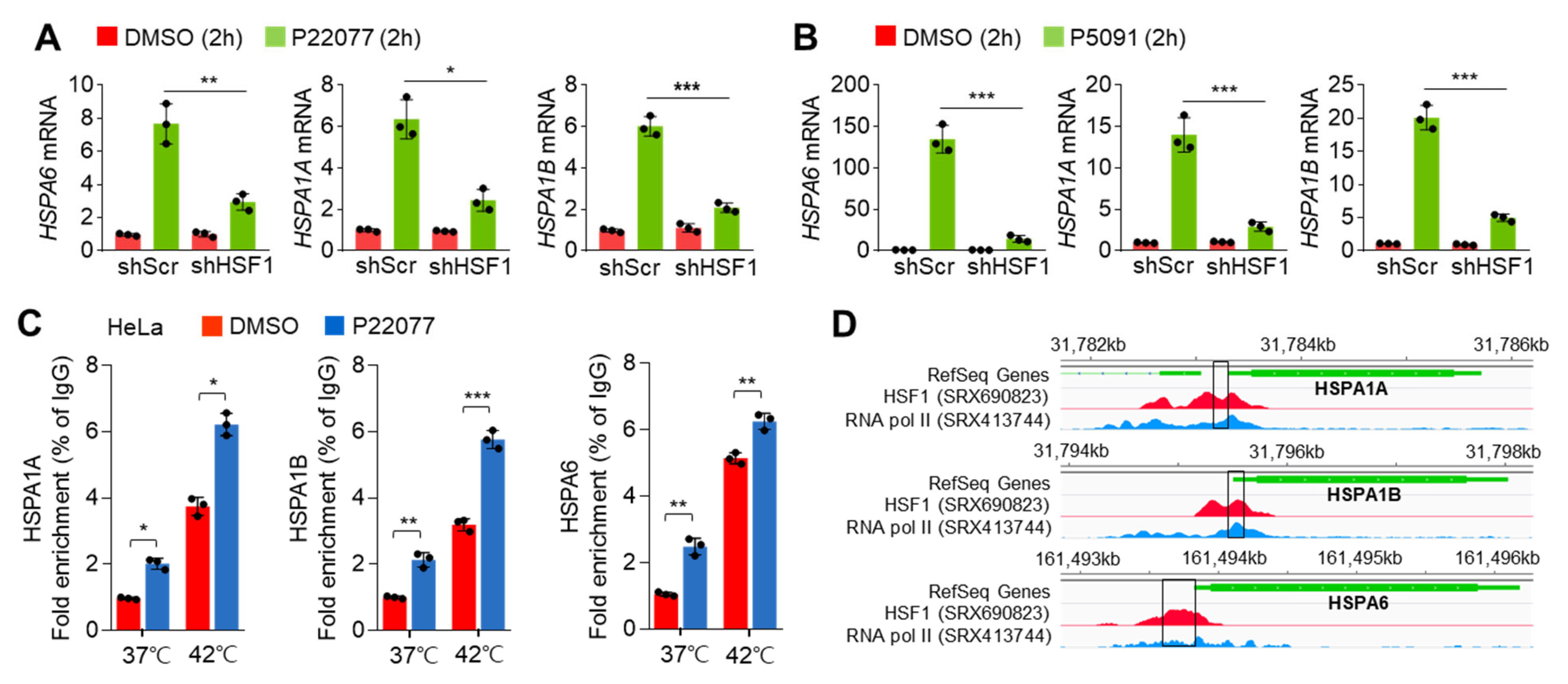
2.3. HSF1 Is Involved in the Expression of HSR- and UPR-Related Genes in Response to USP7i
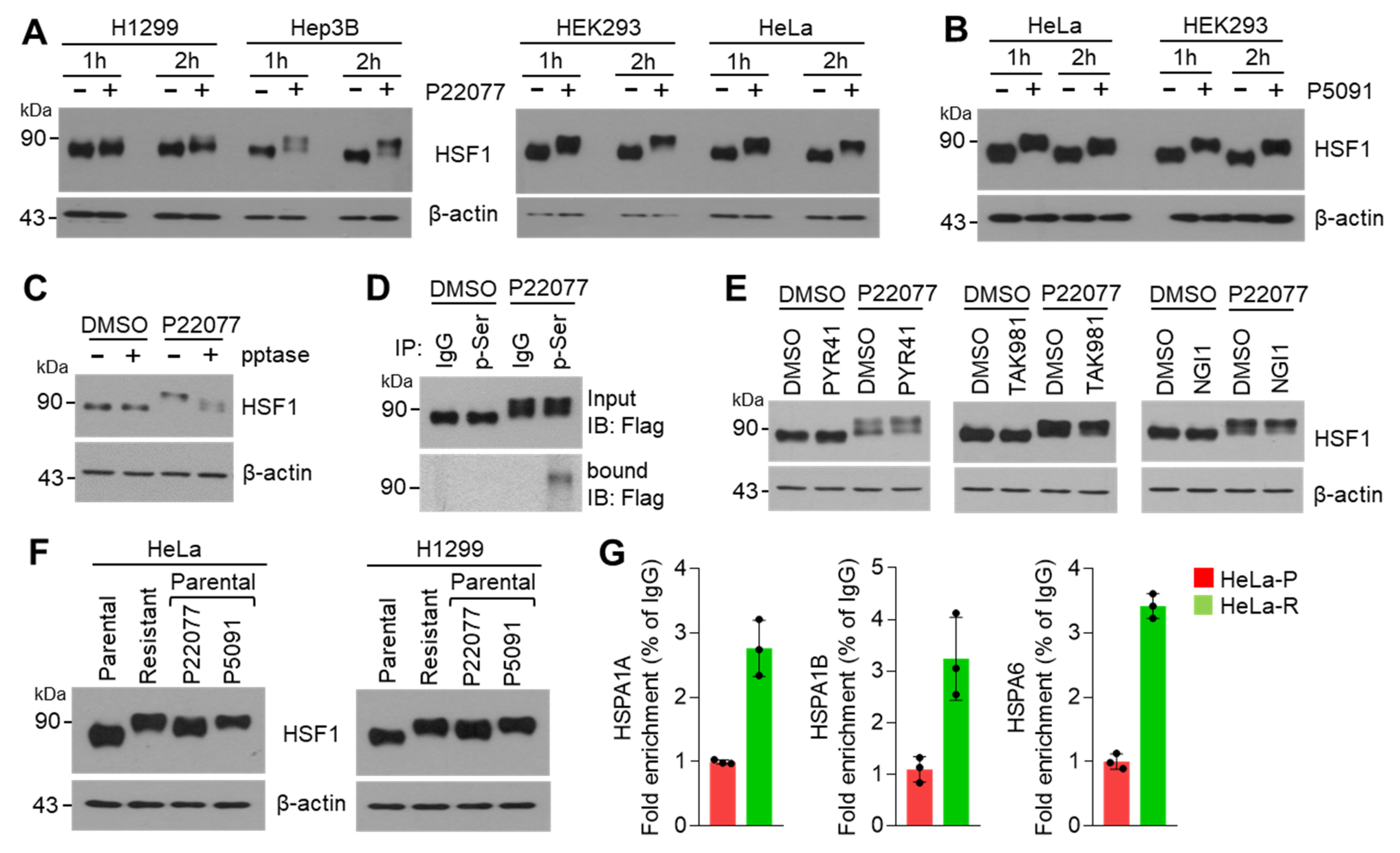
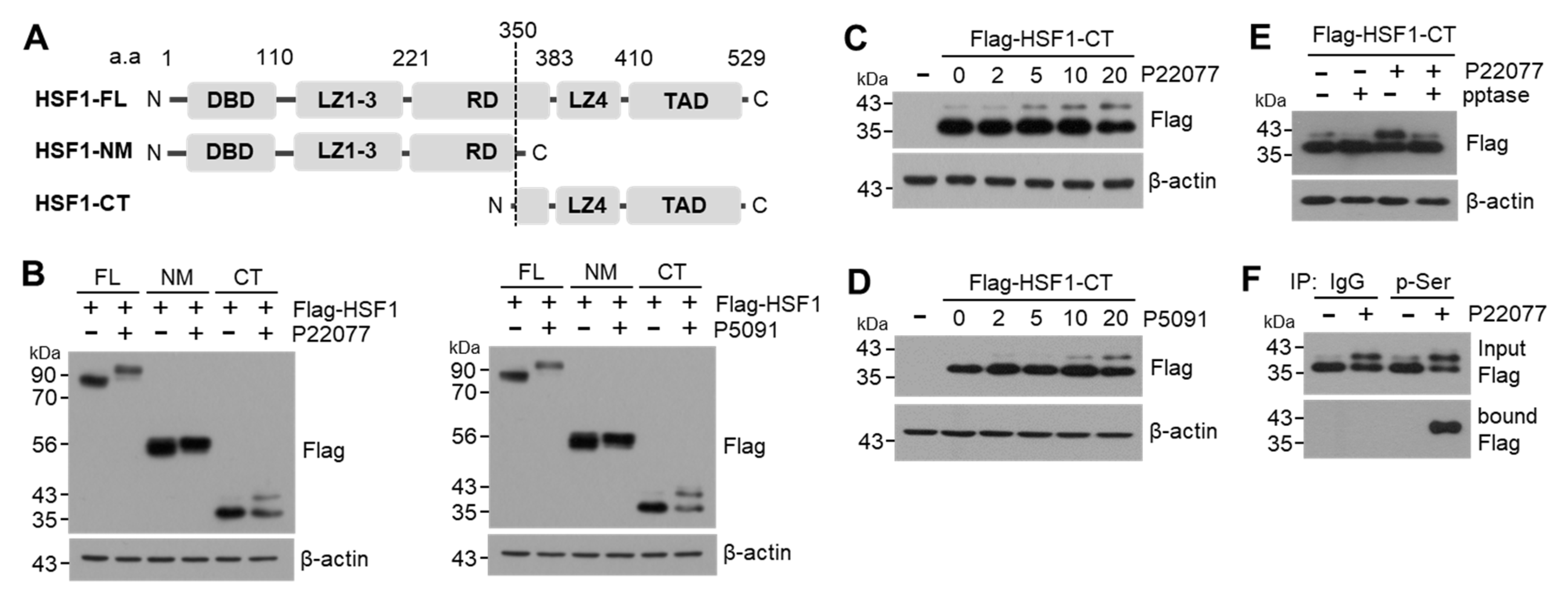
2.4. USP7i Leads to HSF1 Phosphorylation
2.5. C-Terminal Domain of HSF1 Is Phosphorylated in Response to USP7i
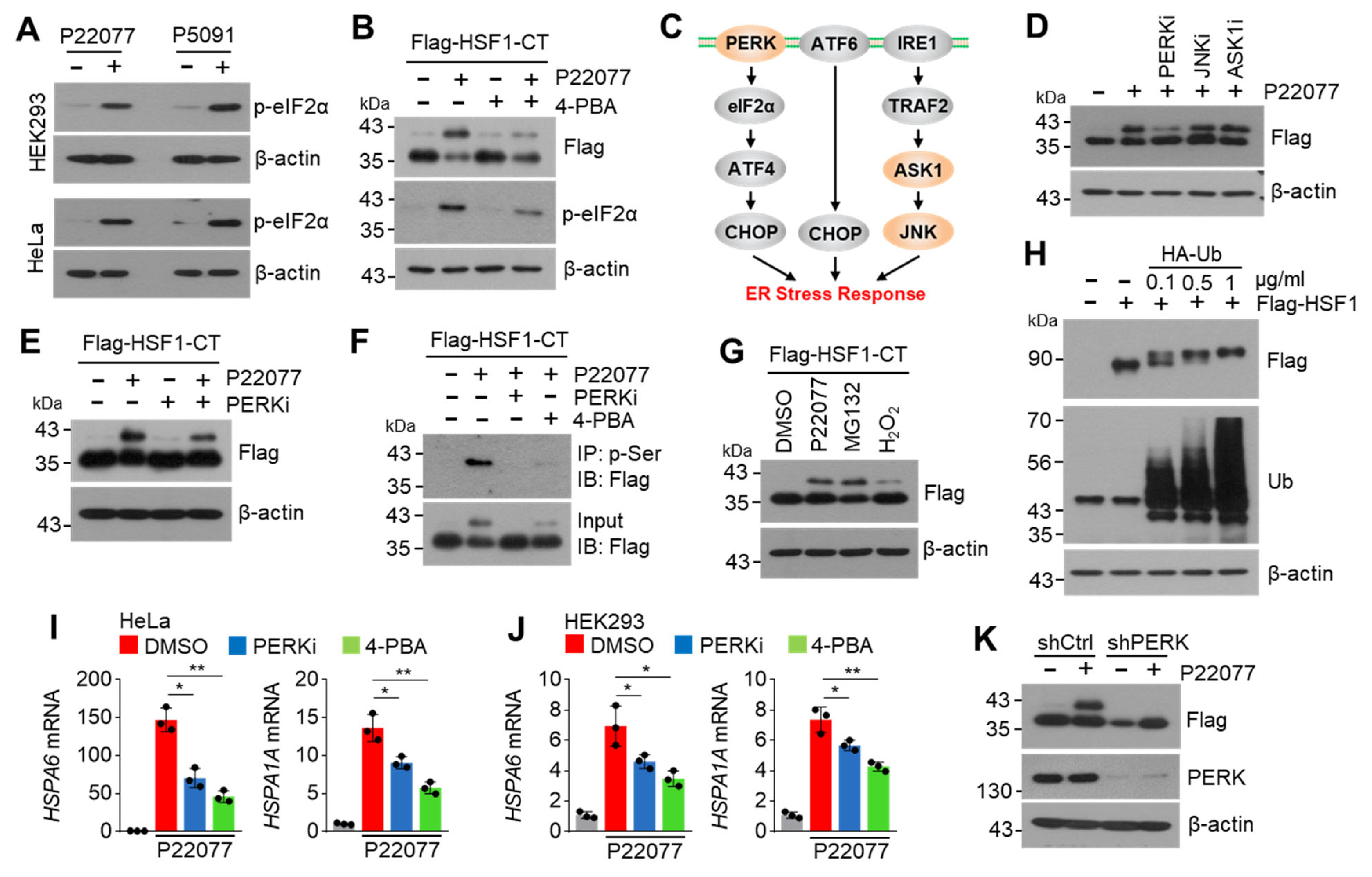
2.6. PERK Is Involved in the USP7i-Mediated HSF1 Phosphorylation
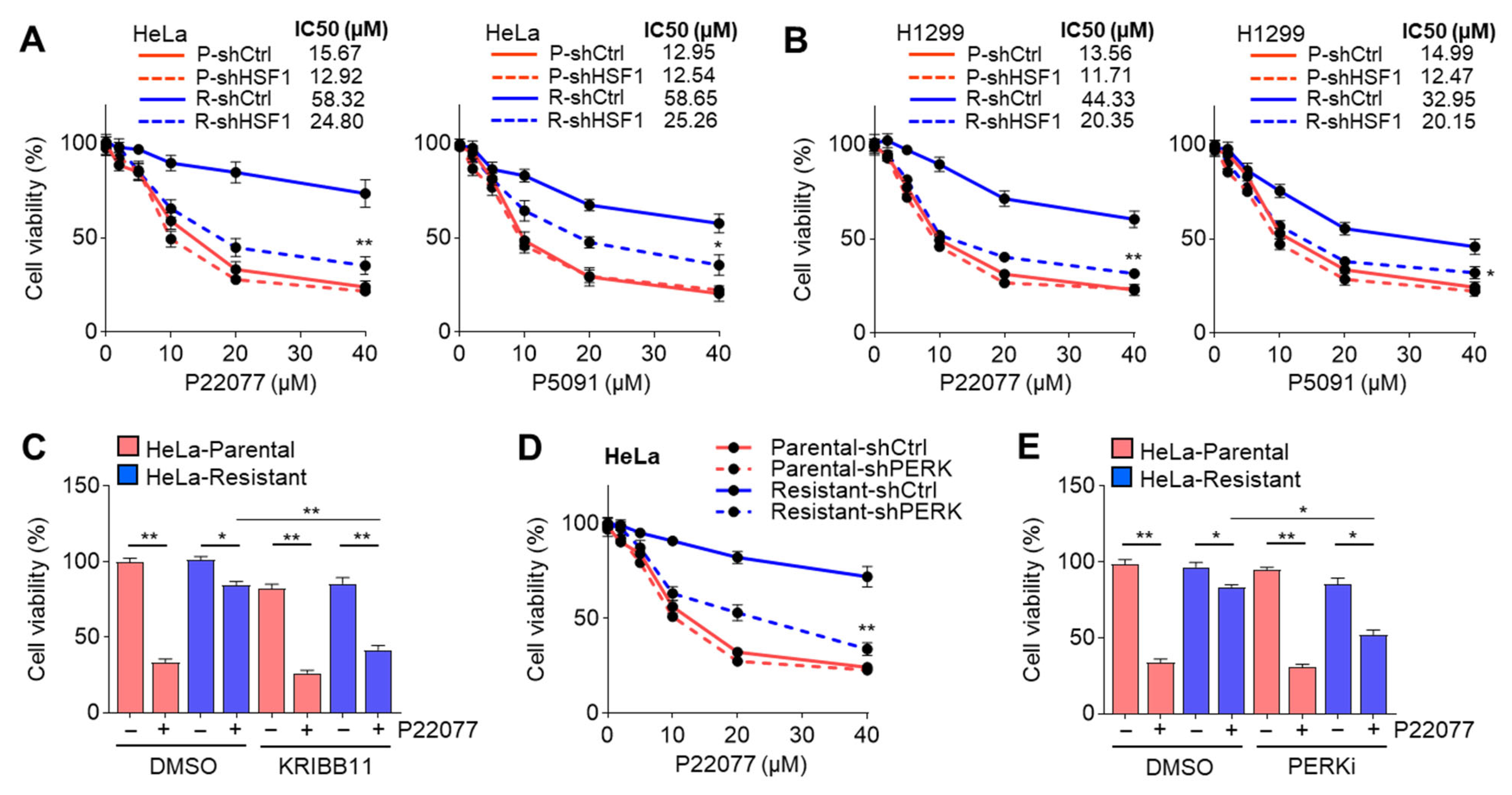
2.7. Suppression of the PERK–HSF1 Axis Sensitizes Cells to the Anticancer Effect of USP7i
3. Discussion
4. Materials and Methods
4.1. RNA-Sequencing and Quantitative Real-Time PCR (qRT-PCR)
4.2. Co-Immunoprecipitation and Western Blotting
4.3. Cell Viability Assay
4.4. Cell Culture, Drug Treatment, and Chemical Reagents
4.5. Plasmids, Transfection, and Lentiviral Transduction
4.6. Chromatin Immunoprecipitation and Polymerase Chain Reaction (ChIP-PCR)
4.7. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Saha, G.; Roy, S.; Basu, M.; Ghosh, M.K. USP7—A crucial regulator of cancer hallmarks. Biochim. Biophys. Acta Rev. Cancer 2023, 1878, 188903. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.T.; Lin, J.; Liu, Y.E.; Chen, Y.C.; Liu, S.T.; Hsu, K.W.; Chen, D.R.; Wu, H.T. USP7 Induces Chemoresistance in Triple-Negative Breast Cancer via Deubiquitination and Stabilization of ABCB1. Cells 2022, 11, 3294. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Zhang, J.; Xu, C.; Zhang, S.; Bian, S.; Jiang, F.; Ni, W.; Qu, L.; Lu, C.; Ni, R.; et al. Ubiquitin-specific protease 7 is a drug-able target that promotes hepatocellular carcinoma and chemoresistance. Cancer Cell Int. 2020, 20, 28. [Google Scholar] [CrossRef]
- Liu, J.; Zhou, T.; Dong, X.; Guo, Q.; Zheng, L.; Wang, X.; Zhang, N.; Li, D.; Ren, L.; Yi, F.; et al. De-ubiquitination of SAMHD1 by USP7 promotes DNA damage repair to overcome oncogenic stress and affect chemotherapy sensitivity. Oncogene 2023, 42, 1843–1856. [Google Scholar] [CrossRef]
