
Gluing GAP to RAS Mutants: A New Approach to an Old Problem in Cancer Drug Development

 , ,
, ,  and
and 
Abstract
1. Introduction
2. Results
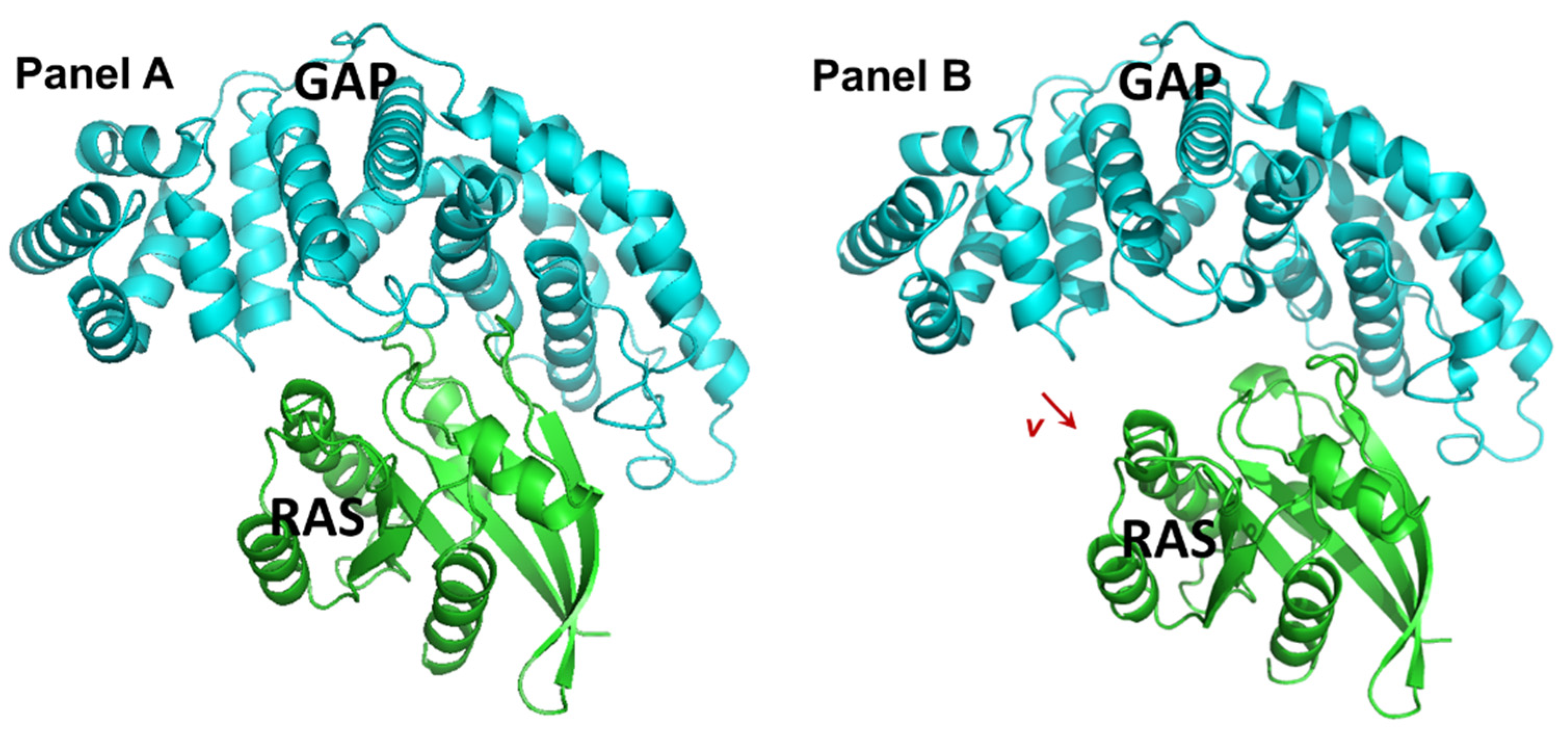
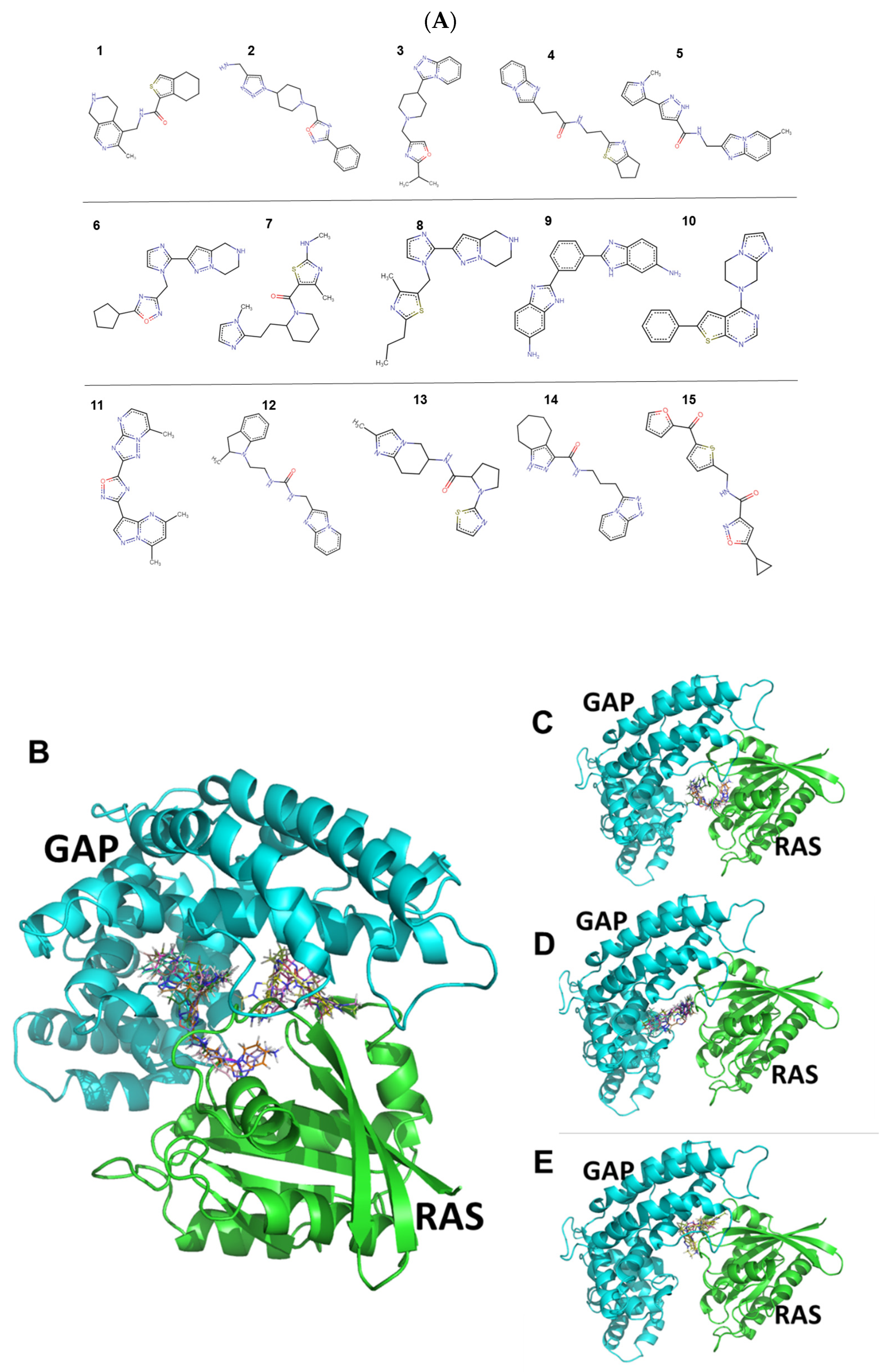
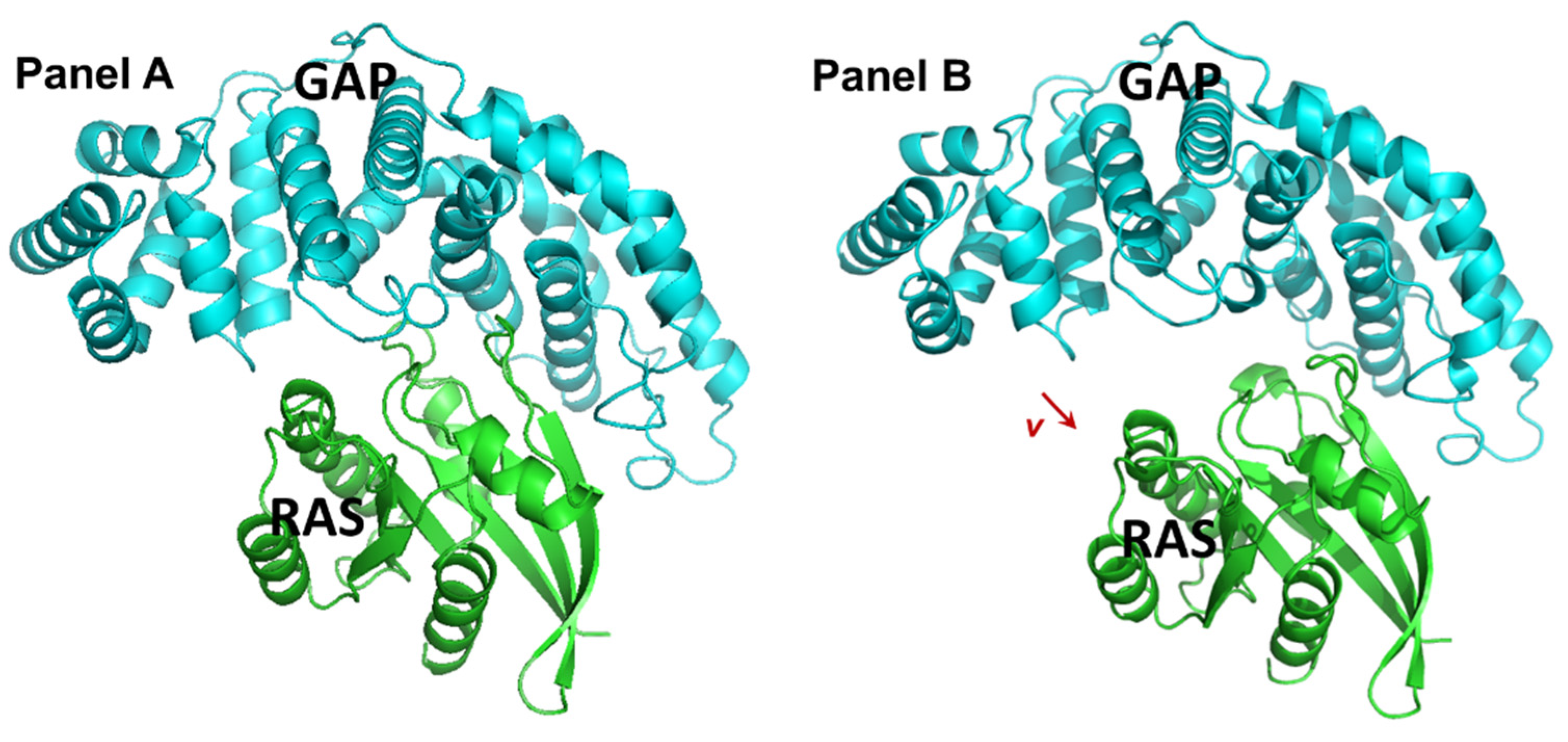
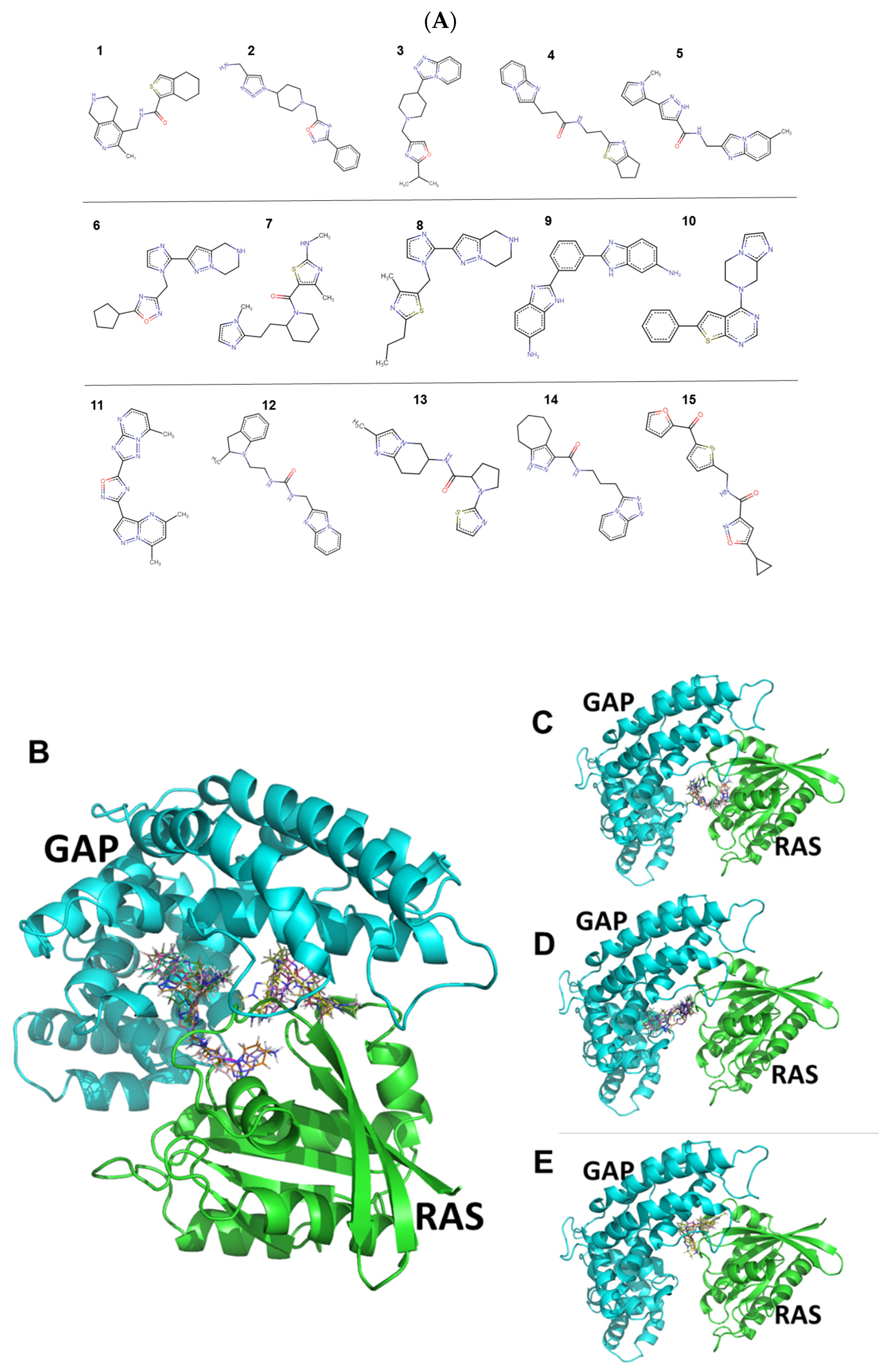
2.1. Identification and Characteristics of the Candidate Molecules from the Gluing Docking Strategy
2.2. In Vitro Antiproliferative Activity of the Compounds and Selectivity toward KRASG12D Mutation
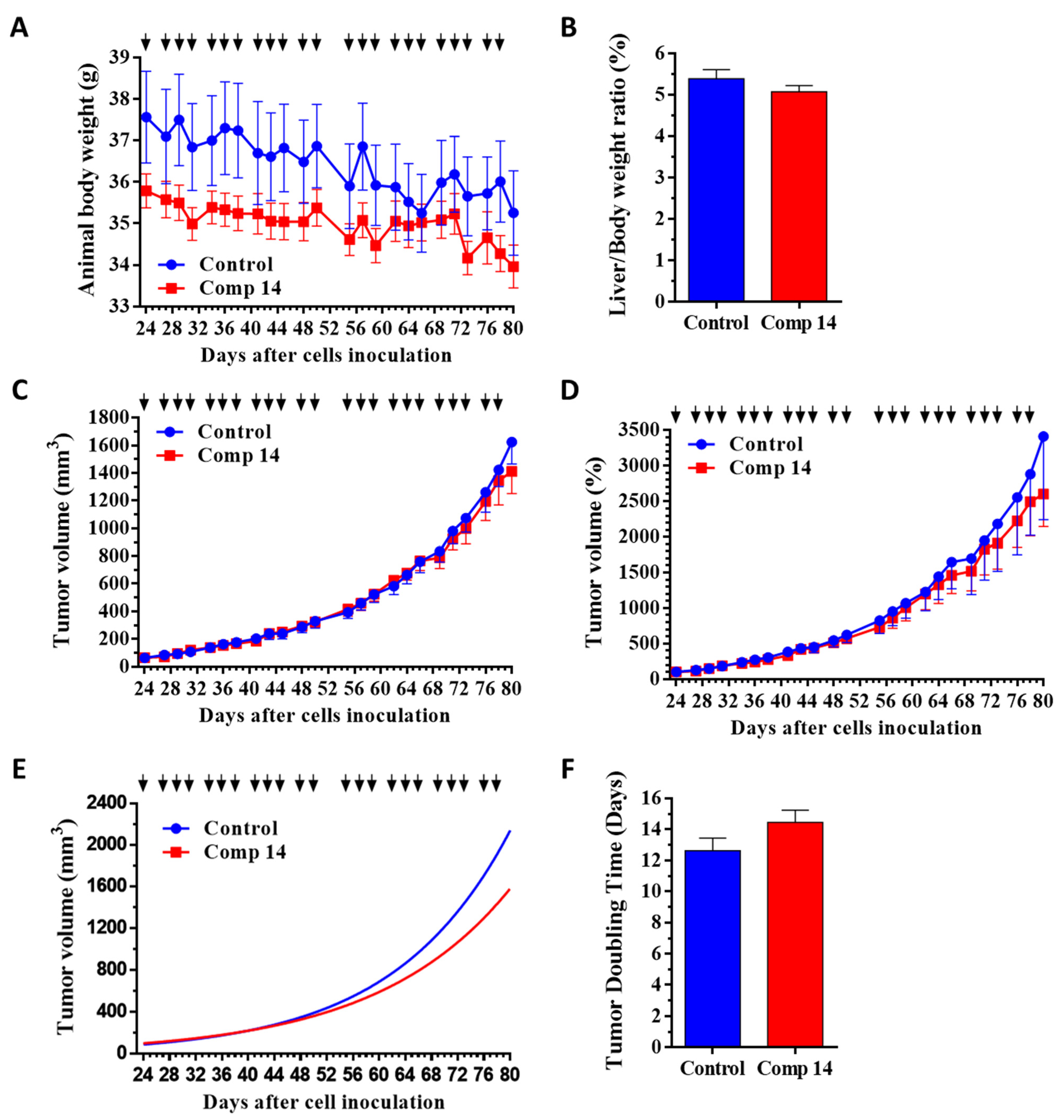
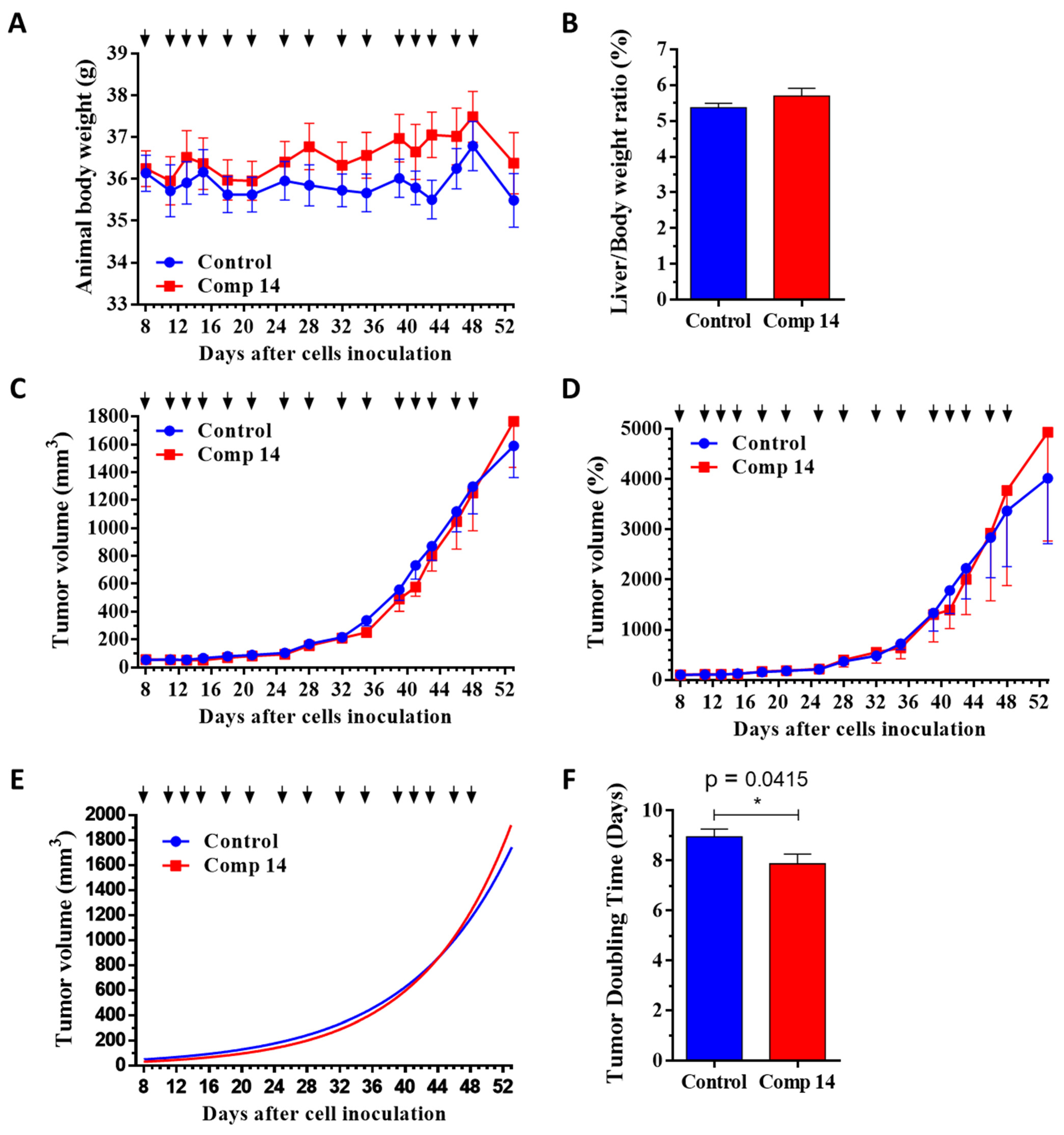
2.3. Compound 14 Shows Selective In Vivo Inhibition of Tumor Growth against KRASG12D Xenograft-Expressing Mice
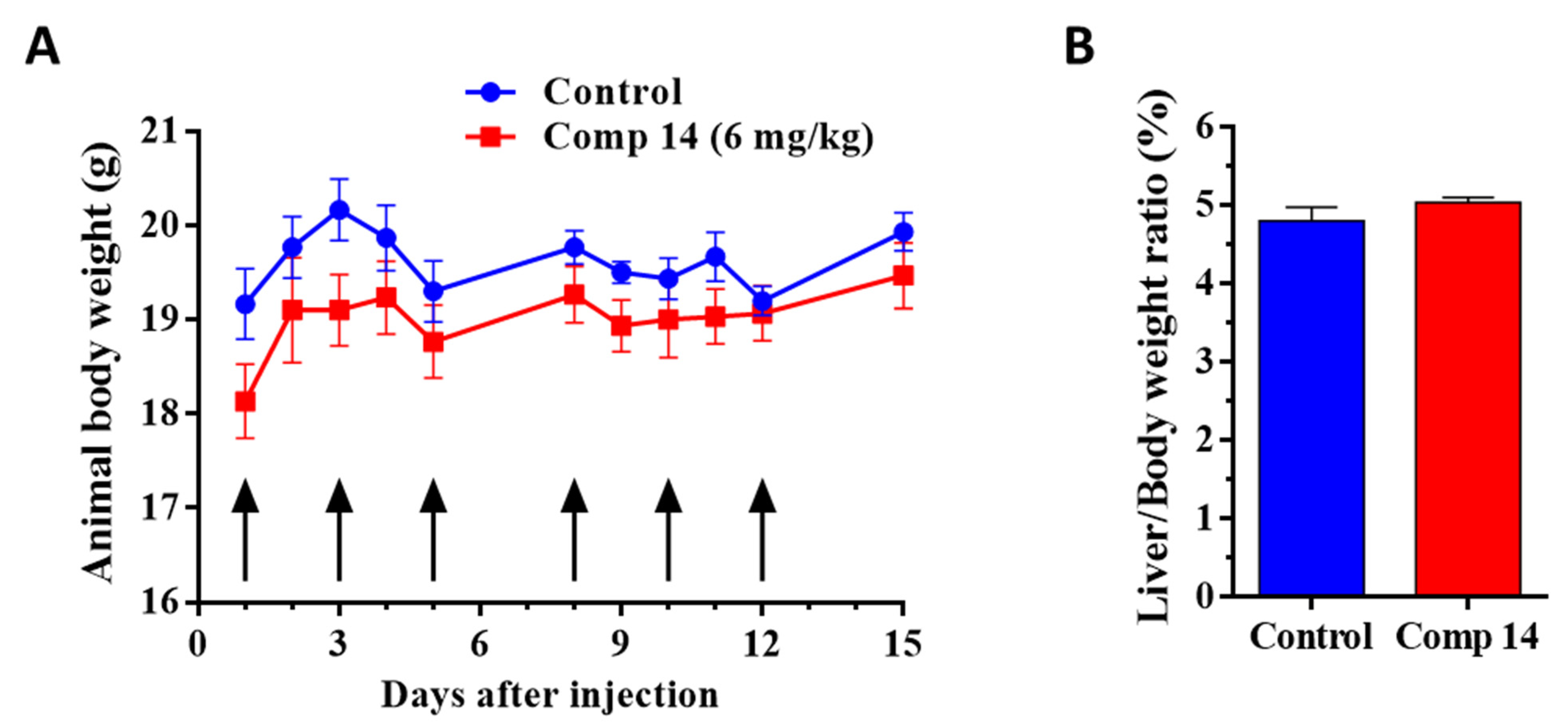
2.3.1. Chronic Toxicity Study of Compound 14 In Vivo
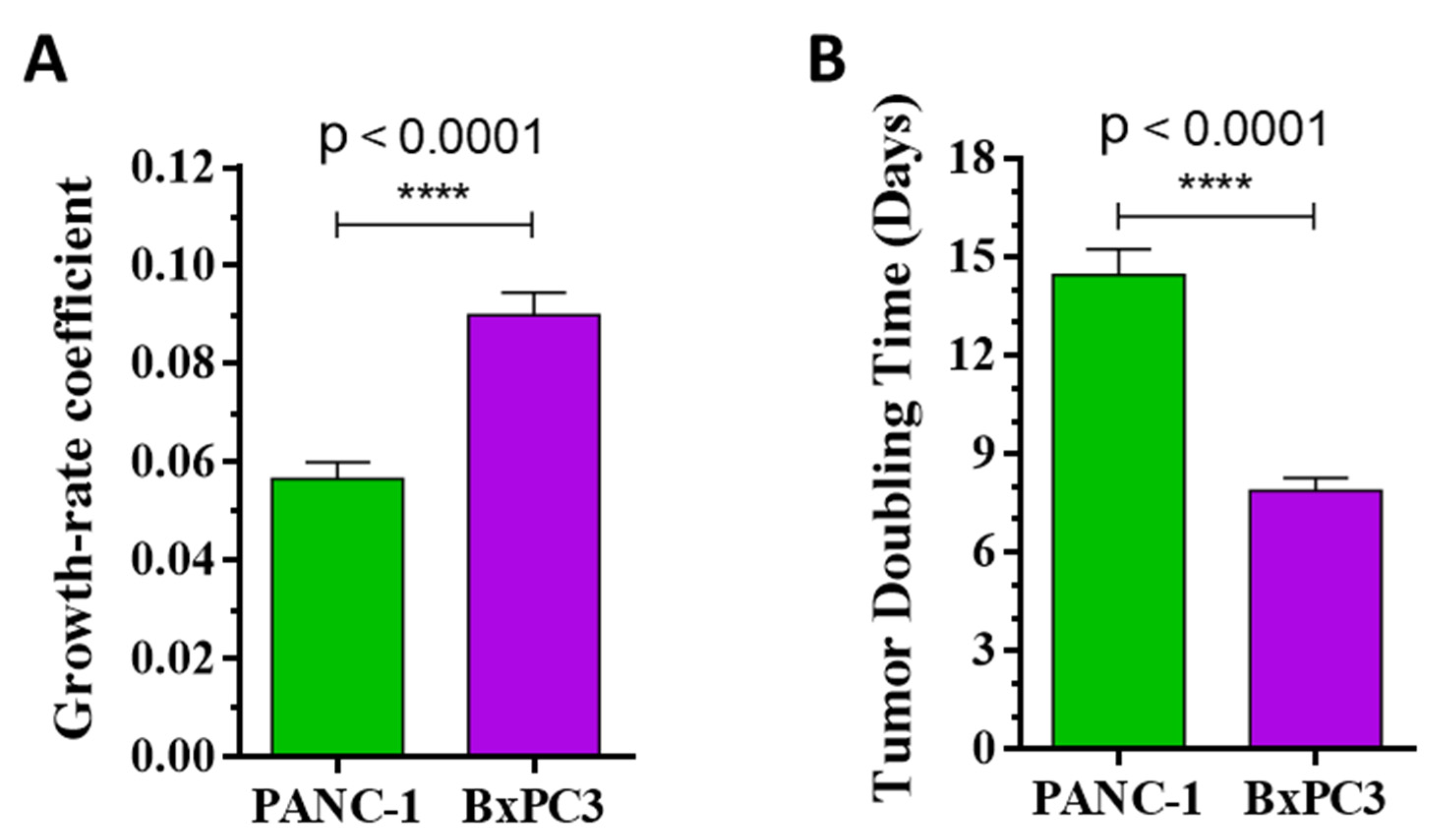
2.3.2. Effect of Compound 14 in Subcutaneous Human Pancreatic PANC-1 (KRASG12D) and BxPC3 (KRASwt) Tumor Models In Vivo
2.4. In Vitro Binding of Compound 14 to the GAP and KRASG12D Proteins
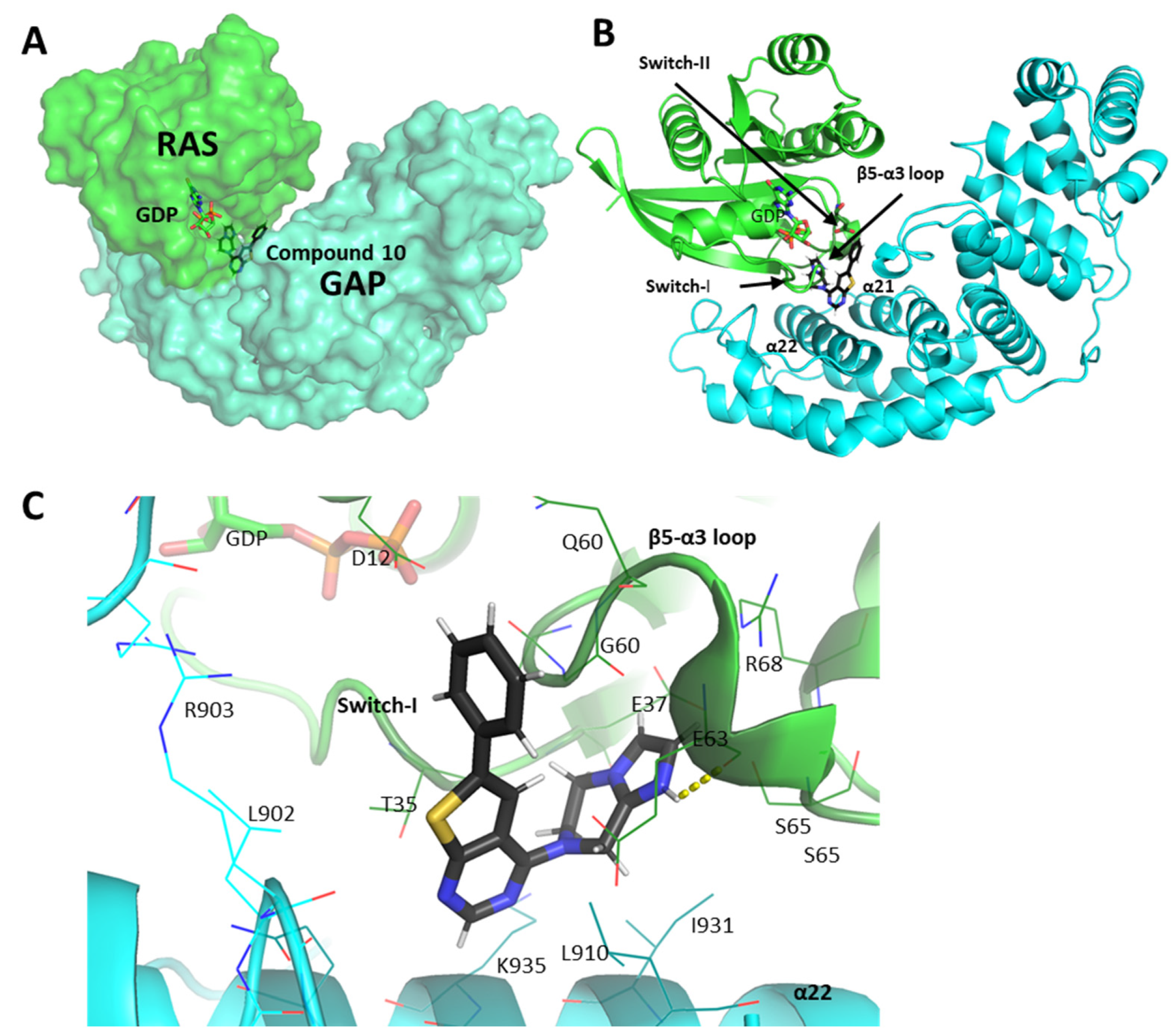
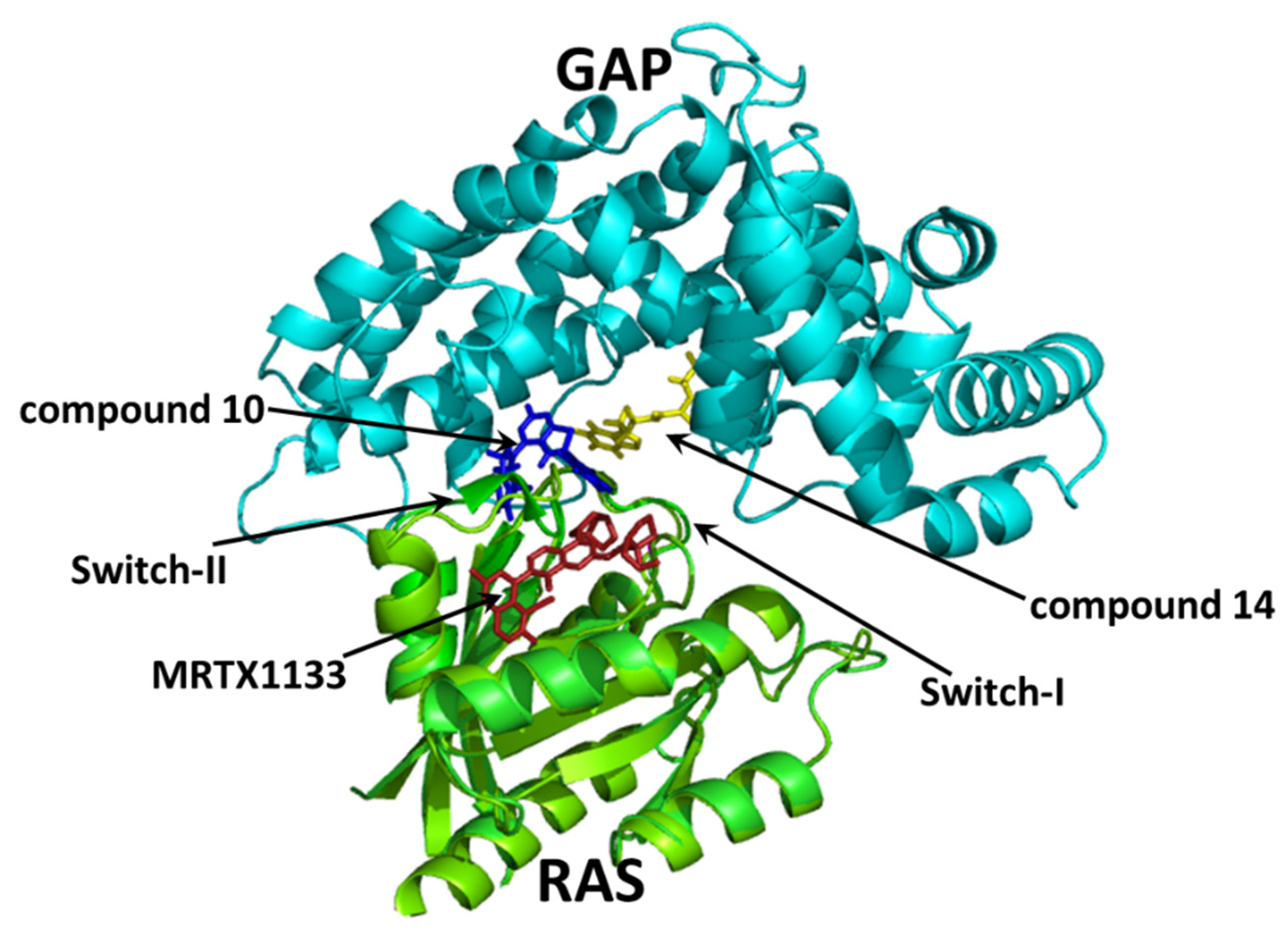
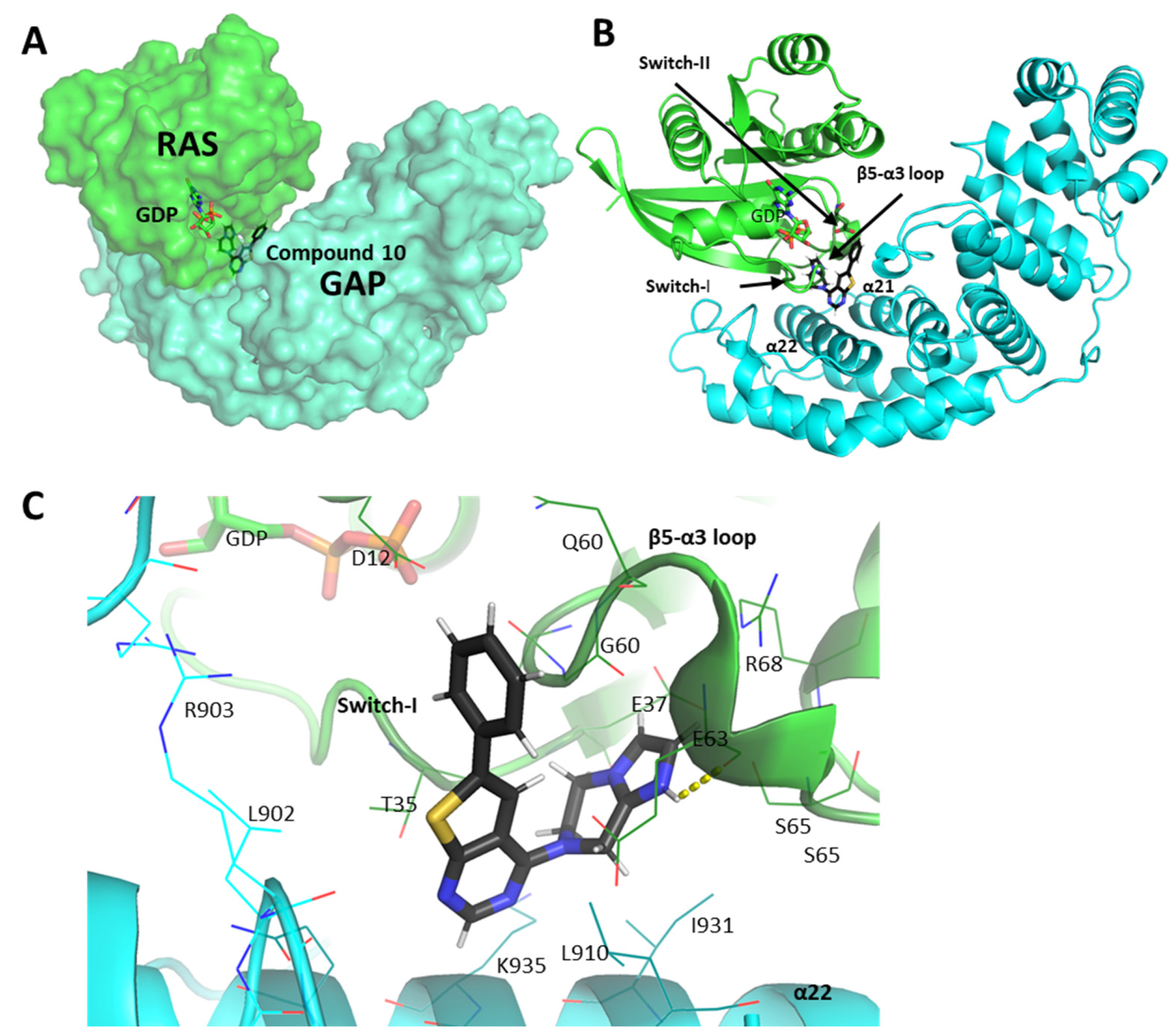
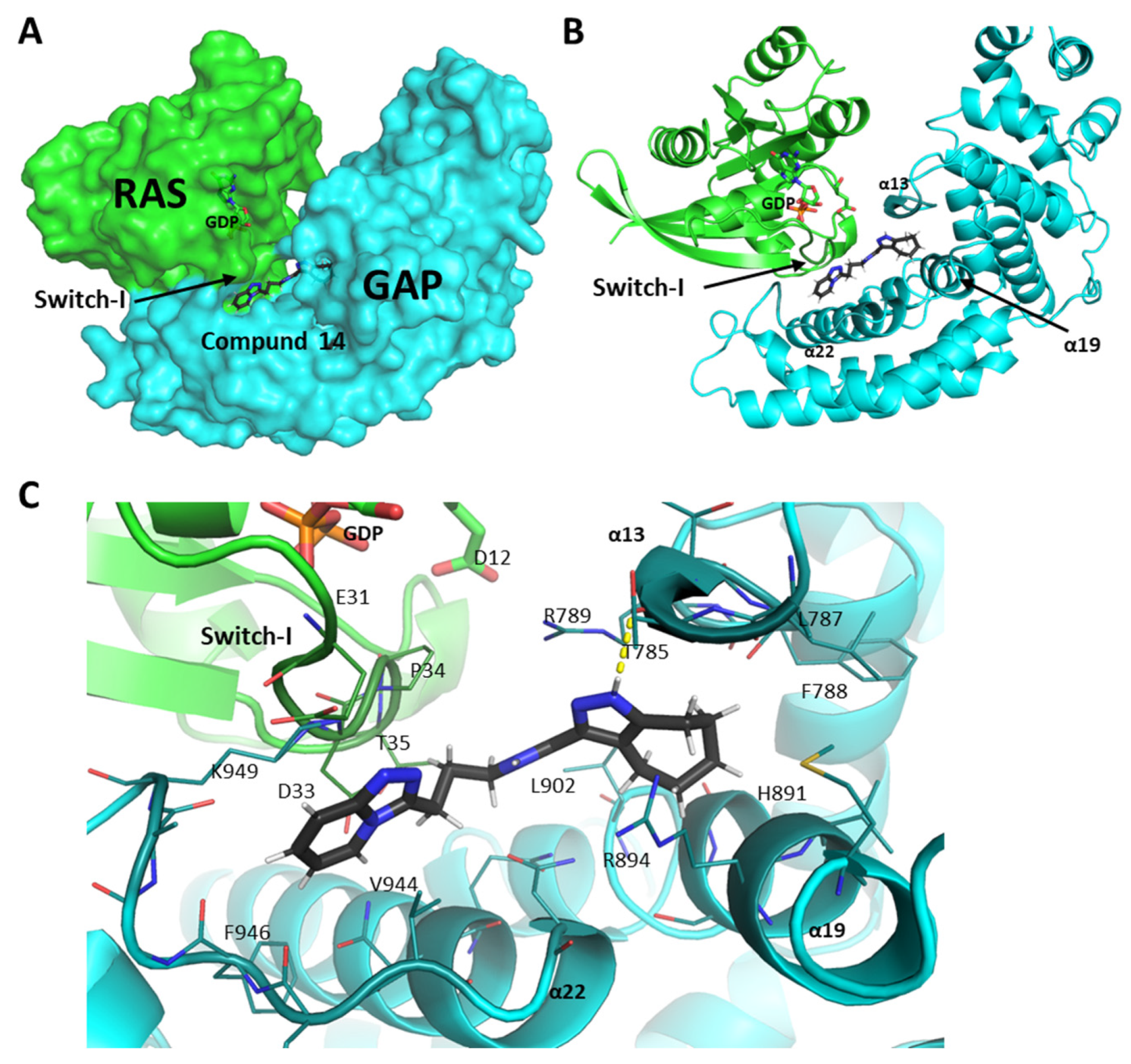
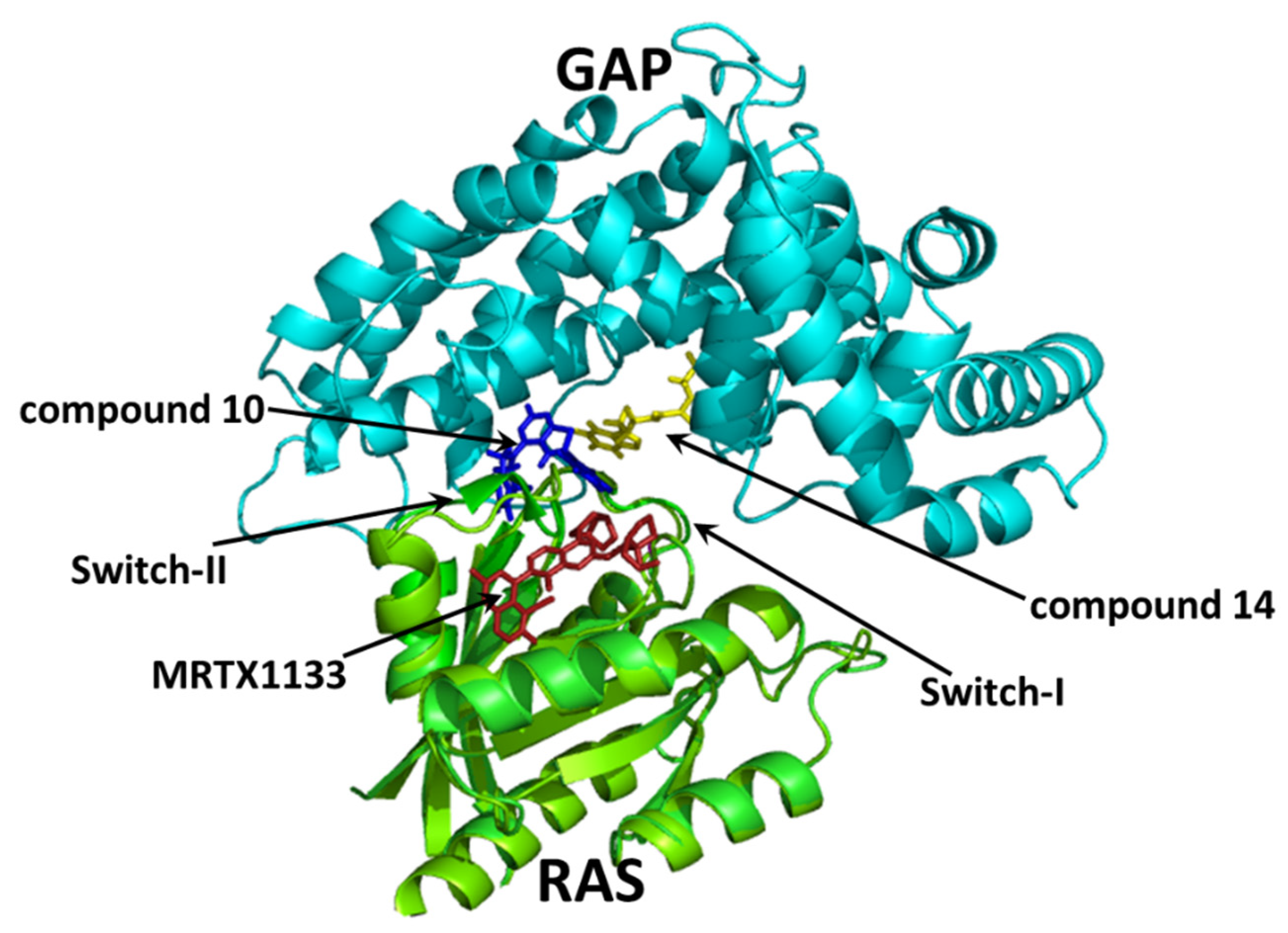
2.5. Molecular Interactions of Compounds 10 and 14 with the Complex of GAP and G12D Mutant KRAS
3. Discussion
4. Methods and Materials
4.1. Identification of Small Molecules Gluing GAP and KRasG12D
