
Application Prospect of Induced Pluripotent Stem Cells in Organoids and Cell Therapy

 ,
,  , , and
, , and
Abstract
1. Background
2. Nonintegrating Way of iPSC Reprogramming
2.1. Adenovirus
2.2. SeV
2.3. Protein
2.4. mRNA/miRNA
2.5. PiggyBac
2.6. Episomal Plasmids
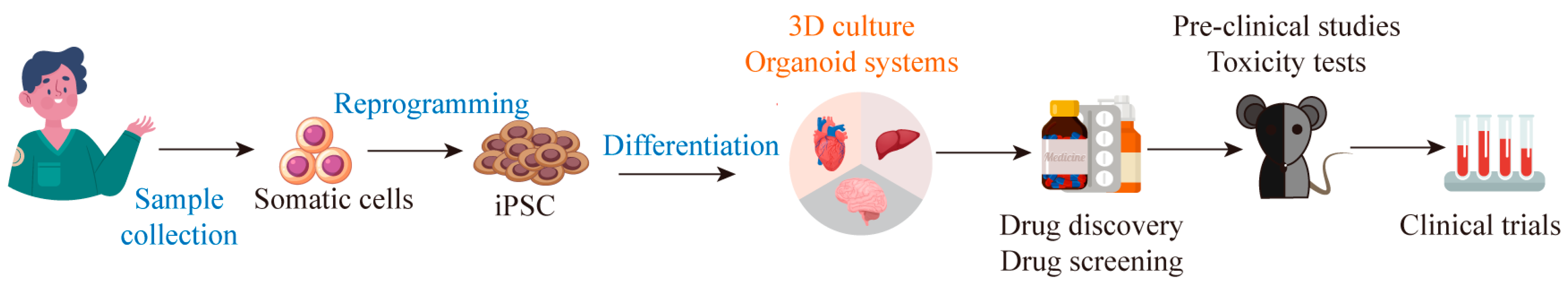
3. Construction of Organoids Based on iPSC Technology
3.1. Derivation of Neural Organoids
3.2. Derivation of Liver Organoids
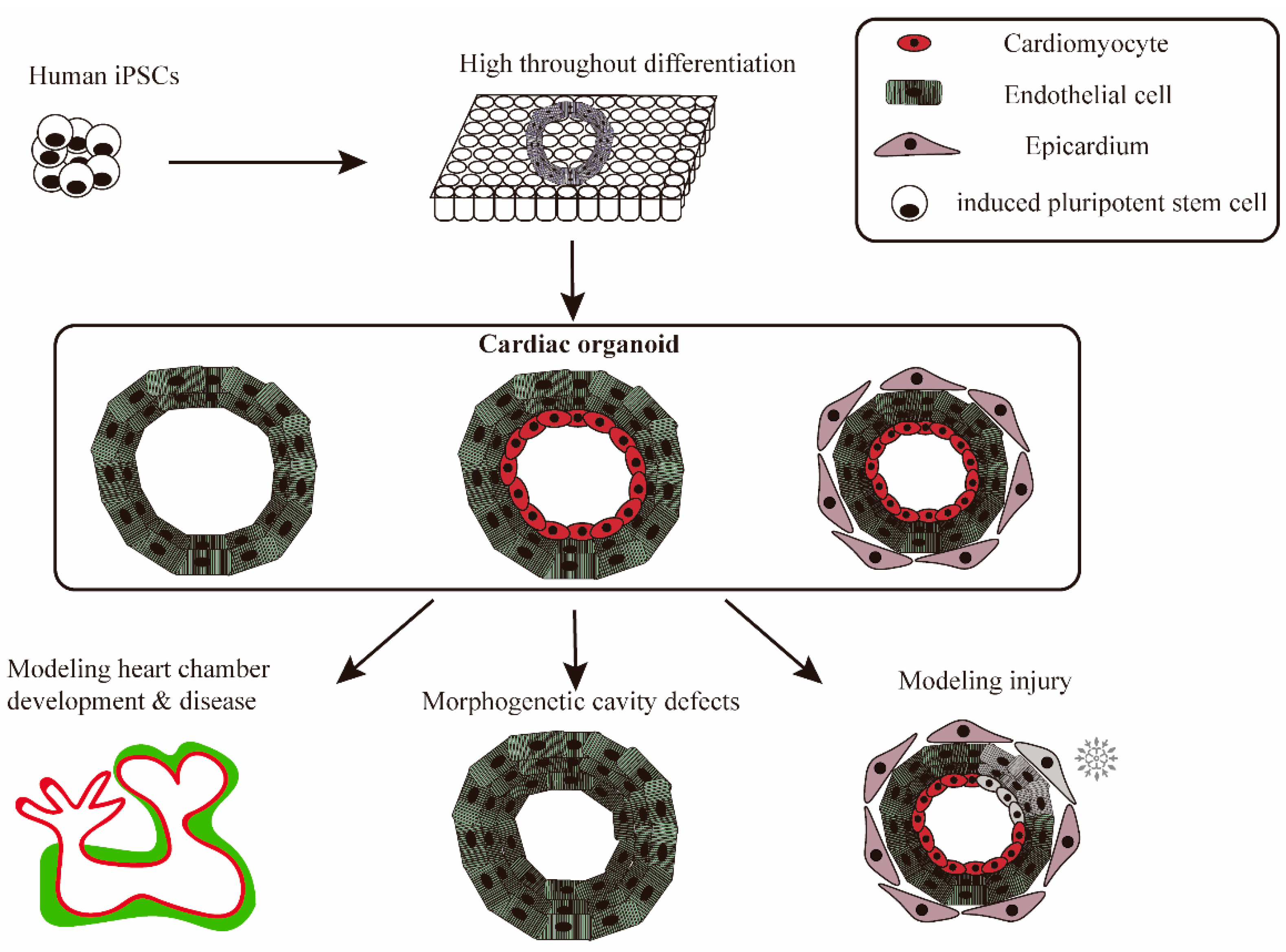
3.3. Derivation of Cardiac Organoids
3.4. Derivation of Cancer Organoids
4. Screening Drugs and Toxicity Detection of iPSC-Derived Organoids
4.1. Neural Organoids
4.2. Liver Organoids
4.3. Cardiac Organoids
5. Cell Therapy Based on iPSC Technology
5.1. The Advent of iPSC-Derived DA Neurons Offers an Opportunity to Treat Neurodegenerative Disease
5.2. iPSC-Derived Hepatocytes and Immune Cells Are Effective in the Treatment of Liver Diseases
5.3. iPSC-Derived Cardiac Patches and Extracellular Vesicles May Be a New Approach to Treating Heart Failure
5.4. iPSC-Derived Immune Cells Are a New Method of Tumor Immunotherapy
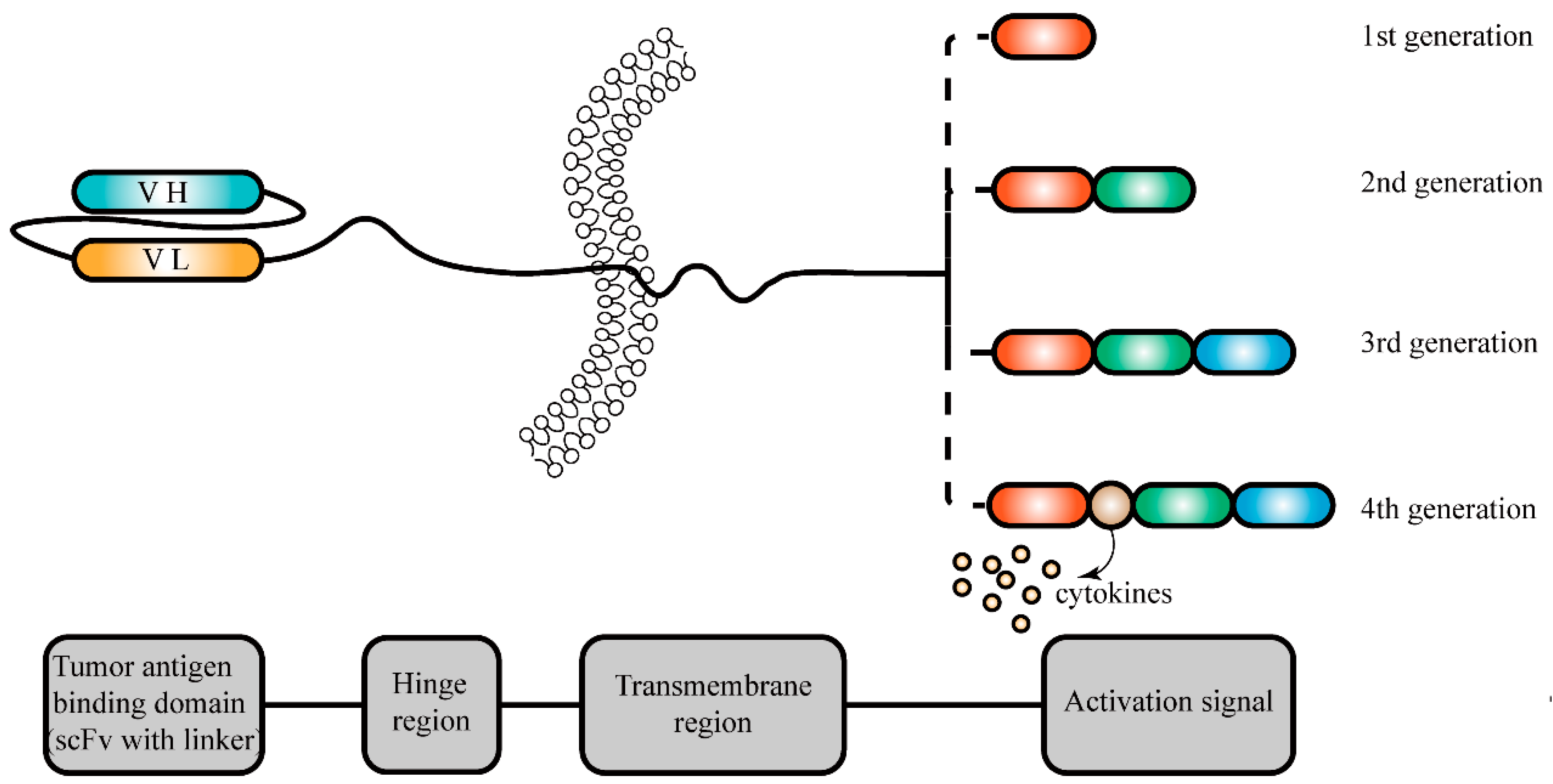
5.4.1. T Cells
5.4.2. NK Cells
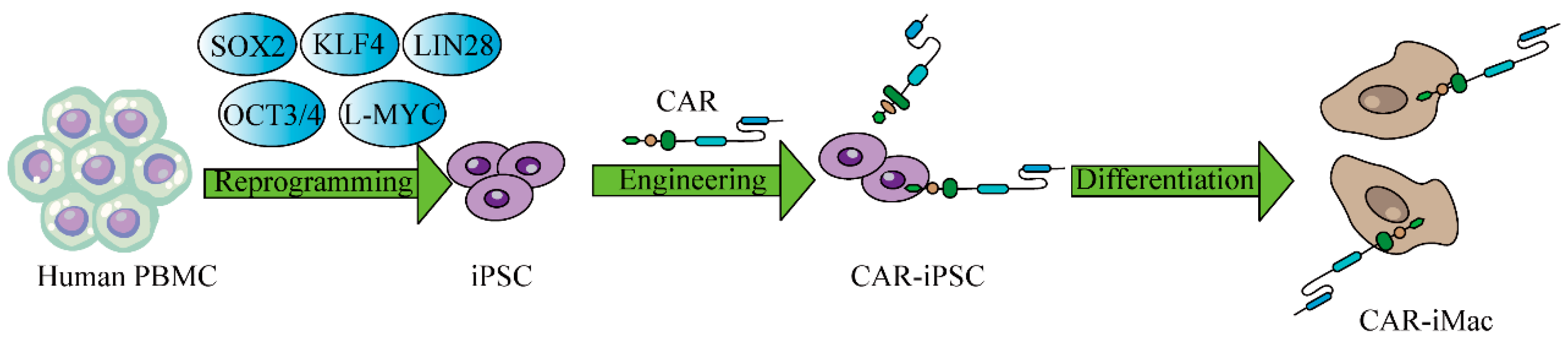
5.4.3. Macrophages
6. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Yoshida, Y.; Yamanaka, S. Induced Pluripotent Stem Cells 10 Years Later: For Cardiac Applications. Circ. Res. 2017, 120, 1958–1968. [Google Scholar] [CrossRef] [PubMed]
- Okano, H.; Morimoto, S. iPSC-based disease modeling and drug discovery in cardinal neurodegenerative disorders. Cell Stem Cell 2022, 29, 189–208. [Google Scholar] [CrossRef] [PubMed]
- Andrews, M.G.; Kriegstein, A.R. Challenges of Organoid Research. Annu. Rev. Neurosci. 2022, 45, 23–39. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.Y.; Wu, S.; Wang, D.; Chu, C.; Hong, Y.; Tao, M.; Hu, H.; Xu, M.; Guo, X.; Liu, Y. Human organoids in basic research and clinical applications. Signal Transduct. Target. Ther. 2022, 7, 168. [Google Scholar] [CrossRef] [PubMed]
- Rowe, R.G.; Daley, G.Q. Induced pluripotent stem cells in disease modelling and drug discovery. Nat. Rev. Genet. 2019, 20, 377–388. [Google Scholar] [CrossRef] [PubMed]
- Halevy, T.; Urbach, A. Comparing ESC and iPSC-Based Models for Human Genetic Disorders. J. Clin. Med. 2014, 3, 1146–1162. [Google Scholar] [CrossRef]
- Karagiannis, P.; Takahashi, K.; Saito, M.; Yoshida, Y.; Okita, K.; Watanabe, A.; Inoue, H.; Yamashita, J.K.; Todani, M.; Nak-agawa, M.; et al. Induced Pluripotent Stem Cells and Their Use in Human Models of Disease and Development. Physiol. Rev. 2019, 99, 79–114. [Google Scholar] [CrossRef]
- Stadtfeld, M.; Nagaya, M.; Utikal, J.; Weir, G.; Hochedlinger, K. Induced pluripotent stem cells generated without viral inte-gration. Science 2008, 322, 945–949. [Google Scholar] [CrossRef]
- Mitsui, K.; Takahashi, T.; Ide, K.; Matsuda, E.; Kosai, K.I. Optimization of adenoviral gene transfer in human pluripotent stem cells. Biochem. Biophys. Res. Commun. 2021, 541, 78–83. [Google Scholar] [CrossRef]
- Bayart, E.; Cohen-Haguenauer, O. Technological overview of iPS induction from human adult somatic cells. Curr. Gene Ther. 2013, 13, 73–92. [Google Scholar] [CrossRef]
- Zhou, W.; Freed, C.R. Adenoviral gene delivery can reprogram human fibroblasts to induced pluripotent stem cells. Stem Cells 2009, 27, 2667–2674. [Google Scholar] [CrossRef] [PubMed]
- Schlaeger, T.M. Nonintegrating Human Somatic Cell Reprogramming Methods. Adv. Biochem. Eng. Biotechnol. 2018, 163, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Haridhasapavalan, K.K.; Borgohain, M.P.; Dey, C.; Saha, B.; Narayan, G.; Kumar, S.; Thummer, R.P. An insight into non-integrative gene delivery approaches to generate transgene-free induced pluripotent stem cells. Gene 2019, 686, 146–159. [Google Scholar] [CrossRef]
- Okumura, T.; Horie, Y.; Lai, C.Y.; Lin, H.T.; Shoda, H.; Natsumoto, B.; Fujio, K.; Kumaki, E.; Okano, T.; Ono, S.; et al. Robust and highly efficient hiPSC generation from patient non-mobilized peripheral blood-derived CD34(+) cells using the auto-erasable Sendai virus vector. Stem Cell Res. Ther. 2019, 10, 185. [Google Scholar] [CrossRef]
- Seo, B.J.; Hong, Y.J.; Do, J.T. Cellular Reprogramming Using Protein and Cell-Penetrating Peptides. Int. J. Mol. Sci. 2017, 18, 552. [Google Scholar] [CrossRef]
- Cho, H.J.; Lee, C.S.; Kwon, Y.W.; Paek, J.S.; Lee, S.H.; Hur, J.; Lee, E.J.; Roh, T.Y.; Chu, I.S.; Leem, S.H.; et al. Induction of pluripotent stem cells from adult somatic cells by protein-based reprogramming without genetic manipulation. Blood 2010, 116, 386–395. [Google Scholar] [CrossRef] [PubMed]
- Warren, L.; Lin, C. mRNA-Based Genetic Reprogramming. Mol. Ther. J. Am. Soc. Gene Ther. 2019, 27, 729–734. [Google Scholar] [CrossRef] [PubMed]
- Warren, L.; Manos, P.D.; Ahfeldt, T.; Loh, Y.H.; Li, H.; Lau, F.; Ebina, W.; Mandal, P.K.; Smith, Z.D.; Meissner, A.; et al. Highly efficient reprogramming to pluripotency and directed differentiation of human cells with synthetic modified mRNA. Cell Stem Cell 2010, 7, 618–630. [Google Scholar] [CrossRef]
- Wang, A.Y.L. Application of Modified mRNA in Somatic Reprogramming to Pluripotency and Directed Conversion of Cell Fate. Int. J. Mol. Sci. 2021, 22, 8148. [Google Scholar] [CrossRef]
- Woodard, L.E.; Wilson, M.H. piggyBac-ing models and new therapeutic strategies. Trends Biotechnol. 2015, 33, 525–533. [Google Scholar] [CrossRef]
- Woltjen, K.; Michael, I.P.; Mohseni, P.; Desai, R.; Mileikovsky, M.; Hämäläinen, R.; Cowling, R.; Wang, W.; Liu, P.; Gertsenstein, M.; et al. piggyBac transposition reprograms fibroblasts to induced pluripotent stem cells. Nature 2009, 458, 766–770. [Google Scholar] [CrossRef]
- Wang, G.; Yang, L.; Grishin, D.; Rios, X.; Ye, L.Y.; Hu, Y.; Li, K.; Zhang, D.; Church, G.M.; Pu, W.T. Efficient, footprint-free human iPSC genome editing by consolidation of Cas9/CRISPR and piggyBac technologies. Nat. Protoc. 2017, 12, 88–103. [Google Scholar] [CrossRef]
- Itoh, M.; Kawagoe, S.; Tamai, K.; Nakagawa, H.; Asahina, A.; Okano, H.J. Footprint-free gene mutation correction in induced pluripotent stem cell (iPSC) derived from recessive dystrophic epidermolysis bullosa (RDEB) using the CRISPR/Cas9 and pig-gyBac transposon system. J. Dermatol. Sci. 2020, 98, 163–172. [Google Scholar] [CrossRef]
- Rodriguez-Polo, I.; Mißbach, S.; Petkov, S.; Mattern, F.; Maierhofer, A.; Grządzielewska, I.; Tereshchenko, Y.; Urrutia-Cabrera, D.; Haaf, T.; Dressel, R.; et al. A piggyBac-based platform for genome editing and clonal rhesus macaque iPSC line derivation. Sci. Rep. 2021, 11, 15439. [Google Scholar] [CrossRef]
- Okita, K.; Nakagawa, M.; Hong, H.J.; Ichisaka, T.; Yamanaka, S. Generation of Mouse Induced Pluripotent Stem Cells without Viral Vectors. Science 2008, 322, 949–953. [Google Scholar] [CrossRef]
- Hu, W.; He, Y.; Xiong, Y.; Lu, H.; Chen, H.; Hou, L.; Qiu, Z.; Fang, Y.; Zhang, S. Derivation, Expansion, and Motor Neuron Differentiation of Human-Induced Pluripotent Stem Cells with Non-Integrating Episomal Vectors and a Defined Xenogene-ic-free Culture System. Mol. Neurobiol. 2016, 53, 1589–1600. [Google Scholar] [CrossRef]
