
Uncovering the Role of Hydroxycinnamoyl Transferase in Boosting Chlorogenic Acid Accumulation in Carthamus tinctorius Cells under Methyl Jasmonate Elicitation

 , , and
, , and
Abstract

1. Introduction
2. Results
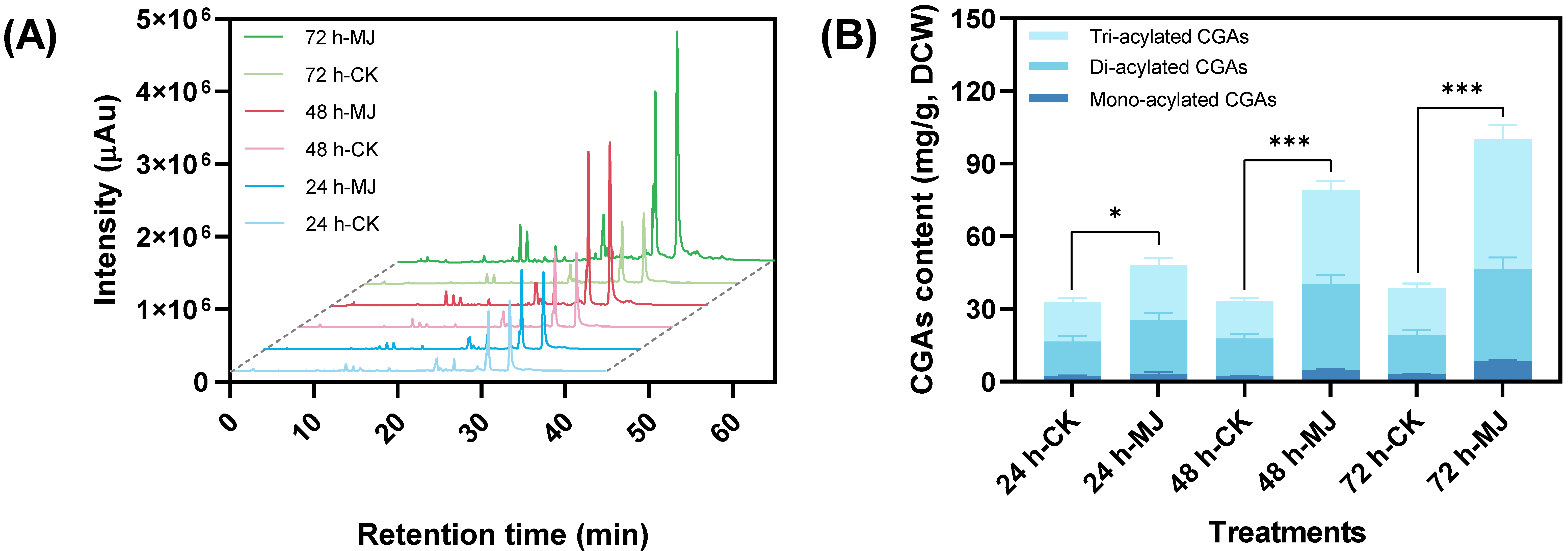
2.1. Temporal Effect of Methyl Jasmonate on Intracellular Metabolites in C. tinctorius Cells
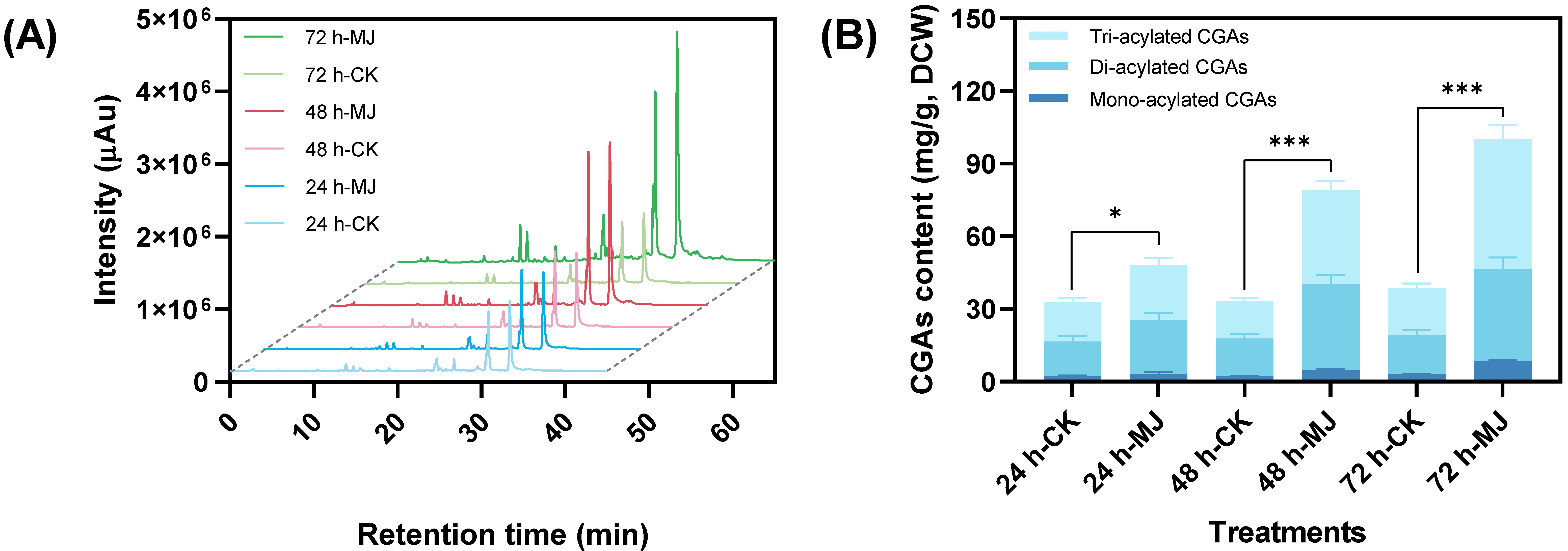
2.1.1. Promoting Effect on Chlorogenic Acid Accumulation
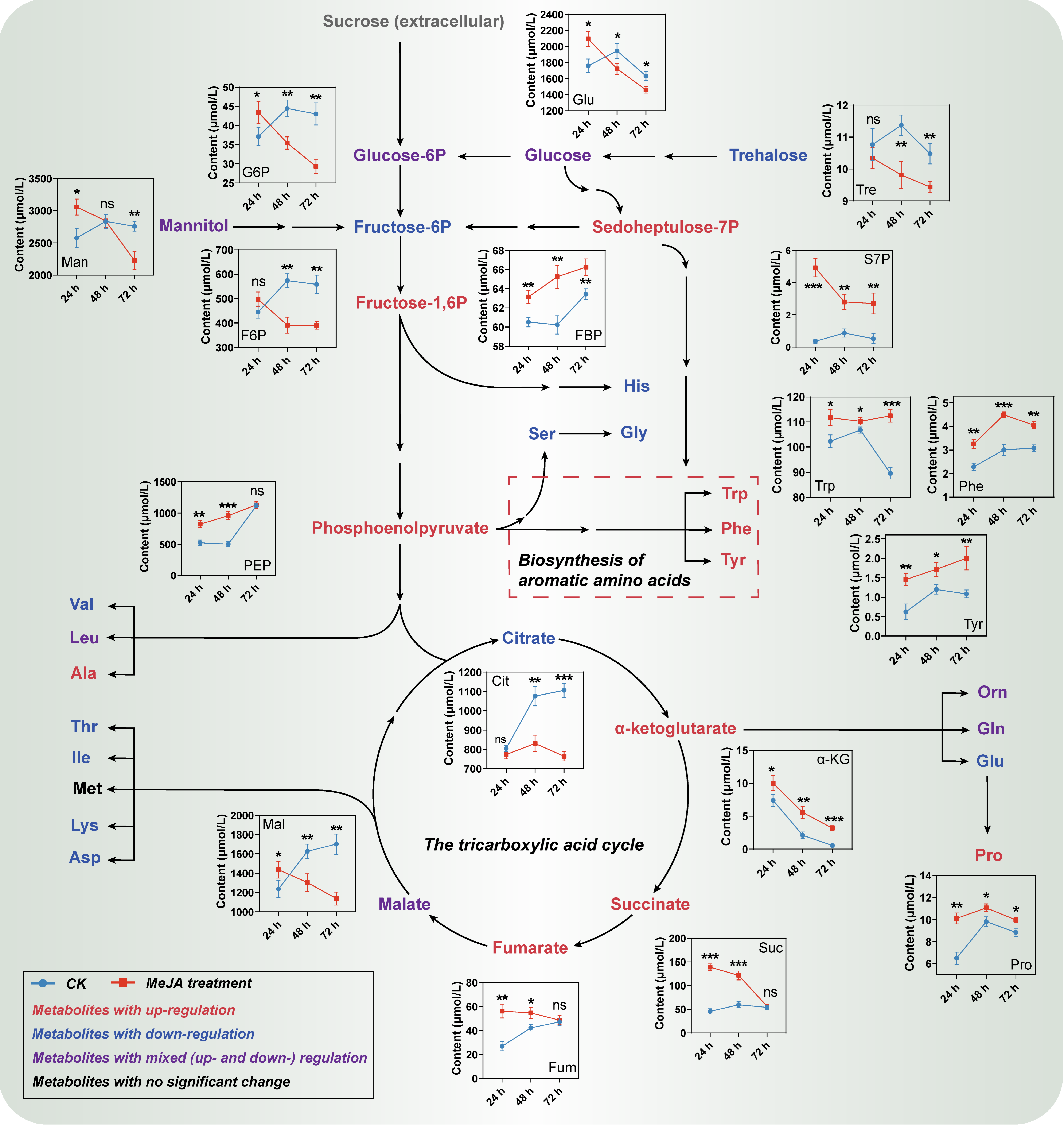
2.1.2. Metabolic Profile Changes of Primary Metabolites
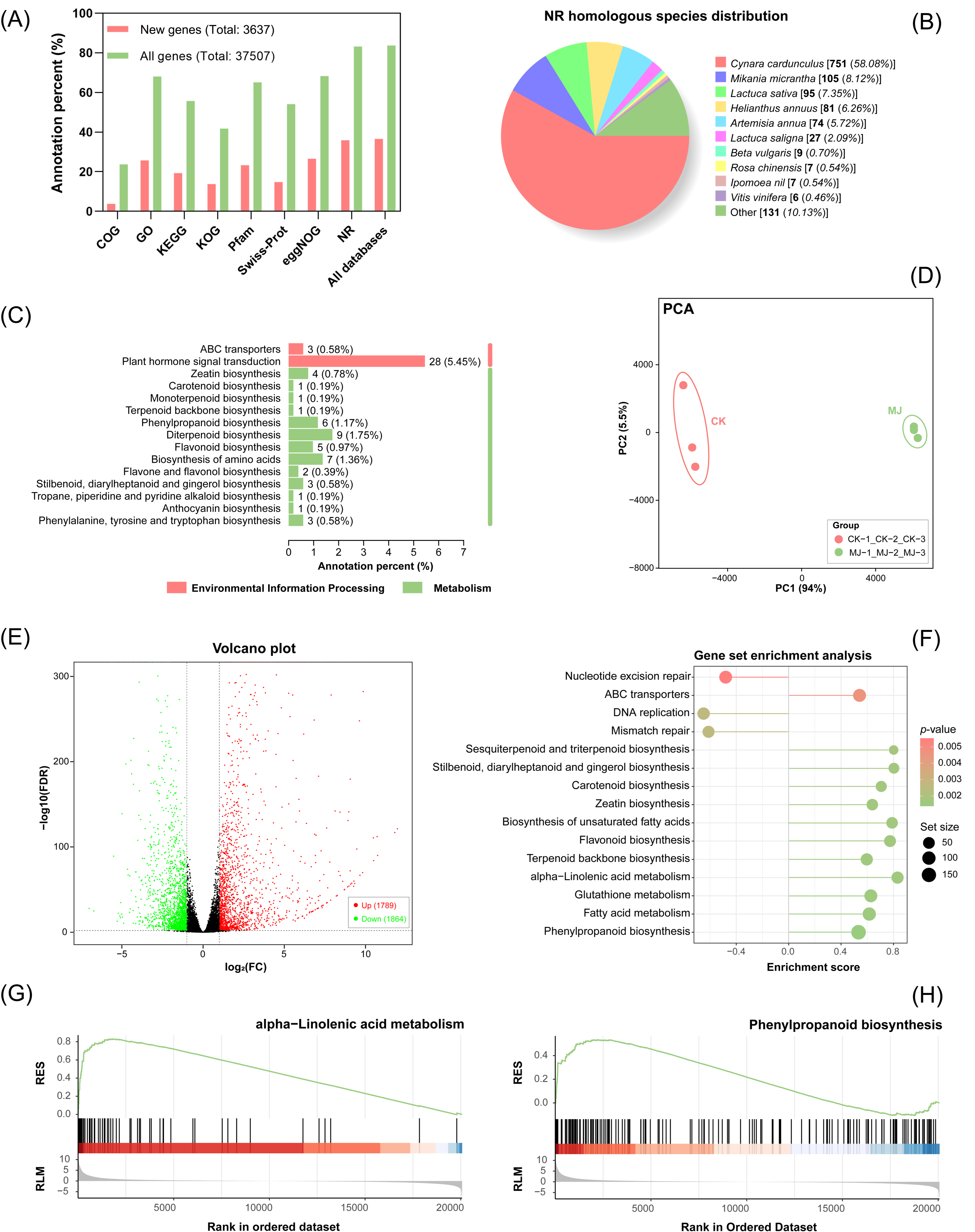
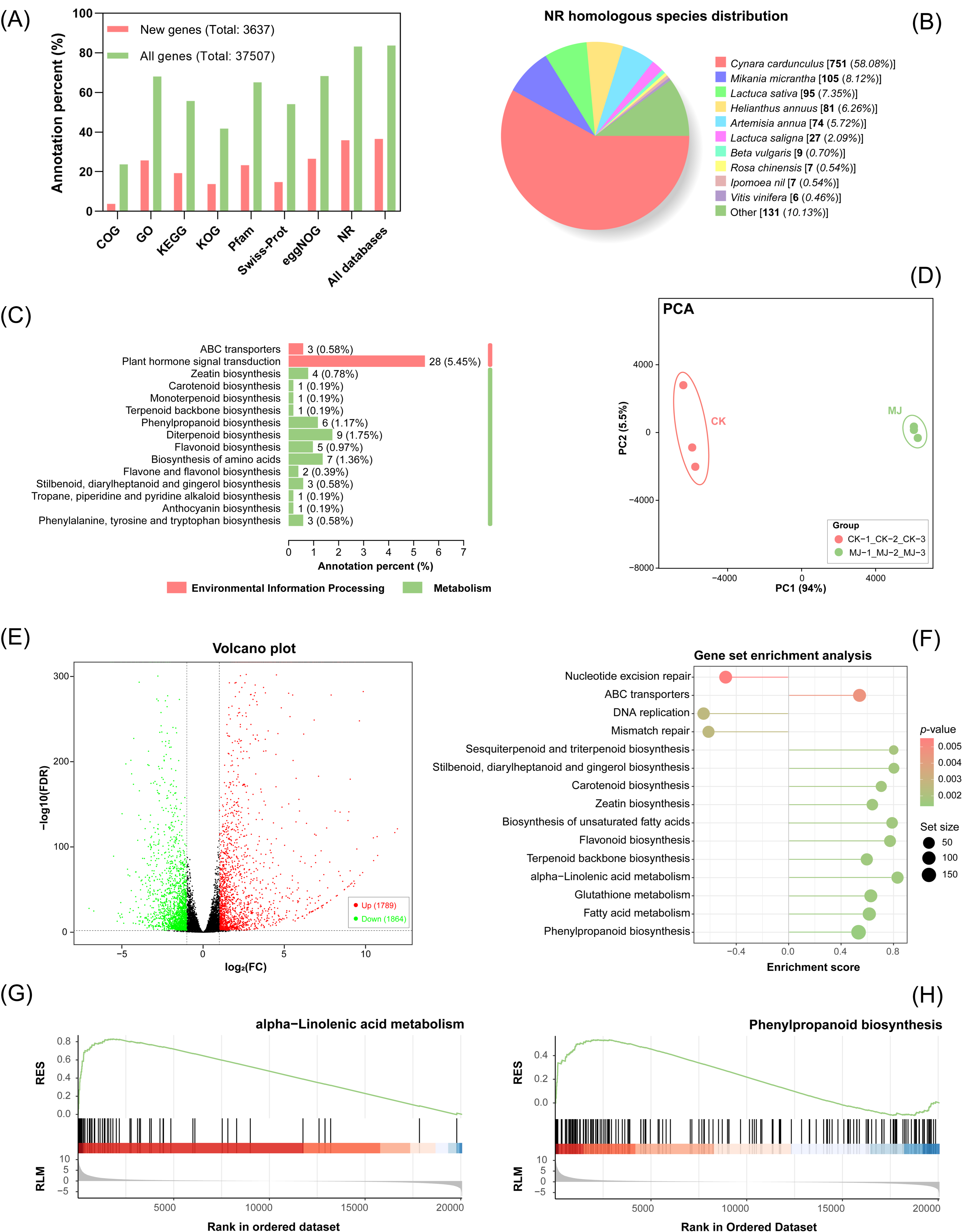
2.2. Overall Transcriptional Alteration of C. tinctorius Cells in Response to MeJA Treatment
2.2.1. Results of Gene Annotation and New Gene Identification
2.2.2. Gene Differential Expression and Gene Set Enrichment Analysis
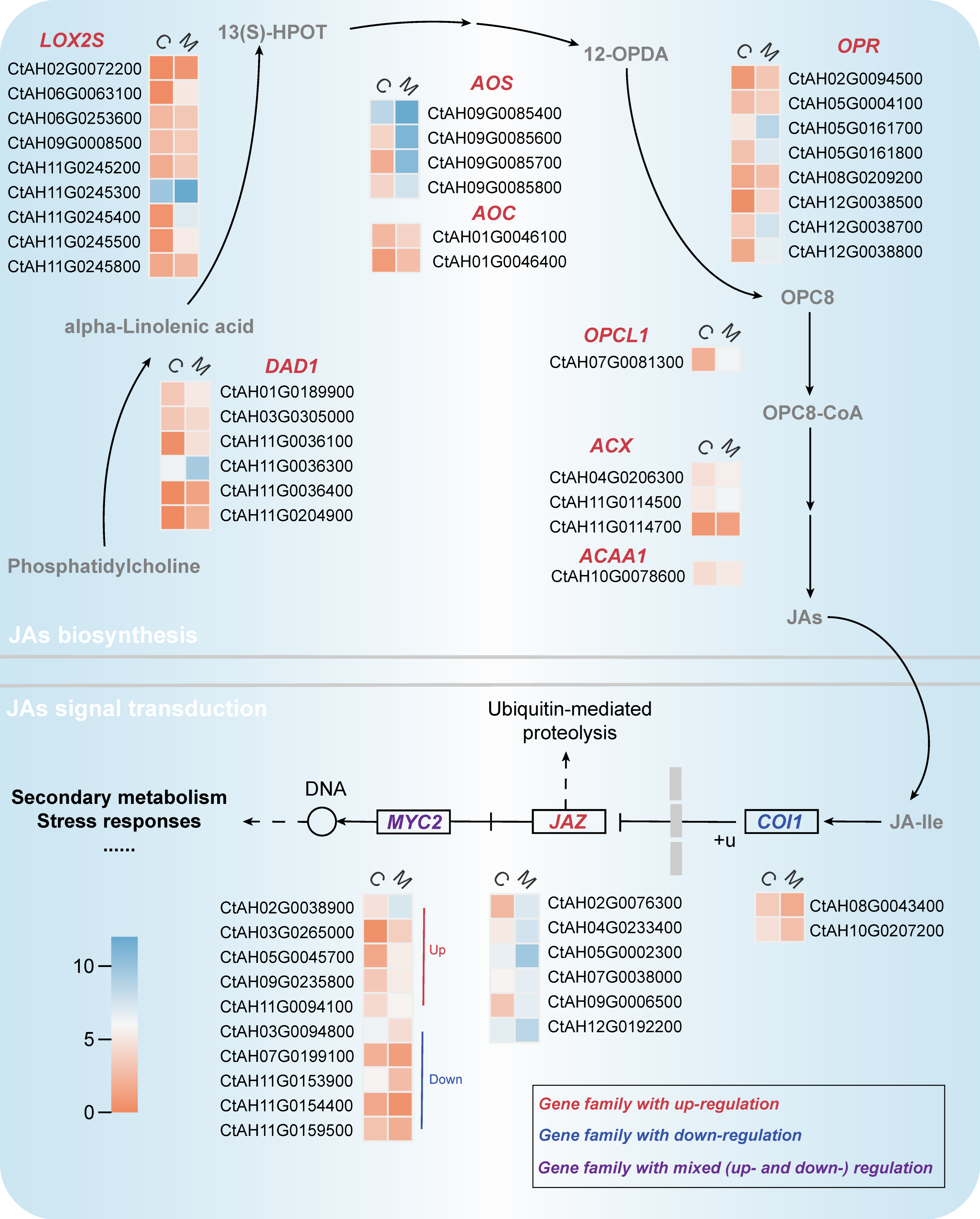
2.3. Transcriptional Differential Analysis of Endogenous Jasmonate Biosynthesis and Its Signaling Pathways in C. tinctorius Cells under MeJA Elicitation
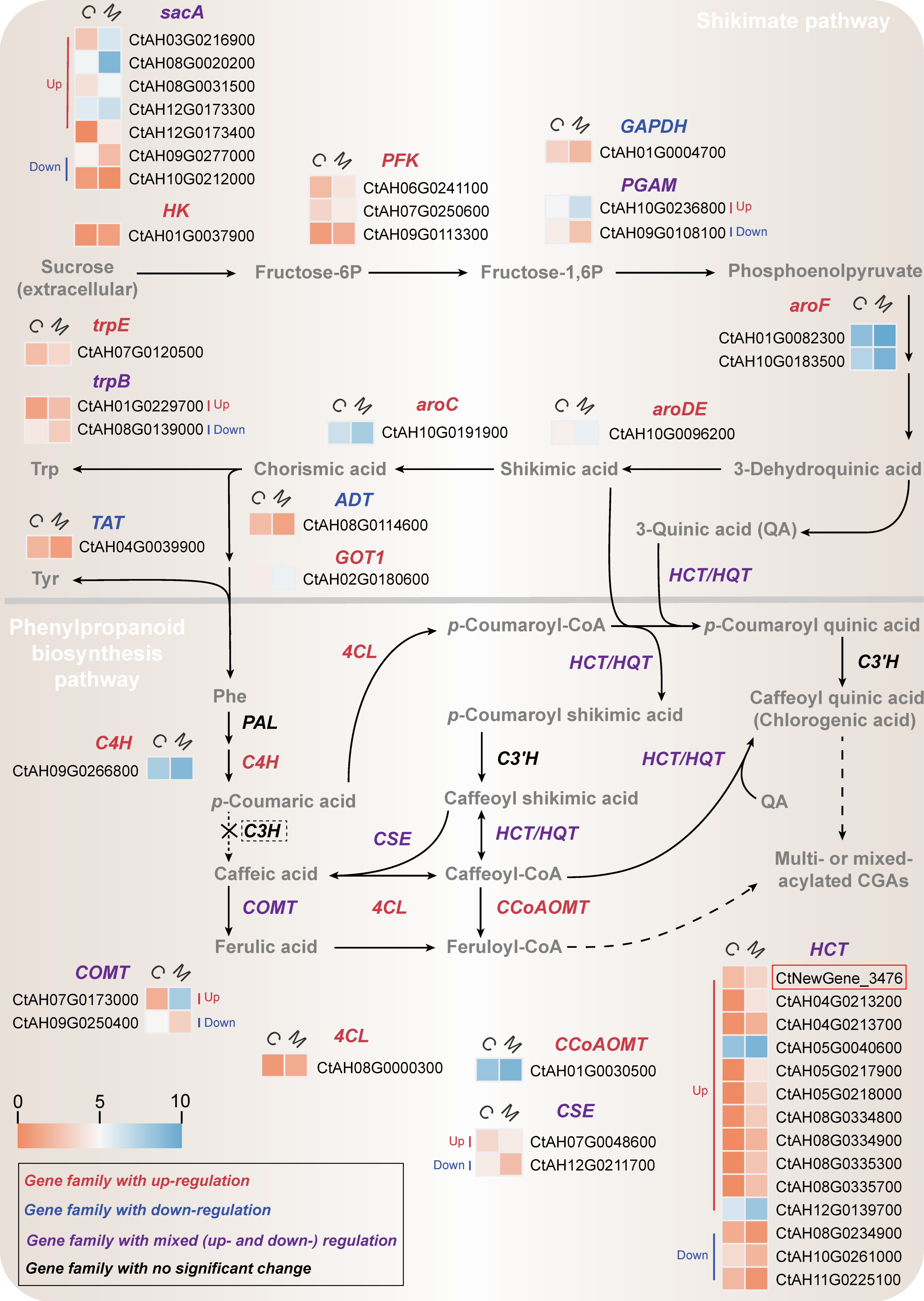
2.4. Transcriptional Differential Analysis of CGA biosynthesis Pathway in MeJA-Induced C. tinctorius Cells
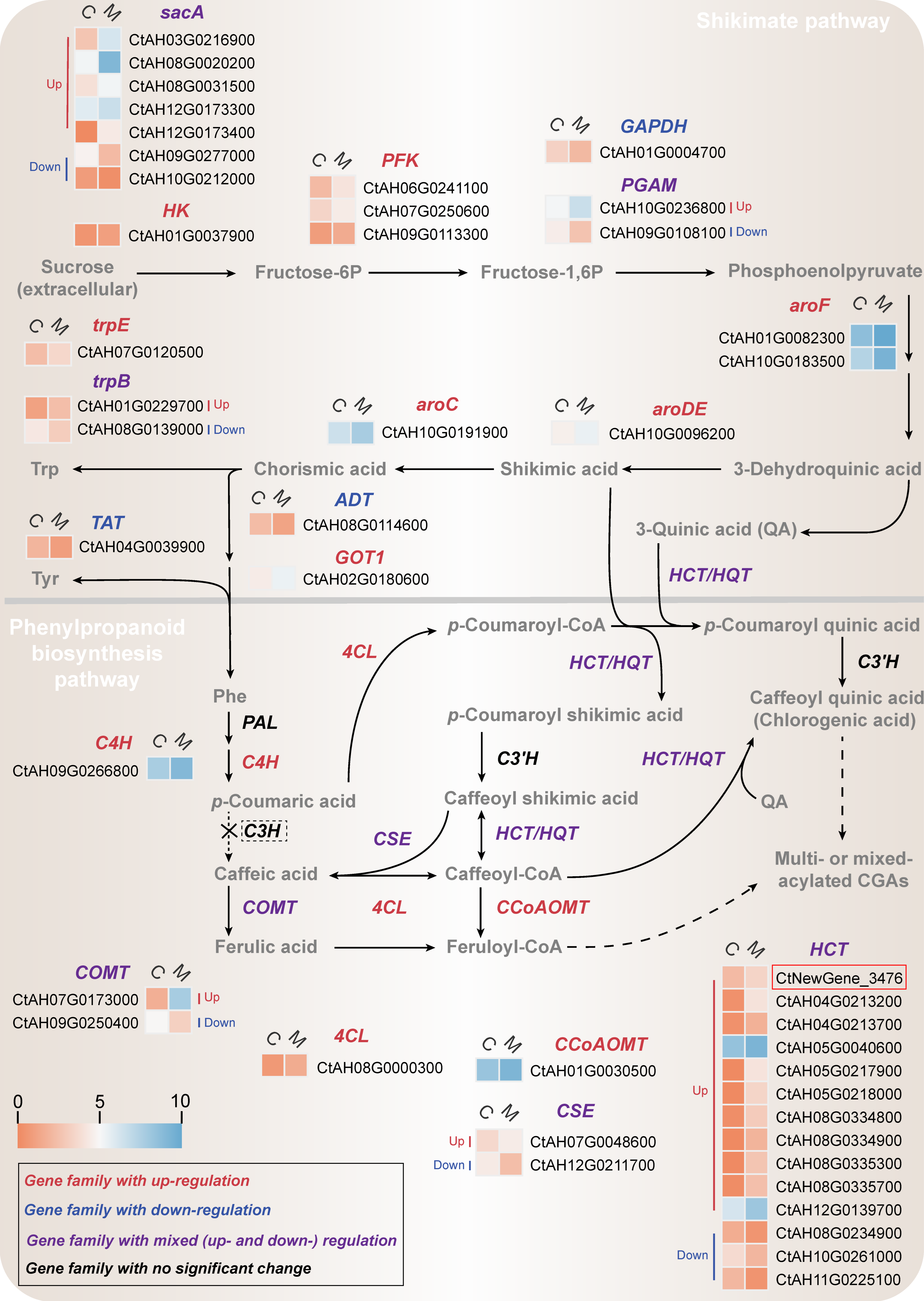
2.4.1. Transcriptional Changes in Shikimate Pathway
2.4.2. Transcriptional Changes in Phenylpropanoid Biosynthesis Pathway
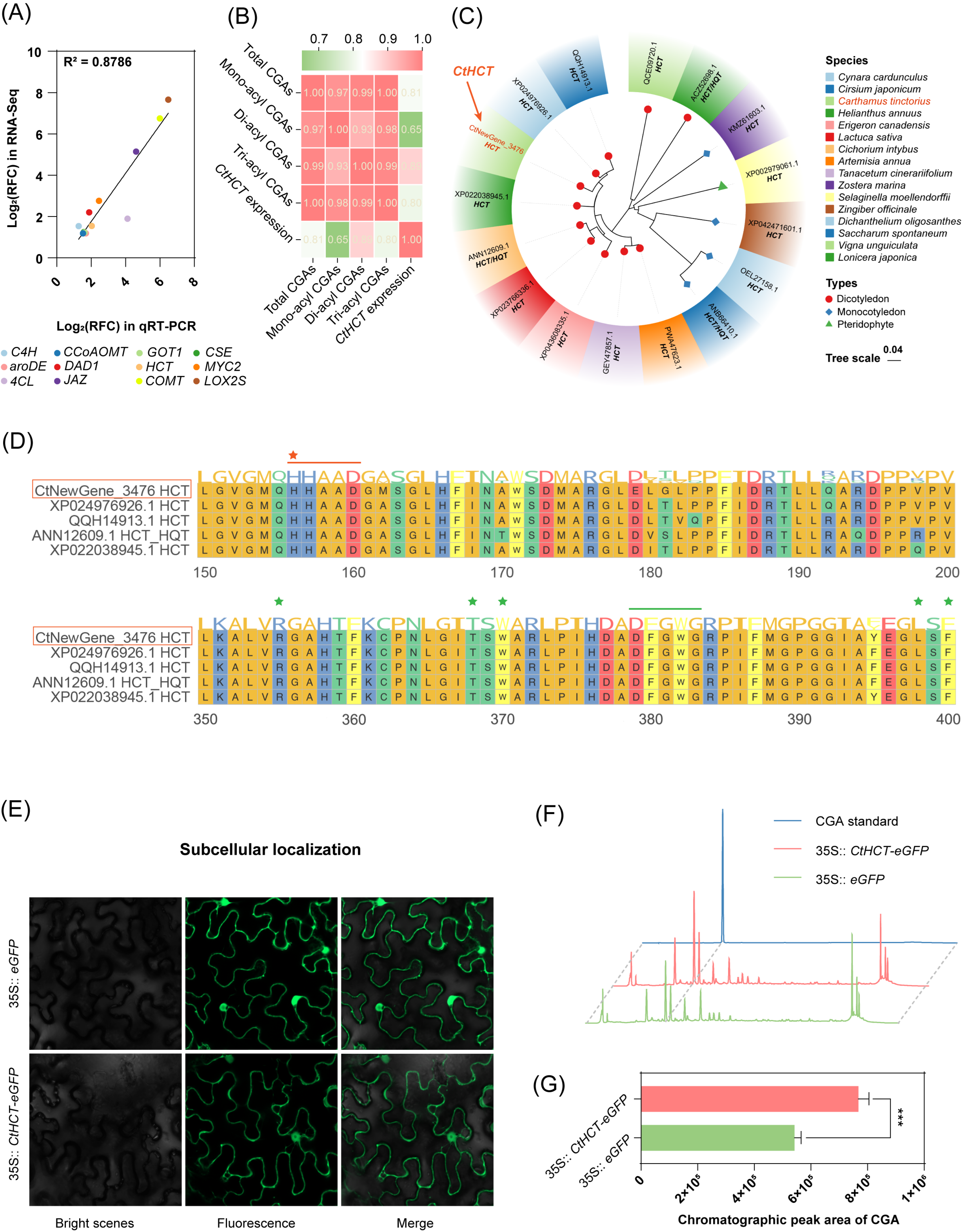
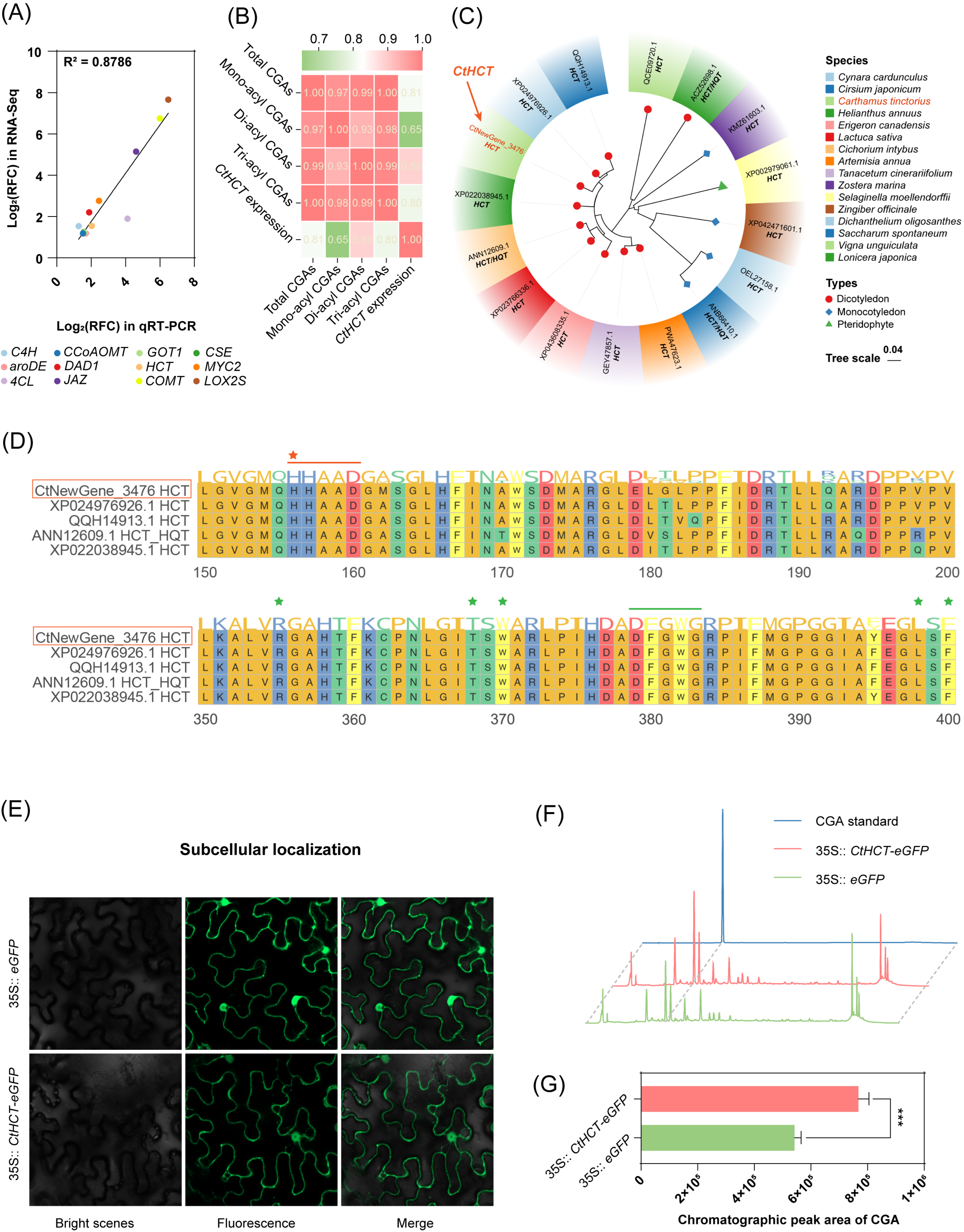
2.4.3. Correlation Analysis between the Newly Annotated CtHCT Expression and CGA Accumulation in MeJA-Treated C. tinctorius Cells
2.5. Analysis of Bioinformatics, Subcellular Localization, and Transient Expression of the Newly Annotated and MeJA-Responsive CtHCT
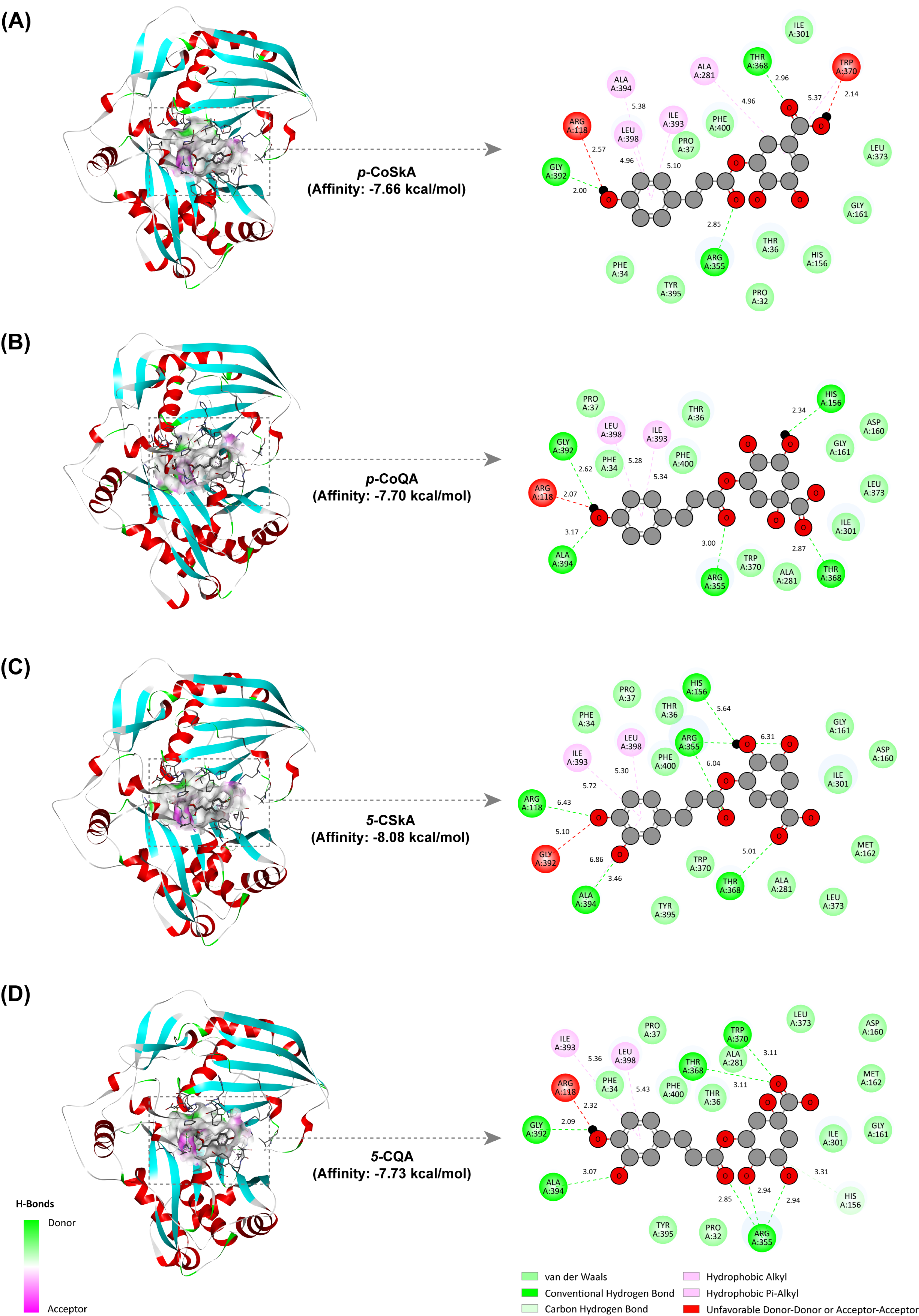
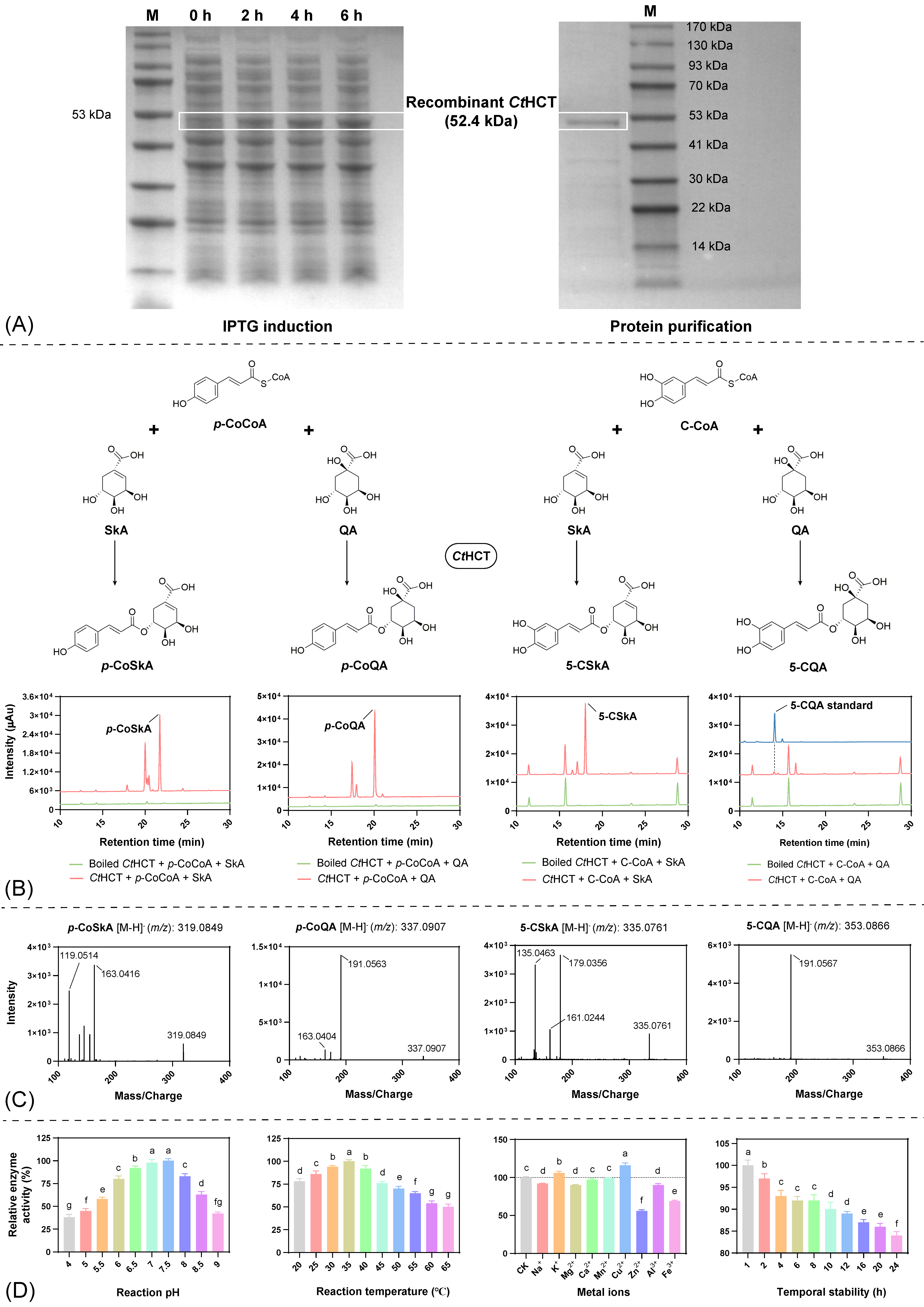
2.6. In Vitro Catalytic Kinetic Analysis of the Recombinant CtHCT Protein
2.6.1. In Vitro Catalytic Activity Assay