4.2. Cell Lines and Culture Conditions
4.3. In Vitro Antiproliferative Activity of Compounds and Calculation of Selectivity for KRASG12D Mutation
4.4. Experimental Animals
4.5. Chronic Toxicity Study of Compound 14
4.6. Mouse Models of Subcutaneous Human Pancreatic Cancers PANC-1 (KRASG12D) and BxPC3 (KRASwt), Doses and Schedule of Compound 14 Treatments, and Measurements
4.7. Protein Expression and Purification
4.8. Differential Scanning Fluorimetry
4.9. Statistical ANALYSIS
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rozenblatt-Rosen, O.; Regev, A.; Oberdoerffer, P.; Nawy, T.; Hupalowska, A.; Rood, J.E.; Ashenberg, O.; Cerami, E.; Coffey, R.J.; Demir, E.; et al. The Human Tumor Atlas Network: Charting Tumor Transitions across Space and Time at Single-Cell Resolution. Cell 2020, 181, 236–249. [Google Scholar] [CrossRef]
- Papke, B.; Der, C.J. Drugging RAS: Know the enemy. Science 2017, 355, 1158–1163. [Google Scholar] [CrossRef]
- Prior, I.A.; Hood, F.E.; Hartley, J.L. The Frequency of Ras Mutations in Cancer. Cancer Res. 2020, 80, 2969–2974. [Google Scholar] [CrossRef] [PubMed]
- Stephen, A.G.; Esposito, D.; Bagni, R.K.; McCormick, F. Dragging ras back in the ring. Cancer Cell 2014, 25, 272–281. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.S.; Lee, A.; Nagasaka, M. CodeBreak 200: Sotorasib Has Not Broken the KRAS(G12C) Enigma Code. Lung Cancer 2023, 14, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Simanshu, D.K.; Nissley, D.V.; McCormick, F. RAS Proteins and Their Regulators in Human Disease. Cell 2017, 170, 17–33. [Google Scholar] [CrossRef] [PubMed]