- Yue, G.; Liao, J.; Wang, Y.; He, L.; Wang, T.; Zhou, G.; Lei, B. Quality evaluation of induced pluripotent stem cell colonies by fusing multi-source features. Comput. Methods Programs Biomed. 2021, 208, 106235. [Google Scholar] [CrossRef] [PubMed]
- Okita, K.; Yamanaka, S. Induced pluripotent stem cells: Opportunities and challenges. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 2011, 366, 2198–2207. [Google Scholar] [CrossRef] [PubMed]
- Maali, A.; Maroufi, F.; Sadeghi, F.; Atashi, A.; Kouchaki, R.; Moghadami, M.; Azad, M. Induced pluripotent stem cell tech-nology: Trends in molecular biology, from genetics to epigenetics. Epigenomics 2021, 13, 631–647. [Google Scholar] [CrossRef] [PubMed]
- VandenDriessche, T.; Ivics, Z.; Izsvák, Z.; Chuah, M.K. Emerging potential of transposons for gene therapy and generation of induced pluripotent stem cells. Blood 2009, 114, 1461–1468. [Google Scholar] [CrossRef] [PubMed]
- Hu, K. Vectorology and factor delivery in induced pluripotent stem cell reprogramming. Stem Cells Dev. 2014, 23, 1301–1315. [Google Scholar] [CrossRef] [PubMed]
- Cowan, C.S.; Renner, M.; De Gennaro, M.; Gross-Scherf, B.; Goldblum, D.; Hou, Y.; Munz, M.; Rodrigues, T.M.; Krol, J.; Szikra, T.; et al. Cell Types of the Human Retina and Its Organoids at Single-Cell Resolution. Cell 2020, 182, 1623–1640.e34. [Google Scholar] [CrossRef] [PubMed]
- Human brain ‘organoids’ grafted into rats take note of light and pattern. Nature 2023, 614, 198. [CrossRef] [PubMed]
- Eisenstein, M. Organoids open fresh paths to biomedical advances. Nature 2022, 612, S34–S35. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; Maynard, A.; Jain, A.; Gerber, T.; Petri, R.; Lin, H.C.; Santel, M.; Ly, K.; Dupré, J.S.; Sidow, L.; et al. Lineage recording in human cerebral organoids. Nat. Methods 2022, 19, 90–99. [Google Scholar] [CrossRef] [PubMed]
- Bender, E. How organoids are advancing the understanding of chronic kidney disease. Nature 2023, 615, S10–S11. [Google Scholar] [CrossRef] [PubMed]
- Pașca, S.P.; Arlotta, P.; Bateup, H.S.; Camp, J.G.; Cappello, S.; Gage, F.H.; Knoblich, J.A.; Kriegstein, A.R.; Lancaster, M.A.; Ming, G.L.; et al. A nomenclature consensus for nervous system organoids and assembloids. Nature 2022, 609, 907–910. [Google Scholar] [CrossRef]
- Doss, M.X.; Sachinidis, A. Current Challenges of iPSC-Based Disease Modeling and Therapeutic Implications. Cells 2019, 8, 403. [Google Scholar] [CrossRef]
- Rauth, S.; Karmakar, S.; Batra, S.K.; Ponnusamy, M.P. Recent advances in organoid development and applications in disease modeling. Biochim. Biophys. Acta Rev. Cancer 2021, 1875, 188527. [Google Scholar] [CrossRef]
- Prinz, M.; Jung, S.; Priller, J. Microglia Biology: One Century of Evolving Concepts. Cell 2019, 179, 292–311. [Google Scholar] [CrossRef]
- Borst, K.; Dumas, A.A.; Prinz, M. Microglia: Immune and non-immune functions. Immunity 2021, 54, 2194–2208. [Google Scholar] [CrossRef] [PubMed]
- Colonna, M.; Butovsky, O. Microglia Function in the Central Nervous System During Health and Neurodegeneration. Annu. Rev. Immunol. 2017, 35, 441–468. [Google Scholar] [CrossRef] [PubMed]
- Wolf, S.A.; Boddeke, H.W.; Kettenmann, H. Microglia in Physiology and Disease. Annu. Rev. Physiol. 2017, 79, 619–643. [Google Scholar] [CrossRef] [PubMed]
- Wright-Jin, E.C.; Gutmann, D.H. Microglia as Dynamic Cellular Mediators of Brain Function. Trends Mol. Med. 2019, 25, 967–979. [Google Scholar] [CrossRef]
- Umpierre, A.D.; Wu, L.J. How microglia sense and regulate neuronal activity. Glia 2021, 69, 1637–1653. [Google Scholar] [CrossRef]
- Ghosh, S.; Castillo, E.; Frias, E.S.; Swanson, R.A. Bioenergetic regulation of microglia. Glia 2018, 66, 1200–1212. [Google Scholar] [CrossRef]
- Augusto-Oliveira, M.; Arrifano, G.P.; Delage, C.I.; Tremblay, M.; Crespo-Lopez, M.E.; Verkhratsky, A. Plasticity of microglia. Biol. Rev. Camb. Philos. Soc. 2022, 97, 217–250. [Google Scholar] [CrossRef]
- Li, Q.; Barres, B.A. Microglia and macrophages in brain homeostasis and disease. Nat. Rev. Immunol. 2018, 18, 225–242. [Google Scholar] [CrossRef]
- Xu, R.; Boreland, A.J.; Li, X.; Erickson, C.; Jin, M.; Atkins, C.; Pang, Z.P.; Daniels, B.P.; Jiang, P. Developing human pluripotent stem cell-based cerebral organoids with a controllable microglia ratio for modeling brain development and pathology. Stem Cell Rep. 2021, 16, 1923–1937. [Google Scholar] [CrossRef]
- Abud, E.M.; Ramirez, R.N.; Martinez, E.S.; Healy, L.M.; Nguyen, C.H.H.; Newman, S.A.; Yeromin, A.V.; Scarfone, V.M.; Marsh, S.E.; Fimbres, C.; et al. iPSC-Derived Human Microglia-like Cells to Study Neurological Diseases. Neuron 2017, 94, 278–293.e9. [Google Scholar] [CrossRef]
- Sabate-Soler, S.; Nickels, S.L.; Saraiva, C.; Berger, E.; Dubonyte, U.; Barmpa, K.; Lan, Y.J.; Kouno, T.; Jarazo, J.; Robertson, G.; et al. Microglia integration into human midbrain organoids leads to increased neuronal maturation and functionality. Glia 2022, 70, 1267–1288. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.Y.; Ting, H.C.; Liu, C.A.; Su, H.L.; Chiou, T.W.; Lin, S.Z.; Harn, H.J.; Ho, T.J. Induced Pluripotent Stem Cell (iPSC)-Based Neurodegenerative Disease Models for Phenotype Recapitulation and Drug Screening. Molecules 2020, 25, 2000. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Ra, E.A.; Kweon, S.H.; Seo, B.A.; Ko, H.S.; Oh, Y.; Lee, G. Advanced human iPSC-based preclinical model for Par-kinson’s disease with optogenetic alpha-synuclein aggregation. Cell Stem Cell 2023, 30, 973–986.e11. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.G.; Jung, D.J.; Jung, Y.K.; Kang, K.S. The Effect of a Novel Mica Nanoparticle, STB-MP, on an Alzheimer’s Disease Patient-Induced PSC-Derived Cortical Brain Organoid Model. Nanomaterials 2023, 13, 893. [Google Scholar] [CrossRef] [PubMed]
- Oun, A.; Sabogal-Guaqueta, A.M.; Galuh, S.; Alexander, A.; Kortholt, A.; Dolga, A.M. The multifaceted role of LRRK2 in Parkinson’s disease: From human iPSC to organoids. Neurobiol. Dis. 2022, 173, 105837. [Google Scholar] [CrossRef] [PubMed]
- Tejchman, A.; Znój, A.; Chlebanowska, P.; Frączek-Szczypta, A.; Majka, M. Carbon Fibers as a New Type of Scaffold for Midbrain Organoid Development. Int. J. Mol. Sci. 2020, 21, 5959. [Google Scholar] [CrossRef]
- Kim, H.; Park, H.J.; Choi, H.; Chang, Y.; Park, H.; Shin, J.; Kim, J.; Lengner, C.J.; Lee, Y.K.; Kim, J. Modeling G2019S-LRRK2 Sporadic Parkinson’s Disease in 3D Midbrain Organoids. Stem Cell Rep. 2019, 12, 518–531. [Google Scholar] [CrossRef]
- Cao, D.; Ge, J.Y.; Wang, Y.; Oda, T.; Zheng, Y.W. Hepatitis B virus infection modeling using multi-cellular organoids derived from human induced pluripotent stem cells. World J. Gastroenterol. 2021, 27, 4784–4801. [Google Scholar] [CrossRef]
- Takebe, T.; Sekine, K.; Kimura, M.; Yoshizawa, E.; Ayano, S.; Koido, M.; Funayama, S.; Nakanishi, N.; Hisai, T.; Kobayashi, T.; et al. Massive and Reproducible Production of Liver Buds Entirely from Human Pluripotent Stem Cells. Cell Rep. 2017, 21, 2661–2670. [Google Scholar] [CrossRef]
- Guo, J.; Duan, L.; He, X.; Li, S.; Wu, Y.; Xiang, G.; Bao, F.; Yang, L.; Shi, H.; Gao, M.; et al. A Combined Model of Human iPSC-Derived Liver Organoids and Hepatocytes Reveals Ferroptosis in DGUOK Mutant mtDNA Depletion Syndrome. Adv. Sci. 2021, 8, 2004680. [Google Scholar] [CrossRef] [PubMed]