- Oliveira, R.I.; Guedes, R.A.; Salvador, J.A.R. Highlights in USP7 inhibitors for cancer treatment. Front. Chem. 2022, 10, 1005727. [Google Scholar] [CrossRef]
- Zhou, J.; Wang, J.; Chen, C.; Yuan, H.; Wen, X.; Sun, H. USP7: Target Validation and Drug Discovery for Cancer Therapy. Med. Chem. 2018, 14, 3–18. [Google Scholar] [CrossRef]
- Vasan, N.; Baselga, J.; Hyman, D.M. A view on drug resistance in cancer. Nature 2019, 575, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Mansoori, B.; Mohammadi, A.; Davudian, S.; Shirjang, S.; Baradaran, B. The Different Mechanisms of Cancer Drug Resistance: A Brief Review. Adv. Pharm. Bull. 2017, 7, 339–348. [Google Scholar] [CrossRef]
- Wu, Q.; Yang, Z.; Nie, Y.; Shi, Y.; Fan, D. Multi-drug resistance in cancer chemotherapeutics: Mechanisms and lab approaches. Cancer Lett. 2014, 347, 159–166. [Google Scholar] [CrossRef]
- Desai, S.; Liu, Z.; Yao, J.; Patel, N.; Chen, J.; Wu, Y.; Ahn, E.E.; Fodstad, O.; Tan, M. Heat shock factor 1 (HSF1) controls chemoresistance and autophagy through transcriptional regulation of autophagy-related protein 7 (ATG7). J. Biol. Chem. 2013, 288, 9165–9176. [Google Scholar] [CrossRef]
- Cyran, A.M.; Zhitkovich, A. Heat Shock Proteins and HSF1 in Cancer. Front. Oncol. 2022, 12, 860320. [Google Scholar] [CrossRef] [PubMed]
- Dai, C.; Sampson, S.B. HSF1: Guardian of Proteostasis in Cancer. Trends Cell Biol. 2016, 26, 17–28. [Google Scholar] [CrossRef]
- Wang, H.; Mi, K. Emerging roles of endoplasmic reticulum stress in the cellular plasticity of cancer cells. Front. Oncol. 2023, 13, 1110881. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Cubillos-Ruiz, J.R. Endoplasmic reticulum stress signals in the tumour and its microenvironment. Nat. Rev. Cancer 2021, 21, 71–88. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Liu, Z.; Liang, M.X.; Fei, Y.J.; Zhang, W.; Wu, Y.; Tang, J.H. Endoplasmic reticulum stress targeted therapy for breast cancer. Cell Commun. Signal 2022, 20, 174. [Google Scholar] [CrossRef] [PubMed]