- Timar, J.; Kashofer, K. Molecular epidemiology and diagnostics of KRAS mutations in human cancer. Cancer Metastasis Rev. 2020, 39, 1029–1038. [Google Scholar] [CrossRef] [PubMed]
- Pylayeva-Gupta, Y.; Grabocka, E.; Bar-Sagi, D. RAS oncogenes: Weaving a tumorigenic web. Nat. Rev. Cancer 2011, 11, 761–774. [Google Scholar] [CrossRef] [PubMed]
- Downward, J. RAS’s cloak of invincibility slips at last? Cancer Cell 2014, 25, 5–6. [Google Scholar] [CrossRef] [PubMed]
- Lito, P.; Solomon, M.; Li, L.S.; Hansen, R.; Rosen, N. Allele-specific inhibitors inactivate mutant KRAS G12C by a trapping mechanism. Science 2016, 351, 604–608. [Google Scholar] [CrossRef]
- Janes, M.R.; Zhang, J.; Li, L.S.; Hansen, R.; Peters, U.; Guo, X.; Chen, Y.; Babbar, A.; Firdaus, S.J.; Darjania, L.; et al. Targeting KRAS Mutant Cancers with a Covalent G12C-Specific Inhibitor. Cell 2018, 172, 578–589. [Google Scholar] [CrossRef]
- Patricelli, M.P.; Janes, M.R.; Li, L.S.; Hansen, R.; Peters, U.; Kessler, L.V.; Chen, Y.; Kucharski, J.M.; Feng, J.; Ely, T.; et al. Selective Inhibition of Oncogenic KRAS Output with Small Molecules Targeting the Inactive State. Cancer Discov. 2016, 6, 316–329. [Google Scholar] [CrossRef] [PubMed]