2.6.2. Reaction Condition Optimization
2.6.3. Enzymatic Kinetic Analysis
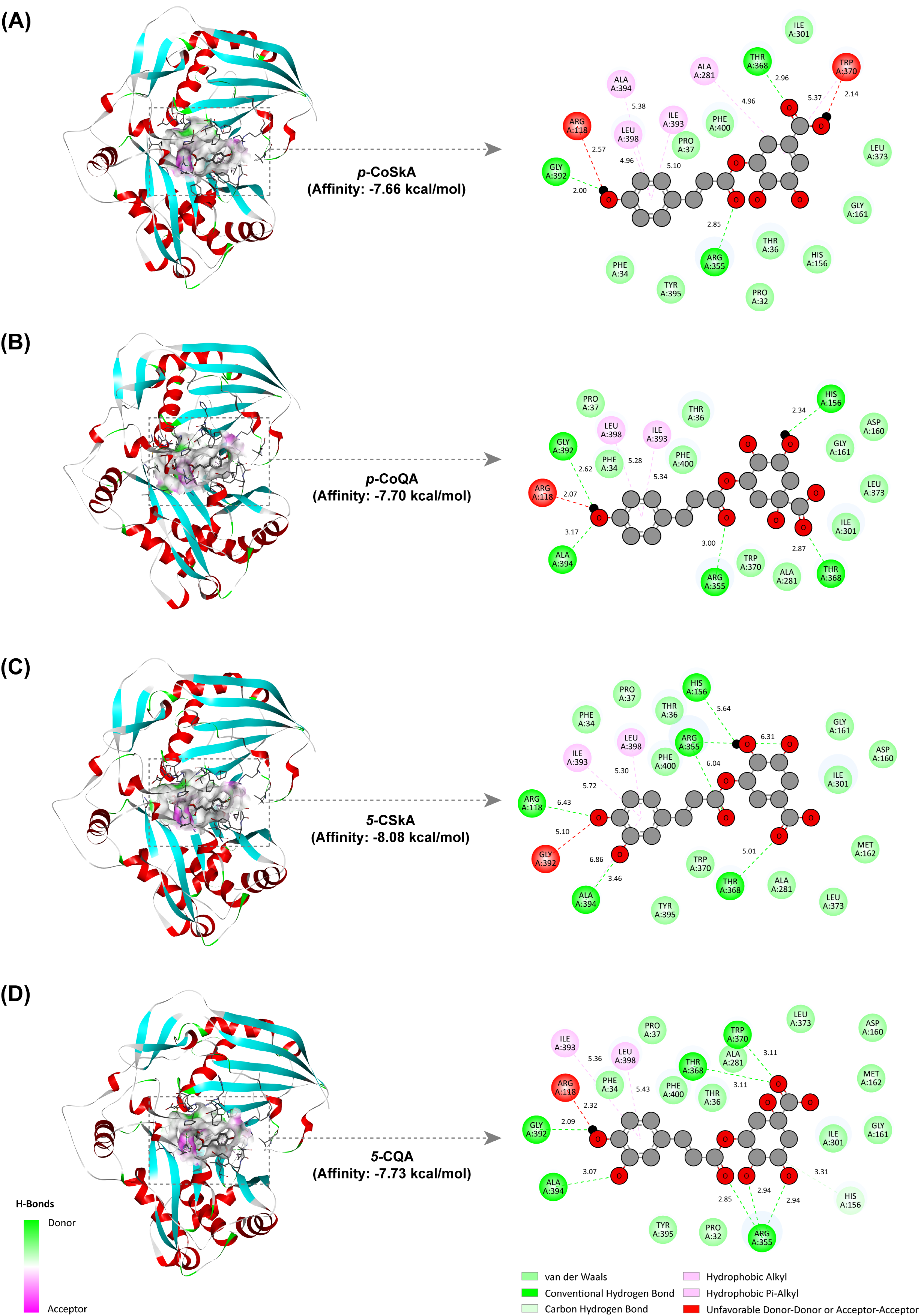
2.7. Revealing the Catalytic Mechanism of the CtHCT Protein by Molecular Docking
3. Discussion
4. Materials and Methods
4.1. Product Induction of C. tinctorius Cells
4.2. Intracellular Metabolites Detection
4.3. Transcriptome Sequencing and Analysis
4.3.1. Transcriptome Sequencing
4.3.2. Gene Annotation and New Gene Identification
4.3.3. Gene Expression and Differential Analysis
4.4. Gene Expression Patterns Validation
4.5. Bioinformatics Analysis Methods
4.6. Subcellular Localization and Transient Transformation
4.7. Prokaryotic Expression and Purification of Protein
4.8. In Vitro Enzyme Activity Assays and Product Identification
4.9. Homologous Modeling and Molecular Docking
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Clifford, M.N.; Jaganath, I.B.; Ludwig, I.A.; Crozier, A. Chlorogenic acids and the acyl-quinic acids: Discovery, biosynthesis, bioavailability and bioactivity. Nat. Prod. Rep. 2017, 34, 1391–1421. [Google Scholar] [CrossRef]
- Lu, H.; Tian, Z.; Cui, Y.; Liu, Z.; Ma, X. Chlorogenic acid: A comprehensive review of the dietary sources, processing effects, bioavailability, beneficial properties, mechanisms of action, and future directions. Compr. Rev. Food Sci. Food Saf. 2020, 19, 3130–3158. [Google Scholar] [CrossRef]
- Liu, W.; Li, J.; Zhang, X.; Zu, Y.; Yang, Y.; Liu, W.; Xu, Z.; Gao, H.; Sun, X.; Jiang, X.; et al. Current Advances in Naturally Occurring Caffeoylquinic Acids: Structure, Bioactivity, and Synthesis. J. Agric. Food Chem. 2020, 68, 10489–10516. [Google Scholar] [CrossRef]