- Rouschop, K.M.; Dubois, L.J.; Keulers, T.G.; van den Beucken, T.; Lambin, P.; Bussink, J.; van der Kogel, A.J.; Koritzinsky, M.; Wouters, B.G. PERK/eIF2alpha signaling protects therapy resistant hypoxic cells through induction of glutathione synthesis and protection against ROS. Proc. Natl. Acad. Sci. USA 2013, 110, 4622–4627. [Google Scholar] [CrossRef] [PubMed]
- Cullinan, S.B.; Zhang, D.; Hannink, M.; Arvisais, E.; Kaufman, R.J.; Diehl, J.A. Nrf2 is a direct PERK substrate and effector of PERK-dependent cell survival. Mol. Cell Biol. 2003, 23, 7198–7209. [Google Scholar] [CrossRef] [PubMed]
- Bobrovnikova-Marjon, E.; Grigoriadou, C.; Pytel, D.; Zhang, F.; Ye, J.; Koumenis, C.; Cavener, D.; Diehl, J.A. PERK promotes cancer cell proliferation and tumor growth by limiting oxidative DNA damage. Oncogene 2010, 29, 3881–3895. [Google Scholar] [CrossRef]
- Bobrovnikova-Marjon, E.; Hatzivassiliou, G.; Grigoriadou, C.; Romero, M.; Cavener, D.R.; Thompson, C.B.; Diehl, J.A. PERK-dependent regulation of lipogenesis during mouse mammary gland development and adipocyte differentiation. Proc. Natl. Acad. Sci. USA 2008, 105, 16314–16319. [Google Scholar] [CrossRef]
- Bu, Y.; Yoshida, A.; Chitnis, N.; Altman, B.J.; Tameire, F.; Oran, A.; Gennaro, V.; Armeson, K.E.; McMahon, S.B.; Wertheim, G.B.; et al. A PERK-miR-211 axis suppresses circadian regulators and protein synthesis to promote cancer cell survival. Nat. Cell Biol. 2018, 20, 104–115. [Google Scholar] [CrossRef]
- Zhu, X.; Li, S.; Xu, B.; Luo, H. Cancer evolution: A means by which tumors evade treatment. Biomed. Pharmacother. 2021, 133, 111016. [Google Scholar] [CrossRef]
- Ciccarelli, M.; Masser, A.E.; Kaimal, J.M.; Planells, J.; Andreasson, C. Genetic inactivation of essential HSF1 reveals an isolated transcriptional stress response selectively induced by protein misfolding. Mol. Biol. Cell 2023, 34, ar101. [Google Scholar] [CrossRef]