- Nyiri, K.; Koppany, G.; Vertessy, B.G. Structure-based inhibitor design of mutant RAS proteins-a paradigm shift. Cancer Metastasis Rev. 2020, 39, 1091–1105. [Google Scholar] [CrossRef] [PubMed]
- Iska, S.; Alley, E.W. Sotorasib as First-Line Treatment for Advanced KRAS G12C-Mutated Non-Small Cell Lung Carcinoma: A Case Report. Case Rep. Oncol. 2023, 16, 177–181. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Allen, S.; Blake, J.F.; Bowcut, V.; Briere, D.M.; Calinisan, A.; Dahlke, J.R.; Fell, J.B.; Fischer, J.P.; Gunn, R.J.; et al. Identification of MRTX1133, a Noncovalent, Potent, and Selective KRAS(G12D) Inhibitor. J. Med. Chem. 2022, 65, 3123–3133. [Google Scholar] [CrossRef] [PubMed]
- Mullard, A. The KRAS crowd targets its next cancer mutations. Nat. Rev. Drug Discov. 2023, 22, 167–171. [Google Scholar] [CrossRef] [PubMed]
- Gulay, K.C.M.; Zhang, X.; Pantazopoulou, V.; Patel, J.; Esparza, E.; Pran Babu, D.S.; Ogawa, S.; Weitz, J.; Ng, I.; Mose, E.S.; et al. Dual inhibition of KRASG12D and pan-ERBB is synergistic in pancreatic ductal adenocarcinoma. Cancer Res. 2023, 83, 3001–3012. [Google Scholar] [CrossRef]
- Gradiz, R.; Silva, H.C.; Carvalho, L.; Botelho, M.F.; Mota-Pinto, A. MIA PaCa-2 and PANC-1—Pancreas ductal adenocarcinoma cell lines with neuroendocrine differentiation and somatostatin receptors. Sci. Rep. 2016, 6, 21648. [Google Scholar] [CrossRef]
- Jiang, W.; Li, H.; Liu, X.; Zhang, J.; Zhang, W.; Li, T.; Liu, L.; Yu, X. Precise and efficient silencing of mutant Kras(G12D) by CRISPR-CasRx controls pancreatic cancer progression. Theranostics 2020, 10, 11507–11519. [Google Scholar] [CrossRef]