- Wang, L.; Pan, X.; Jiang, L.; Chu, Y.; Gao, S.; Jiang, X.; Zhang, Y.; Chen, Y.; Luo, S.; Peng, C. The Biological Activity Mechanism of Chlorogenic Acid and Its Applications in Food Industry: A Review. Front. Nutr. 2022, 9, 943911. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Su, C.; Chen, X.; Wang, Q.; Jiao, W.; Luo, H.; Tang, J.; Wang, W.; Li, S.; Guo, S. Chlorogenic Acids in Cardiovascular Disease: A Review of Dietary Consumption, Pharmacology, and Pharmacokinetics. J. Agric. Food Chem. 2020, 68, 6464–6484. [Google Scholar] [CrossRef] [PubMed]
- Corrigan, H.; Dunne, A.; Purcell, N.; Guo, Y.; Wang, K.; Xuan, H.; Granato, D. Conceptual functional-by-design optimisation of the antioxidant capacity of trans-resveratrol, quercetin, and chlorogenic acid: Application in a functional tea. Food Chem. 2023, 428, 136764. [Google Scholar] [CrossRef]
- Koutroubas, S.D.; Damalas, C.A.; Fotiadis, S. Safflower assimilate remobilization, yield, and oil content in response to nitrogen availability, sowing time, and genotype. Field Crops Res. 2021, 274, 108313. [Google Scholar] [CrossRef]
- Liu, Z.; Du, L.; Liu, N.; Mohsin, A.; Zhu, X.; Sun, H.; Zhou, B.; Yin, Z.; Zhuang, Y.; Guo, M.; et al. Insights into chlorogenic acids’ efficient biosynthesis through Carthamus tinctorius cell suspension cultures and their potential mechanism as α-glucosidase inhibitors. Ind. Crops Prod. 2023, 194, 116337. [Google Scholar] [CrossRef]
- Du, L.; Li, D.; Zhang, J.; Du, J.; Luo, Q.; Xiong, J. Elicitation of Lonicera japonica Thunb suspension cell for enhancement of secondary metabolites and antioxidant activity. Ind. Crops Prod. 2020, 156, 112877. [Google Scholar] [CrossRef]
- Ramabulana, A.-T.; Steenkamp, P.; Madala, N.; Dubery, I.A. Profiling of chlorogenic acids from Bidens pilosa and differentiation of closely related positional isomers with the aid of UHPLC-QTOF-MS/MS-based in-source collision-induced dissociation. Metabolites 2020, 10, 178. [Google Scholar] [CrossRef]