- Fujimoto, M.; Takii, R.; Matsumoto, M.; Okada, M.; Nakayama, K.I.; Nakato, R.; Fujiki, K.; Shirahige, K.; Nakai, A. HSF1 phosphorylation establishes an active chromatin state via the TRRAP-TIP60 complex and promotes tumorigenesis. Nat. Commun. 2022, 13, 4355. [Google Scholar] [CrossRef] [PubMed]
- Chou, S.D.; Prince, T.; Gong, J.; Calderwood, S.K. mTOR is essential for the proteotoxic stress response, HSF1 activation and heat shock protein synthesis. PLoS ONE 2012, 7, e39679. [Google Scholar] [CrossRef] [PubMed]
- Dayalan Naidu, S.; Sutherland, C.; Zhang, Y.; Risco, A.; de la Vega, L.; Caunt, C.J.; Hastie, C.J.; Lamont, D.J.; Torrente, L.; Chowdhry, S.; et al. Heat Shock Factor 1 Is a Substrate for p38 Mitogen-Activated Protein Kinases. Mol. Cell Biol. 2016, 36, 2403–2417. [Google Scholar] [CrossRef] [PubMed]
- San Gil, R.; Ooi, L.; Yerbury, J.J.; Ecroyd, H. The heat shock response in neurons and astroglia and its role in neurodegenerative diseases. Mol. Neurodegener. 2017, 12, 65. [Google Scholar] [CrossRef]
- Lee, G.; Oh, T.I.; Um, K.B.; Yoon, H.; Son, J.; Kim, B.M.; Kim, H.I.; Kim, H.; Kim, Y.J.; Lee, C.S.; et al. Small-molecule inhibitors of USP7 induce apoptosis through oxidative and endoplasmic reticulum stress in cancer cells. Biochem. Biophys. Res. Commun. 2016, 470, 181–186. [Google Scholar] [CrossRef]
- Lee, Y.Y.; Gil, E.S.; Jeong, I.H.; Kim, H.; Jang, J.H.; Choung, Y.H. Heat Shock Factor 1 Prevents Age-Related Hearing Loss by Decreasing Endoplasmic Reticulum Stress. Cells 2021, 10, 2454. [Google Scholar] [CrossRef]
- Lee, A.H.; Iwakoshi, N.N.; Anderson, K.C.; Glimcher, L.H. Proteasome inhibitors disrupt the unfolded protein response in myeloma cells. Proc. Natl. Acad. Sci. USA 2003, 100, 9946–9951. [Google Scholar] [CrossRef]
- Pallepati, P.; Averill-Bates, D.A. Activation of ER stress and apoptosis by hydrogen peroxide in HeLa cells: Protective role of mild heat preconditioning at 40 degrees C. Biochim. Biophys. Acta 2011, 1813, 1987–1999. [Google Scholar] [CrossRef]