- Hallin, J.; Bowcut, V.; Calinisan, A.; Briere, D.M.; Hargis, L.; Engstrom, L.D.; Laguer, J.; Medwid, J.; Vanderpool, D.; Lifset, E.; et al. Anti-tumor efficacy of a potent and selective non-covalent KRAS(G12D) inhibitor. Nat. Med. 2022, 28, 2171–2182. [Google Scholar] [CrossRef]
- Kemp, S.B.; Cheng, N.; Markosyan, N.; Sor, R.; Kim, I.K.; Hallin, J.; Shoush, J.; Quinones, L.; Brown, N.V.; Bassett, J.B.; et al. Efficacy of a Small-Molecule Inhibitor of KrasG12D in Immunocompetent Models of Pancreatic Cancer. Cancer Discov. 2023, 13, 298–311. [Google Scholar] [CrossRef] [PubMed]
- Gao, K.; Oerlemans, R.; Groves, M.R. Theory and applications of differential scanning fluorimetry in early-stage drug discovery. Biophys. Rev. 2020, 12, 85–104. [Google Scholar] [CrossRef] [PubMed]
- Hobbs, G.A.; Der, C.J.; Rossman, K.L. RAS isoforms and mutations in cancer at a glance. J. Cell Sci. 2016, 129, 1287–1292. [Google Scholar] [CrossRef] [PubMed]
- Lietman, C.D.; Johnson, M.L.; McCormick, F.; Lindsay, C.R. More to the RAS Story: KRAS(G12C) Inhibition, Resistance Mechanisms, and Moving Beyond KRAS(G12C). Am. Soc. Clin. Oncol. Educ. Book 2022, 42, 205–217. [Google Scholar] [CrossRef]
- Scheffzek, K.; Ahmadian, M.R.; Kabsch, W.; Wiesmuller, L.; Lautwein, A.; Schmitz, F.; Wittinghofer, A. The Ras-RasGAP complex: Structural basis for GTPase activation and its loss in oncogenic Ras mutants. Science 1997, 277, 333–338. [Google Scholar] [CrossRef] [PubMed]