- Chen, J.; Wang, J.; Wang, R.; Xian, B.; Ren, C.; Liu, Q.; Wu, Q.; Pei, J. Integrated metabolomics and transcriptome analysis on flavonoid biosynthesis in safflower (Carthamus tinctorius L.) under MeJA treatment. BMC Plant Biol. 2020, 20, 353. [Google Scholar] [CrossRef]
- Karami, S.; Sabzalian, M.R.; Basaki, T.; Ghaderi, F.; Jamshidi Goharrizi, K. Phylogenetic relationship and sequence diversity of candidate genes involved in anthocyanin biosynthesis pathway in Carthamus species with contrasting seed coat colors. Physiol. Mol. Biol. Plants 2023, 29, 349–360. [Google Scholar] [CrossRef]
- Wu, Z.; Liu, H.; Zhan, W.; Yu, Z.; Qin, E.; Liu, S.; Yang, T.; Xiang, N.; Kudrna, D.; Chen, Y.; et al. The chromosome-scale reference genome of safflower (Carthamus tinctorius) provides insights into linoleic acid and flavonoid biosynthesis. Plant Biotechnol. J. 2021, 19, 1725–1742. [Google Scholar] [CrossRef]
- Cadena-Zamudio, J.D.; Nicasio-Torres, P.; Monribot-Villanueva, J.L.; Guerrero-Analco, J.A.; Ibarra-Laclette, E. Integrated Analysis of the Transcriptome and Metabolome of Cecropia obtusifolia: A Plant with High Chlorogenic Acid Content Traditionally Used to Treat Diabetes Mellitus. Int. J. Mol. Sci. 2020, 21, 7572. [Google Scholar] [CrossRef] [PubMed]
- Tahmasebi, A.; Ebrahimie, E.; Pakniyat, H.; Ebrahimi, M.; Mohammadi-Dehcheshmeh, M. Insights from the Echinacea purpurea (L.) Moench transcriptome: Global reprogramming of gene expression patterns towards activation of secondary metabolism pathways. Ind. Crops Prod. 2019, 132, 365–376. [Google Scholar] [CrossRef]
- Li, R.; Xu, J.; Qi, Z.; Zhao, S.; Zhao, R.; Ge, Y.; Li, R.; Kong, X.; Wu, Z.; Zhang, X.; et al. High-resolution genome mapping and functional dissection of chlorogenic acid production in Lonicera maackii. Plant Physiol. 2023, 192, 2902–2922. [Google Scholar] [CrossRef]
- Peng, Y.; Pan, R.; Liu, Y.; Medison, M.B.; Shalmani, A.; Yang, X.; Zhang, W. LncRNA-mediated ceRNA regulatory network provides new insight into chlorogenic acid synthesis in sweet potato. Physiol. Plant. 2022, 174, e13826. [Google Scholar] [CrossRef]