- Kulkeaw, K.; Tubsuwan, A.; Tongkrajang, N.; Whangviboonkij, N. Generation of human liver organoids from pluripotent stem cell-derived hepatic endoderms. PeerJ 2020, 8, e9968. [Google Scholar] [CrossRef]
- Nguyen, R.; Da Won Bae, S.; Qiao, L.; George, J. Developing liver organoids from induced pluripotent stem cells (iPSCs): An alternative source of organoid generation for liver cancer research. Cancer Lett. 2021, 508, 13–17. [Google Scholar] [CrossRef]
- Mun, S.J.; Ryu, J.S.; Lee, M.O.; Son, Y.S.; Oh, S.J.; Cho, H.S.; Son, M.Y.; Kim, D.S.; Kim, S.J.; Yoo, H.J.; et al. Generation of expandable human pluripotent stem cell-derived hepatocyte-like liver organoids. J. Hepatol. 2019, 71, 970–985. [Google Scholar] [CrossRef]
- Ouchi, R.; Togo, S.; Kimura, M.; Shinozawa, T.; Koido, M.; Koike, H.; Thompson, W.; Karns, R.A.; Mayhew, C.N.; McGrath, P.S.; et al. Modeling Steatohepatitis in Humans with Pluripotent Stem Cell-Derived Organoids. Cell Metab. 2019, 30, 374–384.e6. [Google Scholar] [CrossRef]
- Guan, Y.; Enejder, A.; Wang, M.; Fang, Z.; Cui, L.; Chen, S.Y.; Wang, J.; Tan, Y.; Wu, M.; Chen, X.; et al. A human multi-lineage hepatic organoid model for liver fibrosis. Nat. Commun. 2021, 12, 6138. [Google Scholar] [CrossRef]
- Toivonen, S.; Lundin, K.; Balboa, D.; Ustinov, J.; Tamminen, K.; Palgi, J.; Trokovic, R.; Tuuri, T.; Otonkoski, T. Activin A and Wnt-dependent specification of human definitive endoderm cells. Exp. Cell Res. 2013, 319, 2535–2544. [Google Scholar] [CrossRef]
- Schmidt, C.; Bladt, F.; Goedecke, S.; Brinkmann, V.; Zschiesche, W.; Sharpe, M.; Gherardi, E.; Birchmeier, C. Scatter fac-tor/hepatocyte growth factor is essential for liver development. Nature 1995, 373, 699–702. [Google Scholar] [CrossRef] [PubMed]
- Kamiya, A.; Kinoshita, T.; Ito, Y.; Matsui, T.; Morikawa, Y.; Senba, E.; Nakashima, K.; Taga, T.; Yoshida, K.; Kishimoto, T.; et al. Fetal liver development requires a paracrine action of oncostatin M through the gp130 signal transducer. EMBO J. 1999, 18, 2127–2136. [Google Scholar] [CrossRef] [PubMed]
- Roy-Chowdhury, N.; Wang, X.; Guha, C.; Roy-Chowdhury, J. Hepatocyte-like cells derived from induced pluripotent stem cells. Hepatol. Int. 2017, 11, 54–69. [Google Scholar] [CrossRef] [PubMed]
- Olgasi, C.; Cucci, A.; Follenzi, A. iPSC-Derived Liver Organoids: A Journey from Drug Screening, to Disease Modeling, Arriving to Regenerative Medicine. Int. J. Mol. Sci. 2020, 21, 6215. [Google Scholar] [CrossRef]
- Torizal, F.G.; Utami, T.; Lau, Q.Y.; Inamura, K.; Nishikawa, M.; Sakai, Y. Dialysis based-culture medium conditioning improved the generation of human induced pluripotent stem cell derived-liver organoid in a high cell density. Sci. Rep. 2022, 12, 20774. [Google Scholar] [CrossRef]
- Sampaziotis, F.; Muraro, D.; Tysoe, O.C.; Sawiak, S.; Beach, T.E.; Godfrey, E.M.; Upponi, S.S.; Brevini, T.; Wesley, B.T.; Gar-cia-Bernardo, J.; et al. Cholangiocyte organoids can repair bile ducts after transplantation in the human liver. Science 2021, 371, 839–846. [Google Scholar] [CrossRef]
- Wu, F.; Wu, D.; Ren, Y.; Huang, Y.; Feng, B.; Zhao, N.; Zhang, T.; Chen, X.; Chen, S.; Xu, A. Generation of hepatobiliary or-ganoids from human induced pluripotent stem cells. J. Hepatol. 2019, 70, 1145–1158. [Google Scholar] [CrossRef]
- Takebe, T.; Sekine, K.; Enomura, M.; Koike, H.; Kimura, M.; Ogaeri, T.; Zhang, R.R.; Ueno, Y.; Zheng, Y.W.; Koike, N.; et al. Vascularized and functional human liver from an iPSC-derived organ bud transplant. Nature 2013, 499, 481–484. [Google Scholar] [CrossRef]
- Rajan, S.A.P.; Aleman, J.; Wan, M.; Pourhabibi Zarandi, N.; Nzou, G.; Murphy, S.; Bishop, C.E.; Sadri-Ardekani, H.; Shupe, T.; Atala, A.; et al. Probing prodrug metabolism and reciprocal toxicity with an integrated and humanized multi-tissue or-gan-on-a-chip platform. Acta Biomater. 2020, 106, 124–135. [Google Scholar] [CrossRef]
- Koike, H.; Iwasawa, K.; Ouchi, R.; Maezawa, M.; Giesbrecht, K.; Saiki, N.; Ferguson, A.; Kimura, M.; Thompson, W.L.; Wells, J.M.; et al. Modelling human hepato-biliary-pancreatic organogenesis from the foregut-midgut boundary. Nature 2019, 574, 112–116. [Google Scholar] [CrossRef] [PubMed]
- Tao, T.; Deng, P.; Wang, Y.; Zhang, X.; Guo, Y.; Chen, W.; Qin, J. Microengineered Multi-Organoid System from hiPSCs to Recapitulate Human Liver-Islet Axis in Normal and Type 2 Diabetes. Adv. Sci. 2022, 9, e2103495. [Google Scholar] [CrossRef] [PubMed]
- Skardal, A.; Shupe, T.; Atala, A. Organoid-on-a-chip and body-on-a-chip systems for drug screening and disease modeling. Drug Discov. Today 2016, 21, 1399–1411. [Google Scholar] [CrossRef] [PubMed]
- Takebe, T.; Zhang, B.; Radisic, M. Synergistic Engineering: Organoids Meet Organs-on-a-Chip. Cell Stem Cell 2017, 21, 297–300. [Google Scholar] [CrossRef]
- Filippo Buono, M.; von Boehmer, L.; Strang, J.; Hoerstrup, S.P.; Emmert, M.Y.; Nugraha, B. Human Cardiac Organoids for Modeling Genetic Cardiomyopathy. Cells 2020, 9, 1733. [Google Scholar] [CrossRef]
- Fang, Y.; Guo, Y.; Wu, B.; Liu, Z.; Ye, M.; Xu, Y.; Ji, M.; Chen, L.; Lu, B.; Nie, K.; et al. Expanding Embedded 3D Bioprinting Capability for Engineering Complex Organs with Freeform Vascular Networks. Adv. Mater. 2023, 35, e2205082. [Google Scholar] [CrossRef] [PubMed]
- Mazzotta, S.; Neves, C.; Bonner, R.J.; Bernardo, A.S.; Docherty, K.; Hoppler, S. Distinctive Roles of Canonical and Noncanonical Wnt Signaling in Human Embryonic Cardiomyocyte Development. Stem Cell Rep. 2016, 7, 764–776. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Sun, J.; Zhong, T.P. Wnt Signaling in Heart Development and Regeneration. Curr. Cardiol. Rep. 2022, 24, 1425–1438. [Google Scholar] [CrossRef]
- Di Sante, M.; Antonucci, S.; Pontarollo, L.; Cappellaro, I.; Segat, F.; Deshwal, S.; Greotti, E.; Grilo, L.F.; Menabò, R.; Di Lisa, F.; et al. Monoamine oxidase A-dependent ROS formation modulates human cardiomyocyte differentiation through AKT and WNT activation. Basic Res. Cardiol. 2023, 118, 4. [Google Scholar] [CrossRef] [PubMed]
- Jeong, M.H.; Kim, H.J.; Pyun, J.H.; Choi, K.S.; Lee, D.I.; Solhjoo, S.; O’Rourke, B.; Tomaselli, G.F.; Jeong, D.S.; Cho, H.; et al. Cdon deficiency causes cardiac remodeling through hyperactivation of WNT/β-catenin signaling. Proc. Natl. Acad. Sci. USA 2017, 114, E1345–E1354. [Google Scholar] [CrossRef]
- Lewis-Israeli, Y.R.; Wasserman, A.H.; Gabalski, M.A.; Volmert, B.D.; Ming, Y.; Ball, K.A.; Yang, W.; Zou, J.; Ni, G.; Pajares, N.; et al. Self-assembling human heart organoids for the modeling of cardiac development and congenital heart disease. Nat. Commun. 2021, 12, 5142. [Google Scholar] [CrossRef]
- Hofbauer, P.; Jahnel, S.M.; Papai, N.; Giesshammer, M.; Deyett, A.; Schmidt, C.; Penc, M.; Tavernini, K.; Grdseloff, N.; Meledeth, C.; et al. Cardioids reveal self-organizing principles of human cardiogenesis. Cell 2021, 184, 3299–3317.e22. [Google Scholar] [CrossRef]
- Ng, W.H.; Johnston, E.K.; Tan, J.J.; Bliley, J.M.; Feinberg, A.W.; Stolz, D.B.; Sun, M.; Wijesekara, P.; Hawkins, F.; Kotton, D.N.; et al. Recapitulating human cardio-pulmonary co-development using simultaneous multilineage differentiation of pluripotent stem cells. eLife 2022, 11, e67872. [Google Scholar] [CrossRef]