- Scheich, C.; Szabadka, Z.; Vértessy, B.; Pütter, V.; Grolmusz, V.; Schade, M. Discovery of novel MDR-Mycobacterium tuberculosis inhibitor by new FRIGATE computational screen. PLoS ONE 2011, 6, e28428. [Google Scholar] [CrossRef] [PubMed]
- Irwin, J.J.; Tang, K.G.; Young, J.; Dandarchuluun, C.; Wong, B.R.; Khurelbaatar, M.; Moroz, Y.S.; Mayfield, J.; Sayle, R.A. ZINC20-A Free Ultralarge-Scale Chemical Database for Ligand Discovery. J. Chem. Inf. Model. 2020, 60, 6065–6073. [Google Scholar] [CrossRef]
- Orgován, Z.; Péczka, N.; Petri, L.; Ábrányi-Balogh, P.; Ranđelović, I.; Tóth, S.; Szakács, G.; Nyíri, K.; Vértessy, B.; Pálfy, G.; et al. Covalent fragment mapping of KRas(G12C) revealed novel chemotypes with in vivo potency. Eur. J. Med. Chem. 2023, 250, 115212. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound Name | IC50 (µM) | Selectivity toward KRASG12D Mutation | IC25 (µM) | Selectivity toward KRASG12D Mutation | ||
|---|---|---|---|---|---|---|
| PANC-1 (KRASG12D) | BxPC3 (KRASwt) | PANC-1 (KRASG12D) | BxPC3 (KRASwt) | |||
| 1 | 53.77 | 48.45 | 0.9 | 17.36 | 30.99 | 1.8 |
| 2 | 69.13 | 35.05 | 0.5 | 16.09 | 19.35 | 1.2 |
| 3 | 95.09 | 42.30 | 0.4 | 31.14 | 24.84 | 0.8 |
| 4 | 93.69 | 39.91 | 0.4 | 28.40 | 22.67 | 0.8 |
| 5 | 118.10 | 30.76 | 0.3 | 32.84 | 14.85 | 0.5 |