- Zhang, C.W.; Wang, Y.N.; Ge, X.L. Lenalidomide use in multiple myeloma (Review). Mol. Clin. Oncol. 2024, 20, 7. [Google Scholar] [CrossRef]
- Cantadori, L.O.; Gaiolla, R.D.; Nunes-Nogueira, V.D.S. Effect of bortezomib on the treatment of multiple myeloma: A systematic review protocol. BMJ Open 2022, 12, e061808. [Google Scholar] [CrossRef]
- Kozalak, G.; Butun, I.; Toyran, E.; Kosar, A. Review on Bortezomib Resistance in Multiple Myeloma and Potential Role of Emerging Technologies. Pharmaceuticals 2023, 16, 111. [Google Scholar] [CrossRef]
- Mitsiades, N.; Mitsiades, C.S.; Poulaki, V.; Chauhan, D.; Fanourakis, G.; Gu, X.; Bailey, C.; Joseph, M.; Libermann, T.A.; Treon, S.P.; et al. Molecular sequelae of proteasome inhibition in human multiple myeloma cells. Proc. Natl. Acad. Sci. USA 2002, 99, 14374–14379. [Google Scholar] [CrossRef]
- Tchenio, T.; Havard, M.; Martinez, L.A.; Dautry, F. Heat shock-independent induction of multidrug resistance by heat shock factor 1. Mol. Cell Biol. 2006, 26, 580–591. [Google Scholar] [CrossRef] [PubMed]
- Vydra, N.; Toma, A.; Glowala-Kosinska, M.; Gogler-Piglowska, A.; Widlak, W. Overexpression of Heat Shock Transcription Factor 1 enhances the resistance of melanoma cells to doxorubicin and paclitaxel. BMC Cancer 2013, 13, 504. [Google Scholar] [CrossRef]
- Shah, S.P.; Nooka, A.K.; Jaye, D.L.; Bahlis, N.J.; Lonial, S.; Boise, L.H. Bortezomib-induced heat shock response protects multiple myeloma cells and is activated by heat shock factor 1 serine 326 phosphorylation. Oncotarget 2016, 7, 59727–59741. [Google Scholar] [CrossRef] [PubMed]
- Dayalan Naidu, S.; Dinkova-Kostova, A.T. Regulation of the mammalian heat shock factor 1. FEBS J. 2017, 284, 1606–1627. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Khaleque, M.A.; Zhao, M.J.; Zhong, R.; Gaestel, M.; Calderwood, S.K. Phosphorylation of HSF1 by MAPK-activated protein kinase 2 on serine 121, inhibits transcriptional activity and promotes HSP90 binding. J. Biol. Chem. 2006, 281, 782–791. [Google Scholar] [CrossRef]