- Silva, A.C.; Matthys, O.B.; Joy, D.A.; Kauss, M.A.; Natarajan, V.; Lai, M.H.; Turaga, D.; Blair, A.P.; Alexanian, M.; Bruneau, B.G.; et al. Co-emergence of cardiac and gut tissues promotes cardiomyocyte maturation within human iPSC-derived organoids. Cell Stem Cell 2021, 28, 2137–2152.e6. [Google Scholar] [CrossRef]
- Chao, H.M.; Chern, E. Patient-derived induced pluripotent stem cells for models of cancer and cancer stem cell research. J. Formos. Med. Assoc. Taiwan Yi Zhi 2018, 117, 1046–1057. [Google Scholar] [CrossRef]
- Kuenzig, M.E.; Fung, S.G.; Marderfeld, L.; Mak, J.W.Y.; Kaplan, G.G.; Ng, S.C.; Wilson, D.C.; Cameron, F.; Henderson, P.; Kotze, P.G.; et al. Twenty-first Century Trends in the Global Epidemiology of Pediatric-Onset Inflammatory Bowel Disease: Systematic Review. Gastroenterology 2022, 162, 1147–1159.e4. [Google Scholar] [CrossRef] [PubMed]
- Gomez, C.L.; Neufeld, K.L. Elevated adenomatous polyposis coli in goblet cells is associated with inflammation in mouse and human colon. Exp. Physiol. 2020, 105, 2154–2167. [Google Scholar] [CrossRef] [PubMed]
- Bopanna, S.; Ananthakrishnan, A.N.; Kedia, S.; Yajnik, V.; Ahuja, V. Risk of colorectal cancer in Asian patients with ulcerative colitis: A systematic review and meta-analysis. Lancet Gastroenterol. Hepatol. 2017, 2, 269–276. [Google Scholar] [CrossRef] [PubMed]
- Pan, Z.; Lin, H.; Fu, Y.; Zeng, F.; Gu, F.; Niu, G.; Fang, J.; Gu, B. Identification of gene signatures associated with ulcerative colitis and the association with immune infiltrates in colon cancer. Front. Immunol. 2023, 14, 1086898. [Google Scholar] [CrossRef]
- Yadav, A.; Jena, A.; Arpitha, K.; Shah, J.; Vaiphei, K.; Dutta, U.; Singh, A.K. Colon Cancer in Ulcerative Colitis: A Mimicker of a Flare of Disease. Inflamm. Bowel Dis. 2022, 28, e116–e117. [Google Scholar] [CrossRef]
- Naydenov, N.G.; Lechuga, S.; Zalavadia, A.; Mukherjee, P.K.; Gordon, I.O.; Skvasik, D.; Vidovic, P.; Huang, E.; Rieder, F.; Ivanov, A.I. P-Cadherin Regulates Intestinal Epithelial Cell Migration and Mucosal Repair, but Is Dispensable for Colitis As-sociated Colon Cancer. Cells 2022, 11, 1467. [Google Scholar] [CrossRef] [PubMed]
- Devall, M.A.; Eaton, S.; Ali, M.W.; Dampier, C.H.; Weisenberger, D.; Powell, S.M.; Li, L.; Casey, G. DNA methylation analysis of normal colon organoids from familial adenomatous polyposis patients reveals novel insight into colon cancer development. Clin. Epigenet. 2022, 14, 104. [Google Scholar] [CrossRef]
- Devall, M.A.; Eaton, S.; Ali, M.W.; Powell, S.M.; Li, L.; Casey, G. Insights into Early Onset Colorectal Cancer through Analysis of Normal Colon Organoids of Familial Adenomatous Polyposis Patients. Cancers 2022, 14, 4138. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, X.; Li, A.; Tang, X. A pan-cancer analysis on the carcinogenic effect of human adenomatous polyposis coli. PLoS ONE 2022, 17, e0265655. [Google Scholar] [CrossRef]
- Richards, S.; Walker, J.; Nakanishi, M.; Belghasem, M.; Lyle, C.; Arinze, N.; Napoleon, M.A.; Ravid, J.D.; Crossland, N.; Zhao, Q.; et al. Haploinsufficiency of Casitas B-Lineage Lymphoma Augments the Progression of Colon Cancer in the Background of Adenomatous Polyposis Coli Inactivation. Am. J. Pathol. 2020, 190, 602–613. [Google Scholar] [CrossRef]
- Janssen, A.W.F.; Duivenvoorde, L.P.M.; Rijkers, D.; Nijssen, R.; Peijnenburg, A.; van der Zande, M.; Louisse, J. Cytochrome P450 expression, induction and activity in human induced pluripotent stem cell-derived intestinal organoids and comparison with primary human intestinal epithelial cells and Caco-2 cells. Arch. Toxicol. 2021, 95, 907–922. [Google Scholar] [CrossRef]
- Crespo, M.; Vilar, E.; Tsai, S.Y.; Chang, K.; Amin, S.; Srinivasan, T.; Zhang, T.; Pipalia, N.H.; Chen, H.J.; Witherspoon, M.; et al. Colonic organoids derived from human induced pluripotent stem cells for modeling colorectal cancer and drug testing. Nat. Med. 2017, 23, 878–884. [Google Scholar] [CrossRef]
- Hirshorn, S.T.; Steele, N.; Zavros, Y. Modeling pancreatic pathophysiology using genome editing of adult stem cell-derived and induced pluripotent stem cell (iPSC)-derived organoids. Am. J. Physiol. Gastrointest. Liver Physiol. 2021, 320, G1142–G1150. [Google Scholar] [CrossRef]
- Wuputra, K.; Ku, C.C.; Kato, K.; Wu, D.C.; Saito, S.; Yokoyama, K.K. Translational models of 3-D organoids and cancer stem cells in gastric cancer research. Stem Cell Res. Ther. 2021, 12, 492. [Google Scholar] [CrossRef]
- Sato, T.; Stange, D.E.; Ferrante, M.; Vries, R.G.; Van Es, J.H.; Van den Brink, S.; Van Houdt, W.J.; Pronk, A.; Van Gorp, J.; Siersema, P.D.; et al. Long-term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett’s epithelium. Gastroenterology 2011, 141, 1762–1772. [Google Scholar] [CrossRef]
- Matano, M.; Date, S.; Shimokawa, M.; Takano, A.; Fujii, M.; Ohta, Y.; Watanabe, T.; Kanai, T.; Sato, T. Modeling colorectal cancer using CRISPR-Cas9-mediated engineering of human intestinal organoids. Nat. Med. 2015, 21, 256–262. [Google Scholar] [CrossRef]
- Hendriks, D.; Pagliaro, A.; Andreatta, F.; Ma, Z.; van Giessen, J.; Massalini, S.; López-Iglesias, C.; van Son, G.J.F.; DeMartino, J.; Damen, J.M.A.; et al. Human fetal brain self-organizes into long-term expanding organoids. Cell 2024, 187, 712–732.e38. [Google Scholar] [CrossRef]
- Norrie, J.L.; Nityanandam, A.; Lai, K.; Chen, X.; Wilson, M.; Stewart, E.; Griffiths, L.; Jin, H.; Wu, G.; Orr, B.; et al. Reti-noblastoma from human stem cell-derived retinal organoids. Nat. Commun. 2021, 12, 4535. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.; Iwagawa, T.; Fukushima, M.; Watanabe, S. Characterization of human-induced pluripotent stem cells carrying homozygous RB1 gene deletion. Genes Cells Devoted Mol. Cell. Mech. 2020, 25, 510–517. [Google Scholar] [CrossRef] [PubMed]
- Rozanska, A.; Cerna-Chavez, R.; Queen, R.; Collin, J.; Zerti, D.; Dorgau, B.; Beh, C.S.; Davey, T.; Coxhead, J.; Hussain, R.; et al. pRB-Depleted Pluripotent Stem Cell Retinal Organoids Recapitulate Cell State Transitions of Retinoblastoma Development and Suggest an Important Role for pRB in Retinal Cell Differentiation. Stem Cells Transl. Med. 2022, 11, 415–433. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, R.; Ishikawa, Y.; Sasamoto, Y.; Katori, R.; Nomura, N.; Ichikawa, T.; Araki, S.; Soma, T.; Kawasaki, S.; Sekiguchi, K.; et al. Co-ordinated ocular development from human iPS cells and recovery of corneal function. Nature 2016, 531, 376–380. [Google Scholar] [CrossRef]
- Dost, A.F.M.; Moye, A.L.; Vedaie, M.; Tran, L.M.; Fung, E.; Heinze, D.; Villacorta-Martin, C.; Huang, J.; Hekman, R.; Kwan, J.H.; et al. Organoids Model Transcriptional Hallmarks of Oncogenic KRAS Activation in Lung Epithelial Progenitor Cells. Cell Stem Cell 2020, 27, 663–678.e8. [Google Scholar] [CrossRef] [PubMed]
- Skylar-Scott, M.A.; Huang, J.Y.; Lu, A.; Ng, A.H.M.; Duenki, T.; Liu, S.; Nam, L.L.; Damaraju, S.; Church, G.M.; Lewis, J.A. Orthogonally induced differentiation of stem cells for the programmatic patterning of vascularized organoids and bioprinted tissues. Nat. Biomed. Eng. 2022, 6, 449–462. [Google Scholar] [CrossRef]
- Centeno, E.G.Z.; Cimarosti, H.; Bithell, A. 2D versus 3D human induced pluripotent stem cell-derived cultures for neuro-degenerative disease modelling. Mol. Neurodegener. 2018, 13, 27. [Google Scholar] [CrossRef]