- Volpi e Silva, N.; Mazzafera, P.; Cesarino, I. Should I stay or should I go: Are chlorogenic acids mobilized towards lignin biosynthesis? Phytochemistry 2019, 166, 112063. [Google Scholar] [CrossRef]
- Alcázar Magaña, A.; Kamimura, N.; Soumyanath, A.; Stevens, J.F.; Maier, C.S. Caffeoylquinic acids: Chemistry, biosynthesis, occurrence, analytical challenges, and bioactivity. Plant J. 2021, 107, 1299–1319. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zheng, X.; Wu, Y.; Zhan, W.; Guo, Y.; Chen, M.; Bai, T.; Jiao, J.; Song, C.; Song, S.; et al. Transcriptome Analysis Identifies Genes Associated with Chlorogenic Acid Biosynthesis during Apple Fruit Development. Horticulturae 2023, 9, 217. [Google Scholar] [CrossRef]
- Xu, D.; Wang, Z.; Zhuang, W.; Wang, T.; Xie, Y. Family characteristics, phylogenetic reconstruction, and potential applications of the plant BAHD acyltransferase family. Front. Plant Sci. 2023, 14, 1218914. [Google Scholar] [CrossRef]
- Legrand, G.; Delporte, M.; Khelifi, C.; Harant, A.; Vuylsteker, C.; Mörchen, M.; Hance, P.; Hilbert, J.-L.; Gagneul, D. Identification and Characterization of Five BAHD Acyltransferases Involved in Hydroxycinnamoyl Ester Metabolism in Chicory. Front. Plant Sci. 2016, 7, 741. [Google Scholar] [CrossRef]
- Kriegshauser, L.; Knosp, S.; Grienenberger, E.; Tatsumi, K.; Gütle, D.D.; Sørensen, I.; Herrgott, L.; Zumsteg, J.; Rose, J.K.C.; Reski, R.; et al. Function of the HYDROXYCINNAMOYL-CoA:SHIKIMATE HYDROXYCINNAMOYL TRANSFERASE is evolutionarily conserved in embryophytes. Plant Cell 2021, 33, 1472–1491. [Google Scholar] [CrossRef]
- Chen, Y.; Yi, N.; Yao, S.b.; Zhuang, J.; Fu, Z.; Ma, J.; Yin, S.; Jiang, X.; Liu, Y.; Gao, L.; et al. CsHCT-Mediated Lignin Synthesis Pathway Involved in the Response of Tea Plants to Biotic and Abiotic Stresses. J. Agric. Food Chem. 2021, 69, 10069–10081. [Google Scholar] [CrossRef]