- Wang, G.; Cao, P.; Fan, Y.; Tan, K. Emerging roles of HSF1 in cancer: Cellular and molecular episodes. Biochim. Biophys. Acta Rev. Cancer 2020, 1874, 188390. [Google Scholar] [CrossRef]
- Olsen, J.V.; Blagoev, B.; Gnad, F.; Macek, B.; Kumar, C.; Mortensen, P.; Mann, M. Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell 2006, 127, 635–648. [Google Scholar] [CrossRef]
- Soncin, F.; Zhang, X.; Chu, B.; Wang, X.; Asea, A.; Ann Stevenson, M.; Sacks, D.B.; Calderwood, S.K. Transcriptional activity and DNA binding of heat shock factor-1 involve phosphorylation on threonine 142 by CK2. Biochem. Biophys. Res. Commun. 2003, 303, 700–706. [Google Scholar] [CrossRef]
- Park, J.; Liu, A.Y. JNK phosphorylates the HSF1 transcriptional activation domain: Role of JNK in the regulation of the heat shock response. J. Cell Biochem. 2001, 82, 326–338. [Google Scholar] [CrossRef]
- Haberzettl, P.; Hill, B.G. Oxidized lipids activate autophagy in a JNK-dependent manner by stimulating the endoplasmic reticulum stress response. Redox Biol. 2013, 1, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Mandula, J.K.; Chang, S.; Mohamed, E.; Jimenez, R.; Sierra-Mondragon, R.A.; Chang, D.C.; Obermayer, A.N.; Moran-Segura, C.M.; Das, S.; Vazquez-Martinez, J.A.; et al. Ablation of the endoplasmic reticulum stress kinase PERK induces paraptosis and type I interferon to promote anti-tumor T cell responses. Cancer Cell 2022, 40, 1145–1160.e9. [Google Scholar] [CrossRef]
- Botrus, G.; Miller, R.M.; Uson Junior, P.L.S.; Kannan, G.; Han, H.; Von Hoff, D.D. Increasing Stress to Induce Apoptosis in Pancreatic Cancer via the Unfolded Protein Response (UPR). Int. J. Mol. Sci. 2022, 24, 577. [Google Scholar] [CrossRef] [PubMed]
- Alasiri, G.; Jiramongkol, Y.; Zona, S.; Fan, L.Y.; Mahmud, Z.; Gong, G.; Lee, H.J.; Lam, E.W. Regulation of PERK expression by FOXO3: A vulnerability of drug-resistant cancer cells. Oncogene 2019, 38, 6382–6398. [Google Scholar] [CrossRef]