- Zhou, Y.; Song, H.; Ming, G.L. Genetics of human brain development. Nat. Rev. Genet. 2024, 25, 26–45. [Google Scholar] [CrossRef]
- Villa, C.; Combi, R.; Conconi, D.; Lavitrano, M. Patient-Derived Induced Pluripotent Stem Cells (iPSCs) and Cerebral Organoids for Drug Screening and Development in Autism Spectrum Disorder: Opportunities and Challenges. Pharmaceutics 2021, 13, 280. [Google Scholar] [CrossRef]
- Lu, K.; Hong, Y.; Tao, M.; Shen, L.; Zheng, Z.; Fang, K.; Yuan, F.; Xu, M.; Wang, C.; Zhu, D.; et al. Depressive patient-derived GABA interneurons reveal abnormal neural activity associated with HTR2C. EMBO Mol. Med. 2023, 15, e16364. [Google Scholar] [CrossRef]
- Velasco, S.; Kedaigle, A.J.; Simmons, S.K.; Nash, A.; Rocha, M.; Quadrato, G.; Paulsen, B.; Nguyen, L.; Adiconis, X.; Regev, A.; et al. Individual brain organoids reproducibly form cell diversity of the human cerebral cortex. Nature 2019, 570, 523–527. [Google Scholar] [CrossRef] [PubMed]
- Mansour, A.A.; Gonçalves, J.T.; Bloyd, C.W.; Li, H.; Fernandes, S.; Quang, D.; Johnston, S.; Parylak, S.L.; Jin, X.; Gage, F.H. An in vivo model of functional and vascularized human brain organoids. Nat. Biotechnol. 2018, 36, 432–441. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Yuan, X.; Jones, Z.; Griffin, K.; Zhou, Y.; Ma, T.; Li, Y. Assembly of Human Stem Cell-Derived Cortical Spheroids and Vascular Spheroids to Model 3-D Brain-like Tissues. Sci. Rep. 2019, 9, 5977. [Google Scholar] [CrossRef]
- Bagley, J.A.; Reumann, D.; Bian, S.; Lévi-Strauss, J.; Knoblich, J.A. Fused cerebral organoids model interactions between brain regions. Nat. Methods 2017, 14, 743–751. [Google Scholar] [CrossRef]
- Costamagna, G.; Comi, G.P.; Corti, S. Advancing Drug Discovery for Neurological Disorders Using iPSC-Derived Neural Or-ganoids. Int. J. Mol. Sci. 2021, 22, 2659. [Google Scholar] [CrossRef]
- Egawa, N.; Kitaoka, S.; Tsukita, K.; Naitoh, M.; Takahashi, K.; Yamamoto, T.; Adachi, F.; Kondo, T.; Okita, K.; Asaka, I.; et al. Drug screening for ALS using patient-specific induced pluripotent stem cells. Sci. Transl. Med. 2012, 4, 145ra104. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, S.; Takahashi, S.; Ito, D.; Daté, Y.; Okada, K.; Kato, C.; Nakamura, S.; Ozawa, F.; Chyi, C.M.; Nishiyama, A.; et al. Phase 1/2a clinical trial in ALS with ropinirole, a drug candidate identified by iPSC drug discovery. Cell Stem Cell 2023, 30, 766–780.e9. [Google Scholar] [CrossRef]
- Ha, J.; Kang, J.S.; Lee, M.; Baek, A.; Kim, S.; Chung, S.K.; Lee, M.O.; Kim, J. Simplified Brain Organoids for Rapid and Robust Modeling of Brain Disease. Front. Cell Dev. Biol. 2020, 8, 594090. [Google Scholar] [CrossRef]
- Pasqua, M.; Di Gesù, R.; Chinnici, C.M.; Conaldi, P.G.; Francipane, M.G. Generation of Hepatobiliary Cell Lineages from Human Induced Pluripotent Stem Cells: Applications in Disease Modeling and Drug Screening. Int. J. Mol. Sci. 2021, 22, 8227. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.J.; Meyer, S.R.; O’Meara, M.J.; Huang, S.; Capeling, M.M.; Ferrer-Torres, D.; Childs, C.J.; Spence, J.R.; Fontana, R.J.; Sexton, J.Z. A human liver organoid screening platform for DILI risk prediction. J. Hepatol. 2023, 78, 998–1006. [Google Scholar] [CrossRef]
- Broutier, L.; Mastrogiovanni, G.; Verstegen, M.M.; Francies, H.E.; Gavarró, L.M.; Bradshaw, C.R.; Allen, G.E.; Arnes-Benito, R.; Sidorova, O.; Gaspersz, M.P.; et al. Human primary liver cancer-derived organoid cultures for disease modeling and drug screening. Nat. Med. 2017, 23, 1424–1435. [Google Scholar] [CrossRef]
- Koui, Y.; Kido, T.; Ito, T.; Oyama, H.; Chen, S.W.; Katou, Y.; Shirahige, K.; Miyajima, A. An In Vitro Human Liver Model by iPSC-Derived Parenchymal and Non-parenchymal Cells. Stem Cell Rep. 2017, 9, 490–498. [Google Scholar] [CrossRef]
- Jiang, S.; Xu, F.; Jin, M.; Wang, Z.; Xu, X.; Zhou, Y.; Wang, J.; Gu, L.; Fan, H.; Fan, Y.; et al. Development of a high-throughput micropatterned agarose scaffold for consistent and reproducible hPSC-derived liver organoids. Biofabrication 2022, 15, 015006. [Google Scholar] [CrossRef] [PubMed]
- Weng, Y.; Han, S.; Sekyi, M.T.; Su, T.; Mattis, A.N.; Chang, T.T. Self-Assembled Matrigel-Free iPSC-Derived Liver Organoids Demonstrate Wide-Ranging Highly Differentiated Liver Functions. Stem Cells 2023, 41, 126–139. [Google Scholar] [CrossRef]
- Xu, X.; Jiang, S.; Gu, L.; Li, B.; Xu, F.; Li, C.; Chen, P. High-throughput bioengineering of homogenous and functional hu-man-induced pluripotent stem cells-derived liver organoids via micropatterning technique. Front. Bioeng. Biotechnol. 2022, 10, 937595. [Google Scholar] [CrossRef]
- Shinozawa, T.; Kimura, M.; Cai, Y.; Saiki, N.; Yoneyama, Y.; Ouchi, R.; Koike, H.; Maezawa, M.; Zhang, R.R.; Dunn, A.; et al. High-Fidelity Drug-Induced Liver Injury Screen Using Human Pluripotent Stem Cell-Derived Organoids. Gastroenterology 2021, 160, 831–846.e10. [Google Scholar] [CrossRef]
- Kim, H.; Im, I.; Jeon, J.S.; Kang, E.H.; Lee, H.A.; Jo, S.; Kim, J.W.; Woo, D.H.; Choi, Y.J.; Kim, H.J.; et al. Development of human pluripotent stem cell-derived hepatic organoids as an alternative model for drug safety assessment. Biomaterials 2022, 286, 121575. [Google Scholar] [CrossRef]
- Cantillon, D.; Goff, A.; Taylor, S.; Salehi, E.; Fidler, K.; Stoneham, S.; Waddell, S.J. Searching for new therapeutic options for the uncommon pathogen Mycobacterium chimaera: An open drug discovery approach. Lancet Microbe 2022, 3, e382–e391. [Google Scholar] [CrossRef] [PubMed]
- Horlock, D.; Kaye, D.M.; Winbanks, C.E.; Gao, X.M.; Kiriazis, H.; Donner, D.G.; Gregorevic, P.; McMullen, J.R.; Bernardo, B.C. Old Drug, New Trick: Tilorone, a Broad-Spectrum Antiviral Drug as a Potential Anti-Fibrotic Therapeutic for the Diseased Heart. Pharmaceuticals 2021, 14, 263. [Google Scholar] [CrossRef] [PubMed]
- Ang, Y.S.; Rajamani, S.; Haldar, S.M.; Hüser, J. A New Therapeutic Framework for Atrial Fibrillation Drug Development. Circ. Res. 2020, 127, 184–201. [Google Scholar] [CrossRef] [PubMed]
- Bekhite, M.; González-Delgado, A.; Hübner, S.; Haxhikadrija, P.; Kretzschmar, T.; Müller, T.; Wu, J.M.F.; Bekfani, T.; Franz, M.; Wartenberg, M.; et al. The role of ceramide accumulation in human induced pluripotent stem cell-derived cardiomyocytes on mitochondrial oxidative stress and mitophagy. Free Radic. Biol. Med. 2021, 167, 66–80. [Google Scholar] [CrossRef]
- Magdy, T.; Jouni, M.; Kuo, H.H.; Weddle, C.J.; Lyra-Leite, D.; Fonoudi, H.; Romero-Tejeda, M.; Gharib, M.; Javed, H.; Fajardo, G.; et al. Identification of Drug Transporter Genomic Variants and Inhibitors That Protect Against Doxorubicin-Induced Cardio-toxicity. Circulation 2022, 145, 279–294. [Google Scholar] [CrossRef] [PubMed]
- Ergir, E.; Oliver-De La Cruz, J.; Fernandes, S.; Cassani, M.; Niro, F.; Pereira-Sousa, D.; Vrbský, J.; Vinarský, V.; Perestrelo, A.R.; Debellis, D.; et al. Generation and maturation of human iPSC-derived 3D organotypic cardiac microtissues in long-term culture. Sci. Rep. 2022, 12, 17409. [Google Scholar] [CrossRef]