| 6 | 93.52 | 45.08 | 0.5 | 41.55 | 27.30 | 0.7 |
| 7 | 138.60 | 40.55 | 0.3 | 52.20 | 24.16 | 0.5 |
| 8 | 90.71 | 43.73 | 0.5 | 26.84 | 27.07 | 1.0 |
| 9 | 74.12 | 23.23 | 0.3 | 31.14 | 13.44 | 0.4 |
| 10 | 2.2 ± 0.4 * | 3.8 ± 0.1 | 1.7 | 0.05 ± 0.01 ## | 0.5 ± 0.05 | 10.6 |
| 11 | 117.30 | 43.18 | 0.4 | 50.28 | 25.30 | 0.5 |
| 12 | 93.11 | 42.97 | 0.5 | 31.41 | 25.12 | 0.8 |
| 13 | 103.20 | 47.54 | 0.5 | 41.12 | 29.96 | 0.7 |
| 14 | 5.5 ± 1.3 * | 8.2 ± 0.6 | 1.5 | 1.2 ± 0.4 ### | 4.5 ± 0.4 | 3.8 |
| 15 | 78.40 | 55.75 | 0.7 | 20.72 | 35.15 | 1.7 |
| MRTX-1133 | 18.3 ± 2.4 ns | 20.6 ± 4.0 | 1.1 | 5.7 ± 2.4 # | 12.3 ± 2.7 | 2.2 |
| Noncancerous Cell Line | Compound 10 | Compound 14 | ||
|---|---|---|---|---|
| IC50 (µM) | Selectivity toward KRASG12D Mutation | IC50 (µM) | Selectivity toward KRASG12D Mutation | |
| HUVEC-TERT | 0.3 ± 0.04 + | 0.1 | 8.6 ± 0.7 * | 1.6 |
| CCD-986Sk | 6.7 ± 1.1 * | 3.0 | 25.6 ± 3.0 ** | 4.7 |
| Parameter | Control | Comp 14 | ||
|---|---|---|---|---|
| PANC-1 | BxPC3 | PANC-1 | BxPC3 | |
| Animal body weight | −6.1 | −1.8 | −5.1 | +0.4 |
| Liver/Body weight ratio | −5.8 | +5.8 | ||
| Tumor volume in mm3 | −13.1 | +11.1 | ||
| Tumor volume in % | −23.8 | +22.8 | ||
| Tumor volume by nonlinear fitting | −26.3 (**** to BxPC3) | +9.9 | ||
| Tumor doubling time | +14.5 (**** to BxPC3) | −12.1 (* to control) | ||
| Melting Point, °C | |
|---|---|
| GAP | 42.5 ± 0.20 |
| GAP + compound 14 | 40.5 ± 0.15 |