- Salaroglio, I.C.; Panada, E.; Moiso, E.; Buondonno, I.; Provero, P.; Rubinstein, M.; Kopecka, J.; Riganti, C. PERK induces resistance to cell death elicited by endoplasmic reticulum stress and chemotherapy. Mol. Cancer 2017, 16, 91. [Google Scholar] [CrossRef] [PubMed]
- Akman, M.; Belisario, D.C.; Salaroglio, I.C.; Kopecka, J.; Donadelli, M.; De Smaele, E.; Riganti, C. Hypoxia, endoplasmic reticulum stress and chemoresistance: Dangerous liaisons. J. Exp. Clin. Cancer Res. 2021, 40, 28. [Google Scholar] [CrossRef]
- Li, X.; Liang, M.; Jiang, J.; He, R.; Wang, M.; Guo, X.; Shen, M.; Qin, R. Combined inhibition of autophagy and Nrf2 signaling augments bortezomib-induced apoptosis by increasing ROS production and ER stress in pancreatic cancer cells. Int. J. Biol. Sci. 2018, 14, 1291–1305. [Google Scholar] [CrossRef]
- Gentleman, R.C.; Carey, V.J.; Bates, D.M.; Bolstad, B.; Dettling, M.; Dudoit, S.; Ellis, B.; Gautier, L.; Ge, Y.; Gentry, J.; et al. Bioconductor: Open software development for computational biology and bioinformatics. Genome Biol. 2004, 5, R80. [Google Scholar] [CrossRef] [PubMed]
- Oh, T.I.; Lee, M.; Lee, Y.M.; Kim, G.H.; Lee, D.; You, J.S.; Kim, S.H.; Choi, M.; Jang, H.; Park, Y.M.; et al. PGC1alpha Loss Promotes Lung Cancer Metastasis through Epithelial-Mesenchymal Transition. Cancers 2021, 13, 1772. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.; Oh, T.; Bahk, Y.Y.; Kim, G.H.; Kan, S.Y.; Shin, D.H.; Kim, J.H.; Lim, J.H. HSF1 Regulates Mevalonate and Cholesterol Biosynthesis Pathways. Cancers 2019, 11, 1363. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Grammatikakis, N.; Siganou, A.; Calderwood, S.K. Regulation of molecular chaperone gene transcription involves the serine phosphorylation, 14-3-3 epsilon binding, and cytoplasmic sequestration of heat shock factor 1. Mol. Cell Biol. 2003, 23, 6013–6026. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward Sequences (5′-3′) | Reverse Sequences (5′-3′) |