- Sallam, K.; Thomas, D.; Gaddam, S.; Lopez, N.; Beck, A.; Beach, L.; Rogers, A.J.; Zhang, H.; Chen, I.Y.; Ameen, M.; et al. Modeling Effects of Immunosuppressive Drugs on Human Hearts Using Induced Pluripotent Stem Cell-Derived Cardiac Or-ganoids and Single-Cell RNA Sequencing. Circulation 2022, 145, 1367–1369. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, M.W.; Ting, C.Y.; Chan, D.Z.H.; Cheng, Y.C.; Lee, Y.C.; Hsu, C.C.; Huang, C.Y.; Hsieh, P.C.H. Utility of iPSC-Derived Cells for Disease Modeling, Drug Development, and Cell Therapy. Cells 2022, 11, 1853. [Google Scholar] [CrossRef] [PubMed]
- Song, B.; Cha, Y.; Ko, S.; Jeon, J.; Lee, N.; Seo, H.; Park, K.J.; Lee, I.H.; Lopes, C.; Feitosa, M.; et al. Human autologous iPSC-derived dopaminergic progenitors restore motor function in Parkinson’s disease models. J. Clin. Investig. 2020, 130, 904–920. [Google Scholar] [CrossRef] [PubMed]
- Hiller, B.M.; Marmion, D.J.; Thompson, C.A.; Elliott, N.A.; Federoff, H.; Brundin, P.; Mattis, V.B.; McMahon, C.W.; Kordower, J.H. Optimizing maturity and dose of iPSC-derived dopamine progenitor cell therapy for Parkinson’s disease. NPJ Regen. Med. 2022, 7, 24. [Google Scholar] [CrossRef] [PubMed]
- Schweitzer, J.S.; Song, B.; Herrington, T.M.; Park, T.Y.; Lee, N.; Ko, S.; Jeon, J.; Cha, Y.; Kim, K.; Li, Q.; et al. Personalized iPSC-Derived Dopamine Progenitor Cells for Parkinson’s Disease. N. Engl. J. Med. 2020, 382, 1926–1932. [Google Scholar] [CrossRef]
- Kikuchi, T.; Morizane, A.; Doi, D.; Magotani, H.; Onoe, H.; Hayashi, T.; Mizuma, H.; Takara, S.; Takahashi, R.; Inoue, H.; et al. Human iPS cell-derived dopaminergic neurons function in a primate Parkinson’s disease model. Nature 2017, 548, 592–596. [Google Scholar] [CrossRef]
- Doi, D.; Magotani, H.; Kikuchi, T.; Ikeda, M.; Hiramatsu, S.; Yoshida, K.; Amano, N.; Nomura, M.; Umekage, M.; Morizane, A.; et al. Pre-clinical study of induced pluripotent stem cell-derived dopaminergic progenitor cells for Parkinson’s disease. Nat. Commun. 2020, 11, 3369. [Google Scholar] [CrossRef]
- Fernandes, H.J.R.; Patikas, N.; Foskolou, S.; Field, S.F.; Park, J.E.; Byrne, M.L.; Bassett, A.R.; Metzakopian, E. Single-Cell Transcriptomics of Parkinson’s Disease Human In Vitro Models Reveals Dopamine Neuron-Specific Stress Responses. Cell Rep. 2020, 33, 108263. [Google Scholar] [CrossRef]
- Lyu, Y.; Su, Z.; Ye, G.; He, X.; Liu, Y.; Yin, Q.; Xie, F.; Xu, L.; Chen, Y.; Long, D. MiR-210-5p promotes the differentiation of human induced pluripotent stem cells into dopaminergic neural precursors by targeting SMAD4 and SUFU and treats par-kinsonian rats. Exp. Gerontol. 2023, 179, 112243. [Google Scholar] [CrossRef]
- Brot, S.; Thamrin, N.P.; Bonnet, M.L.; Francheteau, M.; Patrigeon, M.; Belnoue, L.; Gaillard, A. Long-Term Evaluation of In-tranigral Transplantation of Human iPSC-Derived Dopamine Neurons in a Parkinson’s Disease Mouse Model. Cells 2022, 11, 1596. [Google Scholar] [CrossRef]
- Xu, Z.; Chu, X.; Jiang, H.; Schilling, H.; Chen, S.; Feng, J. Induced dopaminergic neurons: A new promise for Parkinson’s disease. Redox Biol. 2017, 11, 606–612. [Google Scholar] [CrossRef] [PubMed]
- Paul, K.C.; Krolewski, R.C.; Lucumi Moreno, E.; Blank, J.; Holton, K.M.; Ahfeldt, T.; Furlong, M.; Yu, Y.; Cockburn, M.; Thompson, L.K.; et al. A pesticide and iPSC dopaminergic neuron screen identifies and classifies Parkinson-relevant pesticides. Nat. Commun. 2023, 14, 2803. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Jiang, H.; Li, H.; Li, L.; Yan, Z.; Feng, J. Generation of human A9 dopaminergic pacemakers from induced pluripotent stem cells. Mol. Psychiatry 2022, 27, 4407–4418. [Google Scholar] [CrossRef] [PubMed]
- Povero, D.; Pinatel, E.M.; Leszczynska, A.; Goyal, N.P.; Nishio, T.; Kim, J.; Kneiber, D.; de Araujo Horcel, L.; Eguchi, A.; Or-donez, P.M.; et al. Human induced pluripotent stem cell-derived extracellular vesicles reduce hepatic stellate cell activation and liver fibrosis. JCI Insight 2019, 4, e125652. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.S.; Jeong, I.S.; Park, Y.J.; Kim, S.W. HGF and IL-10 expressing ALB::GFP reporter cells generated from iPSCs show robust anti-fibrotic property in acute fibrotic liver model. Stem Cell Res. Ther. 2020, 11, 332. [Google Scholar] [CrossRef] [PubMed]
- Monckton, C.P.; Brougham-Cook, A.; Underhill, G.H.; Khetani, S.R. Modulation of human iPSC-derived hepatocyte phenotype via extracellular matrix microarrays. Acta Biomater. 2022, 153, 216–230. [Google Scholar] [CrossRef] [PubMed]
- Song, Z.; Cai, J.; Liu, Y.; Zhao, D.; Yong, J.; Duo, S.; Song, X.; Guo, Y.; Zhao, Y.; Qin, H.; et al. Efficient generation of hepato-cyte-like cells from human induced pluripotent stem cells. Cell Res. 2009, 19, 1233–1242. [Google Scholar] [CrossRef] [PubMed]
- Pouyanfard, S.; Meshgin, N.; Cruz, L.S.; Diggle, K.; Hashemi, H.; Pham, T.V.; Fierro, M.; Tamayo, P.; Fanjul, A.; Kisseleva, T.; et al. Human induced pluripotent stem cell-derived macrophages ameliorate liver fibrosis. Stem Cells 2021, 39, 1701–1717. [Google Scholar] [CrossRef]
- Yeung, E.; Fukunishi, T.; Bai, Y.; Bedja, D.; Pitaktong, I.; Mattson, G.; Jeyaram, A.; Lui, C.; Ong, C.S.; Inoue, T.; et al. Cardiac regeneration using human-induced pluripotent stem cell-derived biomaterial-free 3D-bioprinted cardiac patch in vivo. J. Tissue Eng. Regen. Med. 2019, 13, 2031–2039. [Google Scholar] [CrossRef]
- Lancaster, J.J.; Sanchez, P.; Repetti, G.G.; Juneman, E.; Pandey, A.C.; Chinyere, I.R.; Moukabary, T.; LaHood, N.; Daugherty, S.L.; Goldman, S. Human Induced Pluripotent Stem Cell-Derived Cardiomyocyte Patch in Rats with Heart Failure. Ann. Thorac. Surg. 2019, 108, 1169–1177. [Google Scholar] [CrossRef]
- Ikeda, G.; Santoso, M.R.; Tada, Y.; Li, A.M.; Vaskova, E.; Jung, J.H.; O’Brien, C.; Egan, E.; Ye, J.; Yang, P.C. Mitochondria-Rich Extracellular Vesicles From Autologous Stem Cell-Derived Cardiomyocytes Restore Energetics of Ischemic Myocardium. J. Am. Coll. Cardiol. 2021, 77, 1073–1088. [Google Scholar] [CrossRef]
- Jiang, X.; Yang, Z.; Dong, M. Cardiac repair in a murine model of myocardial infarction with human induced pluripotent stem cell-derived cardiomyocytes. Stem Cell Res. Ther. 2020, 11, 297. [Google Scholar] [CrossRef]
- Kurtzwald-Josefson, E.; Zeevi-Levin, N.; Rubchevsky, V.; Bechar Erdman, N.; Schwartz Rohaker, O.; Nahum, O.; Hochhauser, E.; Ben-Avraham, B.; Itskovitz-Eldor, J.; Aravot, D.; et al. Cardiac Fibroblast-Induced Pluripotent Stem Cell-Derived Exosomes as a Potential Therapeutic Mean for Heart Failure. Int. J. Mol. Sci. 2020, 21, 7215. [Google Scholar] [CrossRef]
- Dudley, M.E.; Wunderlich, J.R.; Robbins, P.F.; Yang, J.C.; Hwu, P.; Schwartzentruber, D.J.; Topalian, S.L.; Sherry, R.; Restifo, N.P.; Hubicki, A.M.; et al. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lym-phocytes. Science 2002, 298, 850–854. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Yang, J.C.; Sherry, R.M.; Kammula, U.S.; Hughes, M.S.; Phan, G.Q.; Citrin, D.E.; Restifo, N.P.; Robbins, P.F.; Wunderlich, J.R.; et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2011, 17, 4550–4557. [Google Scholar] [CrossRef]
- Dudley, M.E.; Yang, J.C.; Sherry, R.; Hughes, M.S.; Royal, R.; Kammula, U.; Robbins, P.F.; Huang, J.; Citrin, D.E.; Leitman, S.F.; et al. Adoptive cell therapy for patients with metastatic melanoma: Evaluation of intensive myeloablative chemoradiation preparative regimens. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2008, 26, 5233–5239. [Google Scholar] [CrossRef]