- D’Orso, F.; Hill, L.; Appelhagen, I.; Lawrenson, T.; Possenti, M.; Li, J.; Harwood, W.; Morelli, G.; Martin, C. Exploring the metabolic and physiological roles of HQT in S. lycopersicum by gene editing. Front. Plant Sci. 2023, 14, 1124959. [Google Scholar] [CrossRef]
- Liu, Z.; Mohsin, A.; Wang, Z.; Zhu, X.; Zhuang, Y.; Cao, L.; Guo, M.; Yin, Z. Enhanced biosynthesis of chlorogenic acid and its derivatives in methyl-jasmonate-treated Gardenia jasminoides cells: A study on metabolic and transcriptional responses of cells. Front. Bioeng. Biotechnol. 2021, 8, 604957. [Google Scholar] [CrossRef]
- Chen, Y.; Sun, Y.; Liu, Z.; Dong, F.; Li, Y.; Wang, Y. Genome-scale modeling for Bacillus coagulans to understand the metabolic characteristics. Biotechnol. Bioeng. 2020, 117, 3545–3558. [Google Scholar] [CrossRef] [PubMed]
- Demidova, E.; Globa, E.; Klushin, A.; Kochkin, D.; Nosov, A. Effect of Methyl Jasmonate on the Growth and Biosynthesis of C13- and C14-Hydroxylated Taxoids in the Cell Culture of Yew (Taxus wallichiana Zucc.) of Different Ages. Biomolecules 2023, 13, 969. [Google Scholar] [CrossRef] [PubMed]
- An, D.; Wu, C.-H.; Wang, M.; Wang, M.; Chang, G.-N.; Chang, X.-J.; Lian, M.-L. Methyl jasmonate elicits enhancement of bioactive compound synthesis in adventitious root co-culture of Echinacea purpurea and Echinacea pallida. Vitr. Cell. Dev. Biol.-Plant 2022, 58, 181–187. [Google Scholar] [CrossRef]
- Ma, Y.; Sun, S.; Shang, X.; Keller, E.T.; Chen, M.; Zhou, X. Integrative differential expression and gene set enrichment analysis using summary statistics for scRNA-seq studies. Nat. Commun. 2020, 11, 1585. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, G. Jasmonates: Biosynthesis, perception and signal transduction. Essays Biochem. 2020, 64, 501–512. [Google Scholar] [CrossRef]