|---|---|---|
| USP7 | CGAGGACATGGAGATGGAAG | GTTGTGTCCATCACTCAGGG |
| HSPA6 | GCGCAAAATGCAAGACAAGTG | GAGAAGATGGGGCGACAGATT |
| HSPA1B | AGGCCAACAAGATCACCATC | TCGTCCTCCGCTTTGTACTT |
| HSPA1A | GCCGAGAAGGACGAGTTTGA | TCCGCTGATGATGGGGTTAC |
| HMOX | AAGATTGCCCAGAAAGCCCTGGAC | AACTGTCGCCACCAGAAAGCTGAG |
| DNAJB1 | GACCCTCATGCCATGTTTGC | CCCATAGGGAAGCCAGAGAAT |
| β-actin | ACGAGGCCCAGAGCAAGAG | TCTCCAAGTCGTCCCAGTTG |
| Gene | Forward Sequences (5′-3′) | Reverse Sequences (5′-3′) |
|---|---|---|
| HSPA1A | GGCGAAACCCCTGGAATATTCCCGA | AGCCTTGGGACAACGGGAG |
| HSPA6 | GGAAGGTGCGGGAAGGTTCG | TTCTTGTCGGATGCTGGA |
| HSPA1B | GGTCCGCTTCGTCTTTCG | CTCTGTGGGCTCCGCTCT |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lim, C.-H.; Fang, X.-Q.; Kang, H.; Oh, T.; Lee, S.; Kim, Y.-S.; Lim, J.-H. ER Stress-Activated HSF1 Governs Cancer Cell Resistance to USP7 Inhibitor-Based Chemotherapy through the PERK Pathway. Int. J. Mol. Sci. 2024, 25, 2768. https://doi.org/10.3390/ijms25052768
Lim C-H, Fang X-Q, Kang H, Oh T, Lee S, Kim Y-S, Lim J-H. ER Stress-Activated HSF1 Governs Cancer Cell Resistance to USP7 Inhibitor-Based Chemotherapy through the PERK Pathway. International Journal of Molecular Sciences. 2024; 25(5):2768. https://doi.org/10.3390/ijms25052768
Chicago/Turabian StyleLim, Chang-Hoon, Xue-Quan Fang, Hyeji Kang, Taerim Oh, Seonghoon Lee, Young-Seon Kim, and Ji-Hong Lim. 2024. "ER Stress-Activated HSF1 Governs Cancer Cell Resistance to USP7 Inhibitor-Based Chemotherapy through the PERK Pathway" International Journal of Molecular Sciences 25, no. 5: 2768. https://doi.org/10.3390/ijms25052768
APA StyleLim, C.-H., Fang, X.-Q., Kang, H., Oh, T., Lee, S., Kim, Y.-S., & Lim, J.-H. (2024). ER Stress-Activated HSF1 Governs Cancer Cell Resistance to USP7 Inhibitor-Based Chemotherapy through the PERK Pathway. International Journal of Molecular Sciences, 25(5), 2768. https://doi.org/10.3390/ijms25052768