- Li, X.; Halldórsdóttir, H.R.; Weller, S.; Colliander, A.; Bak, M.; Kempen, P.; Clergeaud, G.; Andresen, T.L. Enhancing Adoptive Cell Therapy by T Cell Loading of SHP2 Inhibitor Nanocrystals before Infusion. ACS Nano 2022, 16, 10918–10930. [Google Scholar] [CrossRef]
- Mardiana, S.; Solomon, B.J.; Darcy, P.K.; Beavis, P.A. Supercharging adoptive T cell therapy to overcome solid tumor-induced immunosuppression. Sci. Transl. Med. 2019, 11, eaaw2293. [Google Scholar] [CrossRef] [PubMed]
- Krishna, S.; Lowery, F.J.; Copeland, A.R.; Bahadiroglu, E.; Mukherjee, R.; Jia, L.; Anibal, J.T.; Sachs, A.; Adebola, S.O.; Gu-rusamy, D.; et al. Stem-like CD8 T cells mediate response of adoptive cell immunotherapy against human cancer. Science 2020, 370, 1328–1334. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, E.E.; Sekhri, P.; Telaraja, D.; Chen, J.; Chin, S.J.; Chiappinelli, K.B.; Sanchez, C.E.; Bollard, C.M.; Cruz, C.R.Y.; Fer-nandes, R. Engineered tumor-specific T cells using immunostimulatory photothermal nanoparticles. Cytotherapy 2023, 25, 718–727. [Google Scholar] [CrossRef] [PubMed]
- Potenza, A.; Balestrieri, C.; Spiga, M.; Albarello, L.; Pedica, F.; Manfredi, F.; Cianciotti, B.C.; De Lalla, C.; Botrugno, O.A.; Faccani, C.; et al. Revealing and harnessing CD39 for the treatment of colorectal cancer and liver metastases by engineered T cells. Gut 2023, 72, 1887–1903. [Google Scholar] [CrossRef]
- Heitzeneder, S.; Bosse, K.R.; Zhu, Z.; Zhelev, D.; Majzner, R.G.; Radosevich, M.T. CAR T Cells Engineered to Target Low-Density Antigens Show Efficacy and Safety. Cancer Discov. 2022, 12, 595. [Google Scholar] [CrossRef]
- Ma, L.; Hostetler, A.; Morgan, D.M.; Maiorino, L.; Sulkaj, I.; Whittaker, C.A.; Neeser, A.; Pires, I.S.; Yousefpour, P.; Gregory, J.; et al. Vaccine-boosted CAR T crosstalk with host immunity to reject tumors with antigen heterogeneity. Cell 2023, 186, 3148–3165.e20. [Google Scholar] [CrossRef]
- Stein-Thoeringer, C.K.; Saini, N.Y.; Zamir, E.; Blumenberg, V.; Schubert, M.L.; Mor, U.; Fante, M.A.; Schmidt, S.; Hayase, E.; Hayase, T.; et al. A non-antibiotic-disrupted gut microbiome is associated with clinical responses to CD19-CAR-T cell cancer immunotherapy. Nat. Med. 2023, 29, 906–916. [Google Scholar] [CrossRef]
- Good, Z.; Spiegel, J.Y.; Sahaf, B.; Malipatlolla, M.B.; Ehlinger, Z.J.; Kurra, S.; Desai, M.H.; Reynolds, W.D.; Wong Lin, A.; Vandris, P.; et al. Post-infusion CAR T(Reg) cells identify patients resistant to CD19-CAR therapy. Nat. Med. 2022, 28, 1860–1871. [Google Scholar] [CrossRef] [PubMed]
- Vasic, D.; Lee, J.B.; Leung, Y.; Khatri, I.; Na, Y.; Abate-Daga, D.; Zhang, L. Allogeneic double-negative CAR-T cells inhibit tumor growth without off-tumor toxicities. Sci. Immunol. 2022, 7, eabl3642. [Google Scholar] [CrossRef] [PubMed]
- Meyran, D.; Zhu, J.J.; Butler, J.; Tantalo, D.; MacDonald, S.; Nguyen, T.N.; Wang, M.; Thio, N.; D’Souza, C.; Qin, V.M.; et al. T(STEM)-like CAR-T cells exhibit improved persistence and tumor control compared with conventional CAR-T cells in pre-clinical models. Sci. Transl. Med. 2023, 15, eabk1900. [Google Scholar] [CrossRef]
- Sadeqi Nezhad, M.; Abdollahpour-Alitappeh, M.; Rezaei, B.; Yazdanifar, M.; Seifalian, A.M. Induced Pluripotent Stem Cells (iPSCs) Provide a Potentially Unlimited T Cell Source for CAR-T Cell Development and Off-the-Shelf Products. Pharm. Res. 2021, 38, 931–945. [Google Scholar] [CrossRef]
- Wang, Z.; McWilliams-Koeppen, H.P.; Reza, H.; Ostberg, J.R.; Chen, W.; Wang, X.; Huynh, C.; Vyas, V.; Chang, W.C.; Starr, R.; et al. 3D-organoid culture supports differentiation of human CAR(+) iPSCs into highly functional CAR T cells. Cell Stem Cell 2022, 29, 515–527.e8. [Google Scholar] [CrossRef]
- Kaneko, S. Successful organoid-mediated generation of iPSC-derived CAR-T cells. Cell Stem Cell 2022, 29, 493–495. [Google Scholar] [CrossRef]
- Ueda, T.; Kaneko, S. In Vitro Differentiation of T Cell: From CAR-Modified T-iPSC. Methods Mol. Biol. 2019, 2048, 85–91. [Google Scholar] [CrossRef]
- van der Stegen, S.J.C.; Lindenbergh, P.L.; Petrovic, R.M.; Xie, H.; Diop, M.P.; Alexeeva, V.; Shi, Y.; Mansilla-Soto, J.; Hamieh, M.; Eyquem, J.; et al. Generation of T-cell-receptor-negative CD8αβ-positive CAR T cells from T-cell-derived induced pluripotent stem cells. Nat. Biomed. Eng. 2022, 6, 1284–1297. [Google Scholar] [CrossRef]
- Jing, R.; Scarfo, I.; Najia, M.A.; Lummertz da Rocha, E.; Han, A.; Sanborn, M.; Bingham, T.; Kubaczka, C.; Jha, D.K.; Falchetti, M.; et al. EZH1 repression generates mature iPSC-derived CAR T cells with enhanced antitumor activity. Cell Stem Cell 2022, 29, 1181–1196.e6. [Google Scholar] [CrossRef]
- Ueda, T.; Shiina, S.; Iriguchi, S.; Terakura, S.; Kawai, Y.; Kabai, R.; Sakamoto, S.; Watanabe, A.; Ohara, K.; Wang, B.; et al. Optimization of the proliferation and persistency of CAR T cells derived from human induced pluripotent stem cells. Nat. Biomed. Eng. 2023, 7, 24–37. [Google Scholar] [CrossRef]
- Harada, S.; Ando, M.; Ando, J.; Ishii, M.; Yamaguchi, T.; Yamazaki, S.; Toyota, T.; Ohara, K.; Ohtaka, M.; Nakanishi, M.; et al. Dual-antigen targeted iPSC-derived chimeric antigen receptor-T cell therapy for refractory lymphoma. Mol. Ther. J. Am. Soc. Gene Ther. 2022, 30, 534–549. [Google Scholar] [CrossRef]
- Myers, J.A.; Miller, J.S. Exploring the NK cell platform for cancer immunotherapy. Nat. Rev. Clin. Oncol. 2021, 18, 85–100. [Google Scholar] [CrossRef]
- Saetersmoen, M.L.; Hammer, Q.; Valamehr, B.; Kaufman, D.S.; Malmberg, K.J. Off-the-shelf cell therapy with induced plu-ripotent stem cell-derived natural killer cells. Semin. Immunopathol. 2019, 41, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Cooper, M.A.; Fehniger, T.A.; Caligiuri, M.A. The biology of human natural killer-cell subsets. Trends Immunol. 2001, 22, 633–640. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, K.L.; Finlay, D.K. Immunometabolism and natural killer cell responses. Nat. Rev. Immunol. 2019, 19, 282–290. [Google Scholar] [CrossRef] [PubMed]
- Mehta, R.S.; Rezvani, K. Chimeric Antigen Receptor Expressing Natural Killer Cells for the Immunotherapy of Cancer. Front. Immunol. 2018, 9, 283. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, A.; Cui, H.; Caligiuri, M.A.; Yu, J. Chimeric antigen receptor-engineered natural killer cells for cancer immunotherapy. J. Hematol. Oncol. 2020, 13, 168. [Google Scholar] [CrossRef]
- Cichocki, F.; van der Stegen, S.J.C.; Miller, J.S. Engineered and banked iPSCs for advanced NK- and T-cell immunotherapies. Blood 2023, 141, 846–855. [Google Scholar] [CrossRef]
- Cichocki, F.; Bjordahl, R.; Gaidarova, S.; Mahmood, S.; Abujarour, R.; Wang, H.; Tuininga, K.; Felices, M.; Davis, Z.B.; Bendzick, L.; et al. iPSC-derived NK cells maintain high cytotoxicity and enhance in vivo tumor control in concert with T cells and an-ti-PD-1 therapy. Sci. Transl. Med. 2020, 12, eaaz5618. [Google Scholar] [CrossRef]
- Meng, F.; Zhang, S.; Xie, J.; Zhou, Y.; Wu, Q.; Lu, B.; Zhou, S.; Zhao, X.; Li, Y. Leveraging CD16 fusion receptors to remodel the immune response for enhancing anti-tumor immunotherapy in iPSC-derived NK cells. J. Hematol. Oncol. 2023, 16, 62. [Google Scholar] [CrossRef]