- Levsh, O.; Chiang, Y.-C.; Tung, C.F.; Noel, J.P.; Wang, Y.; Weng, J.-K. Dynamic Conformational States Dictate Selectivity toward the Native Substrate in a Substrate-Permissive Acyltransferase. Biochemistry 2016, 55, 6314–6326. [Google Scholar] [CrossRef]
- Madritsch, S.; Burg, A.; Sehr, E.M. Comparing de novo transcriptome assembly tools in di- and autotetraploid non-model plant species. BMC Bioinform. 2021, 22, 146. [Google Scholar] [CrossRef]
- Fan, S.; Ahmad, N.; Libo, J.; Xinyue, Z.; Xintong, M.; Hoang, N.Q.V.; Mallano, A.I.; Nan, W.; Zhuoda, Y.; Xiuming, L.; et al. Genome-wide investigation of Hydroxycinnamoyl CoA: Shikimate Hydroxycinnamoyl Transferase (HCT) gene family in Carthamus tinctorius L. Not. Bot. Horti Agrobot. Cluj-Napoca 2021, 49, 12489. [Google Scholar] [CrossRef]
- Walker, A.M.; Hayes, R.P.; Youn, B.; Vermerris, W.; Sattler, S.E.; Kang, C. Elucidation of the Structure and Reaction Mechanism of Sorghum Hydroxycinnamoyltransferase and Its Structural Relationship to Other Coenzyme A-Dependent Transferases and Synthases. Plant Physiol. 2013, 162, 640–651. [Google Scholar] [CrossRef]
- Chiang, Y.-C.; Levsh, O.; Lam, C.K.; Weng, J.-K.; Wang, Y. Structural and dynamic basis of substrate permissiveness in hydroxycinnamoyltransferase (HCT). PLoS Comput. Biol. 2018, 14, e1006511. [Google Scholar] [CrossRef]
- An, J.; Liu, Y.; Han, J.; He, C.; Chen, M.; Zhu, X.; Hu, W.; Song, W.; Hu, J.; Guan, Y. Transcriptional multiomics reveals the mechanism of seed deterioration in Nicotiana tabacum L. and Oryza sativa L. J. Adv. Res. 2022, 42, 163–176. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.-C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [PubMed]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Chen, Y.; Cai, G.; Cai, R.; Hu, Z.; Wang, H. Tree Visualization By One Table (tvBOT): A web application for visualizing, modifying and annotating phylogenetic trees. Nucleic Acids Res. 2023, 51, W587–W592. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Varying Substrate | Saturating Substrate | Km (µM) 1 | vmax (µm/min) 1 | vmax/Km (min−1) |