| KRASG12D | 55.5 ± 0.30 |
| KRASG12D + compound 14 | 53.0 ± 0.35 |
| GAP + KRASG12D | 43.5 ± 0.10 |
| GAP + KRASG12D + compound 14 | 40.5 ± 0.25 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ranđelović, I.; Nyíri, K.; Koppány, G.; Baranyi, M.; Tóvári, J.; Kigyós, A.; Tímár, J.; Vértessy, B.G.; Grolmusz, V. Gluing GAP to RAS Mutants: A New Approach to an Old Problem in Cancer Drug Development. Int. J. Mol. Sci. 2024, 25, 2572. https://doi.org/10.3390/ijms25052572
Ranđelović I, Nyíri K, Koppány G, Baranyi M, Tóvári J, Kigyós A, Tímár J, Vértessy BG, Grolmusz V. Gluing GAP to RAS Mutants: A New Approach to an Old Problem in Cancer Drug Development. International Journal of Molecular Sciences. 2024; 25(5):2572. https://doi.org/10.3390/ijms25052572
Chicago/Turabian StyleRanđelović, Ivan, Kinga Nyíri, Gergely Koppány, Marcell Baranyi, József Tóvári, Attila Kigyós, József Tímár, Beáta G. Vértessy, and Vince Grolmusz. 2024. "Gluing GAP to RAS Mutants: A New Approach to an Old Problem in Cancer Drug Development" International Journal of Molecular Sciences 25, no. 5: 2572. https://doi.org/10.3390/ijms25052572
APA StyleRanđelović, I., Nyíri, K., Koppány, G., Baranyi, M., Tóvári, J., Kigyós, A., Tímár, J., Vértessy, B. G., & Grolmusz, V. (2024). Gluing GAP to RAS Mutants: A New Approach to an Old Problem in Cancer Drug Development. International Journal of Molecular Sciences, 25(5), 2572. https://doi.org/10.3390/ijms25052572