- Chiu, E.; Felices, M.; Cichocki, F.; Davis, Z.; Wang, H.; Tuninga, K.; Vallera, D.A.; Lee, T.; Bjordahl, R.; Malmberg, K.J.; et al. Anti-NKG2C/IL-15/anti-CD33 killer engager directs primary and iPSC-derived NKG2C+ NK cells to target myeloid leukemia. Mol. Ther. J. Am. Soc. Gene Ther. 2021, 29, 3410–3421. [Google Scholar] [CrossRef]
- Zhu, H.; Blum, R.H.; Bernareggi, D.; Ask, E.H.; Wu, Z.; Hoel, H.J.; Meng, Z.; Wu, C.; Guan, K.L.; Malmberg, K.J.; et al. Metabolic Reprograming via Deletion of CISH in Human iPSC-Derived NK Cells Promotes In Vivo Persistence and Enhances Anti-tumor Activity. Cell Stem Cell 2020, 27, 224–237.e6. [Google Scholar] [CrossRef] [PubMed]
- Ueda, T.; Kumagai, A.; Iriguchi, S.; Yasui, Y.; Miyasaka, T.; Nakagoshi, K.; Nakane, K.; Saito, K.; Takahashi, M.; Sasaki, A.; et al. Non-clinical efficacy, safety and stable clinical cell processing of induced pluripotent stem cell-derived anti-glypican-3 chimeric antigen receptor-expressing natural killer/innate lymphoid cells. Cancer Sci. 2020, 111, 1478–1490. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Hermanson, D.L.; Moriarity, B.S.; Kaufman, D.S. Human iPSC-Derived Natural Killer Cells Engineered with Chimeric Antigen Receptors Enhance Anti-tumor Activity. Cell Stem Cell 2018, 23, 181–192.e5. [Google Scholar] [CrossRef] [PubMed]
- Cichocki, F.; Goodridge, J.P.; Bjordahl, R.; Mahmood, S.; Davis, Z.B.; Gaidarova, S.; Abujarour, R.; Groff, B.; Witty, A.; Wang, H.; et al. Dual antigen-targeted off-the-shelf NK cells show durable response and prevent antigen escape in lymphoma and leu-kemia. Blood 2022, 140, 2451–2462. [Google Scholar] [CrossRef]
- Klaihmon, P.; Kang, X.; Issaragrisil, S.; Luanpitpong, S. Generation and Functional Characterization of Anti-CD19 Chimeric Antigen Receptor-Natural Killer Cells from Human Induced Pluripotent Stem Cells. Int. J. Mol. Sci. 2023, 24, 10508. [Google Scholar] [CrossRef]
- Goulding, J.; Yeh, W.I.; Hancock, B.; Blum, R.; Xu, T.; Yang, B.H.; Chang, C.W.; Groff, B.; Avramis, E.; Pribadi, M.; et al. A chimeric antigen receptor uniquely recognizing MICA/B stress proteins provides an effective approach to target solid tumors. Med 2023, 4, 457–477.e8. [Google Scholar] [CrossRef]
- Hamieh, M.; Dobrin, A.; Cabriolu, A.; van der Stegen, S.J.C.; Giavridis, T.; Mansilla-Soto, J.; Eyquem, J.; Zhao, Z.; Whitlock, B.M.; Miele, M.M.; et al. CAR T cell trogocytosis and cooperative killing regulate tumour antigen escape. Nature 2019, 568, 112–116. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Basar, R.; Wang, G.; Liu, E.; Moyes, J.S.; Li, L.; Kerbauy, L.N.; Uprety, N.; Fathi, M.; Rezvan, A.; et al. KIR-based inhibitory CARs overcome CAR-NK cell trogocytosis-mediated fratricide and tumor escape. Nat. Med. 2022, 28, 2133–2144. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Wang, L.; Guo, H.; Zhang, W. Macrophage membrane-coated nanovesicles for dual-targeted drug delivery to inhibit tumor and induce macrophage polarization. Bioact. Mater. 2023, 23, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Sun, B.; Duan, S.; Han, J.; Barr, T.; Zhang, J.; Bissonnette, M.B.; Kortylewski, M.; He, C.; Chen, J.; et al. YTHDF2 or-chestrates tumor-associated macrophage reprogramming and controls antitumor immunity through CD8+ T cells. Nat. Immunol. 2023, 24, 255–266. [Google Scholar] [CrossRef] [PubMed]
- Wenes, M.; Shang, M.; Di Matteo, M.; Goveia, J.; Martín-Pérez, R.; Serneels, J.; Prenen, H.; Ghesquière, B.; Carmeliet, P.; Mazzone, M. Macrophage Metabolism Controls Tumor Blood Vessel Morphogenesis and Metastasis. Cell Metab. 2016, 24, 701–715. [Google Scholar] [CrossRef] [PubMed]
- Puttock, E.H.; Tyler, E.J.; Manni, M.; Maniati, E.; Butterworth, C.; Burger Ramos, M.; Peerani, E.; Hirani, P.; Gauthier, V.; Liu, Y.; et al. Extracellular matrix educates an immunoregulatory tumor macrophage phenotype found in ovarian cancer metastasis. Nat. Commun. 2023, 14, 2514. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Qiu, G.; Zhu, X.; Wang, Q.; Zhu, C.; Fang, C.; Liu, J.; Zhang, K.; Liu, Y. Macrophage-inherited exosome excise tumor immunosuppression to expedite immune-activated ferroptosis. J. Immunother. Cancer 2023, 11, e006516. [Google Scholar] [CrossRef] [PubMed]
- Zeng, W.; Yu, M.; Chen, T.; Liu, Y.; Yi, Y.; Huang, C.; Tang, J.; Li, H.; Ou, M.; Wang, T.; et al. Polypyrrole Nanoenzymes as Tumor Microenvironment Modulators to Reprogram Macrophage and Potentiate Immunotherapy. Adv. Sci. 2022, 9, e2201703. [Google Scholar] [CrossRef] [PubMed]
- Goossens, P.; Rodriguez-Vita, J.; Etzerodt, A.; Masse, M.; Rastoin, O.; Gouirand, V.; Ulas, T.; Papantonopoulou, O.; Van Eck, M.; Auphan-Anezin, N.; et al. Membrane Cholesterol Efflux Drives Tumor-Associated Macrophage Reprogramming and Tumor Progression. Cell Metab. 2019, 29, 1376–1389.e4. [Google Scholar] [CrossRef]
- Zhang, H.; Xue, C.; Shah, R.; Bermingham, K.; Hinkle, C.C.; Li, W.; Rodrigues, A.; Tabita-Martinez, J.; Millar, J.S.; Cuchel, M.; et al. Functional analysis and transcriptomic profiling of iPSC-derived macrophages and their application in modeling Mendelian disease. Circ. Res. 2015, 117, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Tian, L.; Dai, X.; Yu, H.; Wang, J.; Lei, A.; Zhu, M.; Xu, J.; Zhao, W.; Zhu, Y.; et al. Pluripotent stem cell-derived CAR-macrophage cells with antigen-dependent anti-cancer cell functions. J. Hematol. Oncol. 2020, 13, 153. [Google Scholar] [CrossRef] [PubMed]
- Kleiman, R.J.; Engle, S.J. Human inducible pluripotent stem cells: Realization of initial promise in drug discovery. Cell Stem Cell 2021, 28, 1507–1515. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method | Type | Efficiency | Preparation of Materials | Delivery Procedure | Remove of Exogenous Factors |
|---|---|---|---|---|---|
| Sendai virus [12,13,27] | Virus | +++ | Difficult | Easy | Easy |
| Adenovirus [11,28] | Virus | + | Difficult | Moderate | Moderate |
| Plasmid [15] | DNA | + | Easy | Difficult | Moderate |
| Episomal plasmid [13,29] | DNA | ++ | Easy | Easy | Moderate |
| PiggyBac transposon [30,31] | DNA | ++ | Difficult | Easy | Moderate |
| mRNA [17,19] | RNA | +++ | Difficult | Difficult | Easy |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, T.; Qian, C.; Song, M.; Tang, Y.; Zhou, Y.; Dong, G.; Shen, Q.; Chen, W.; Wang, A.; Shen, S.; et al. Application Prospect of Induced Pluripotent Stem Cells in Organoids and Cell Therapy. Int. J. Mol. Sci. 2024, 25, 2680. https://doi.org/10.3390/ijms25052680
Zhang T, Qian C, Song M, Tang Y, Zhou Y, Dong G, Shen Q, Chen W, Wang A, Shen S, et al. Application Prospect of Induced Pluripotent Stem Cells in Organoids and Cell Therapy. International Journal of Molecular Sciences. 2024; 25(5):2680. https://doi.org/10.3390/ijms25052680
Chicago/Turabian StyleZhang, Teng, Cheng Qian, Mengyao Song, Yu Tang, Yueke Zhou, Guanglu Dong, Qiuhong Shen, Wenxing Chen, Aiyun Wang, Sanbing Shen, and et al. 2024. "Application Prospect of Induced Pluripotent Stem Cells in Organoids and Cell Therapy" International Journal of Molecular Sciences 25, no. 5: 2680. https://doi.org/10.3390/ijms25052680
APA StyleZhang, T., Qian, C., Song, M., Tang, Y., Zhou, Y., Dong, G., Shen, Q., Chen, W., Wang, A., Shen, S., Zhao, Y., & Lu, Y. (2024). Application Prospect of Induced Pluripotent Stem Cells in Organoids and Cell Therapy. International Journal of Molecular Sciences, 25(5), 2680. https://doi.org/10.3390/ijms25052680