|---|---|---|---|---|
| p-Coumaroyl-CoA | Quinic acid | 9.41 | 10.02 | 1.06 |
| Caffeoyl-CoA | Quinic acid | 32.61 | 1.71 | 0.05 |
| Shikimic acid | p-coumaroyl-CoA | 18.38 | 25.12 | 1.37 |
| Quinic acid | p-coumaroyl-CoA | 7.11 | 13.98 | 1.97 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Z.; Zhu, X.; Mohsin, A.; Sun, H.; Du, L.; Yin, Z.; Zhuang, Y.; Guo, M. Uncovering the Role of Hydroxycinnamoyl Transferase in Boosting Chlorogenic Acid Accumulation in Carthamus tinctorius Cells under Methyl Jasmonate Elicitation. Int. J. Mol. Sci. 2024, 25, 2710. https://doi.org/10.3390/ijms25052710
Liu Z, Zhu X, Mohsin A, Sun H, Du L, Yin Z, Zhuang Y, Guo M. Uncovering the Role of Hydroxycinnamoyl Transferase in Boosting Chlorogenic Acid Accumulation in Carthamus tinctorius Cells under Methyl Jasmonate Elicitation. International Journal of Molecular Sciences. 2024; 25(5):2710. https://doi.org/10.3390/ijms25052710
Chicago/Turabian StyleLiu, Zebo, Xiaofeng Zhu, Ali Mohsin, Huijie Sun, Linxiao Du, Zhongping Yin, Yingping Zhuang, and Meijin Guo. 2024. "Uncovering the Role of Hydroxycinnamoyl Transferase in Boosting Chlorogenic Acid Accumulation in Carthamus tinctorius Cells under Methyl Jasmonate Elicitation" International Journal of Molecular Sciences 25, no. 5: 2710. https://doi.org/10.3390/ijms25052710
APA StyleLiu, Z., Zhu, X., Mohsin, A., Sun, H., Du, L., Yin, Z., Zhuang, Y., & Guo, M. (2024). Uncovering the Role of Hydroxycinnamoyl Transferase in Boosting Chlorogenic Acid Accumulation in Carthamus tinctorius Cells under Methyl Jasmonate Elicitation. International Journal of Molecular Sciences, 25(5), 2710. https://doi.org/10.3390/ijms25052710