
Glycyrol Alleviates Acute Kidney Injury by Inhibiting Ferroptsis


Abstract
1. Introduction
2. Results
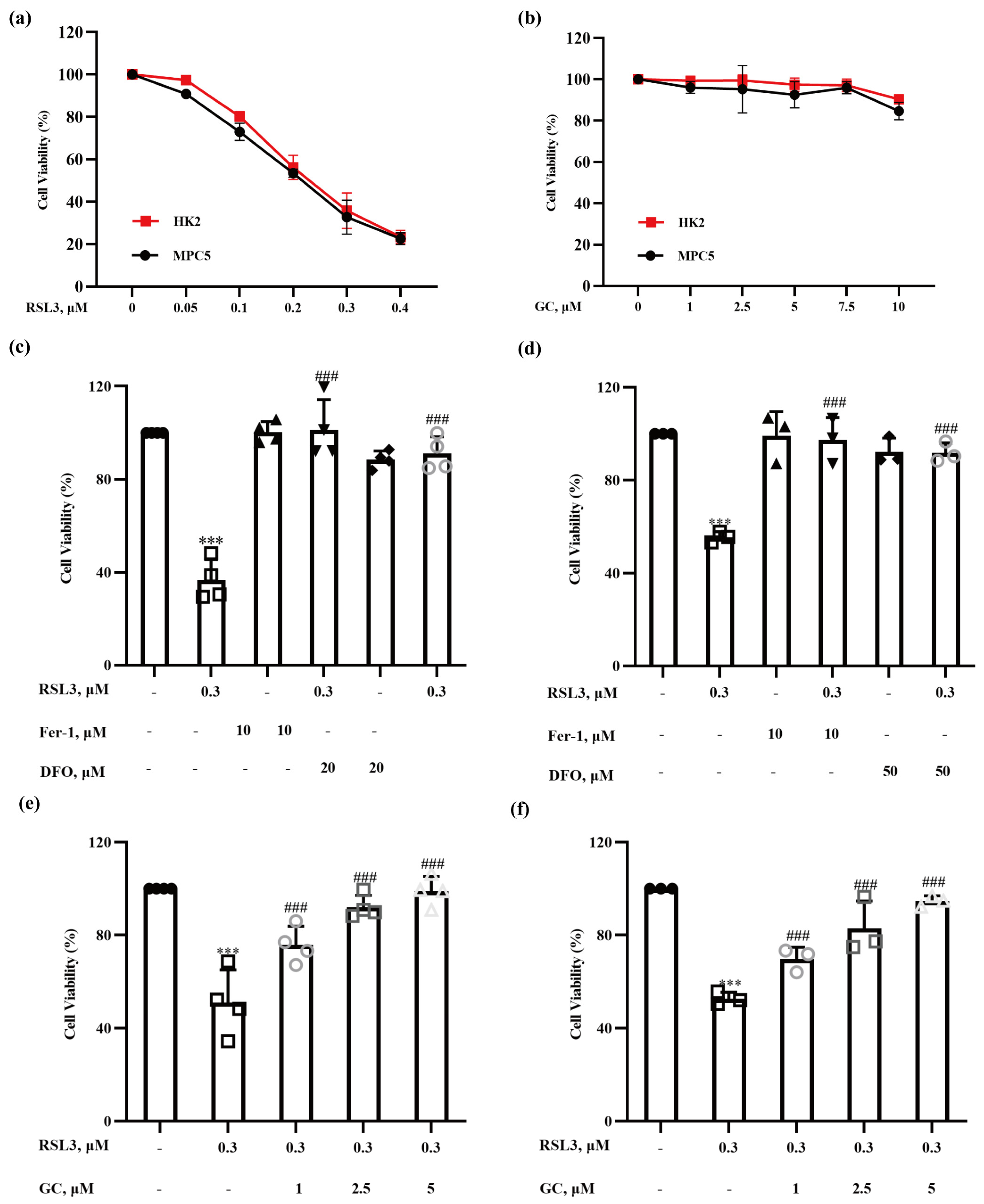
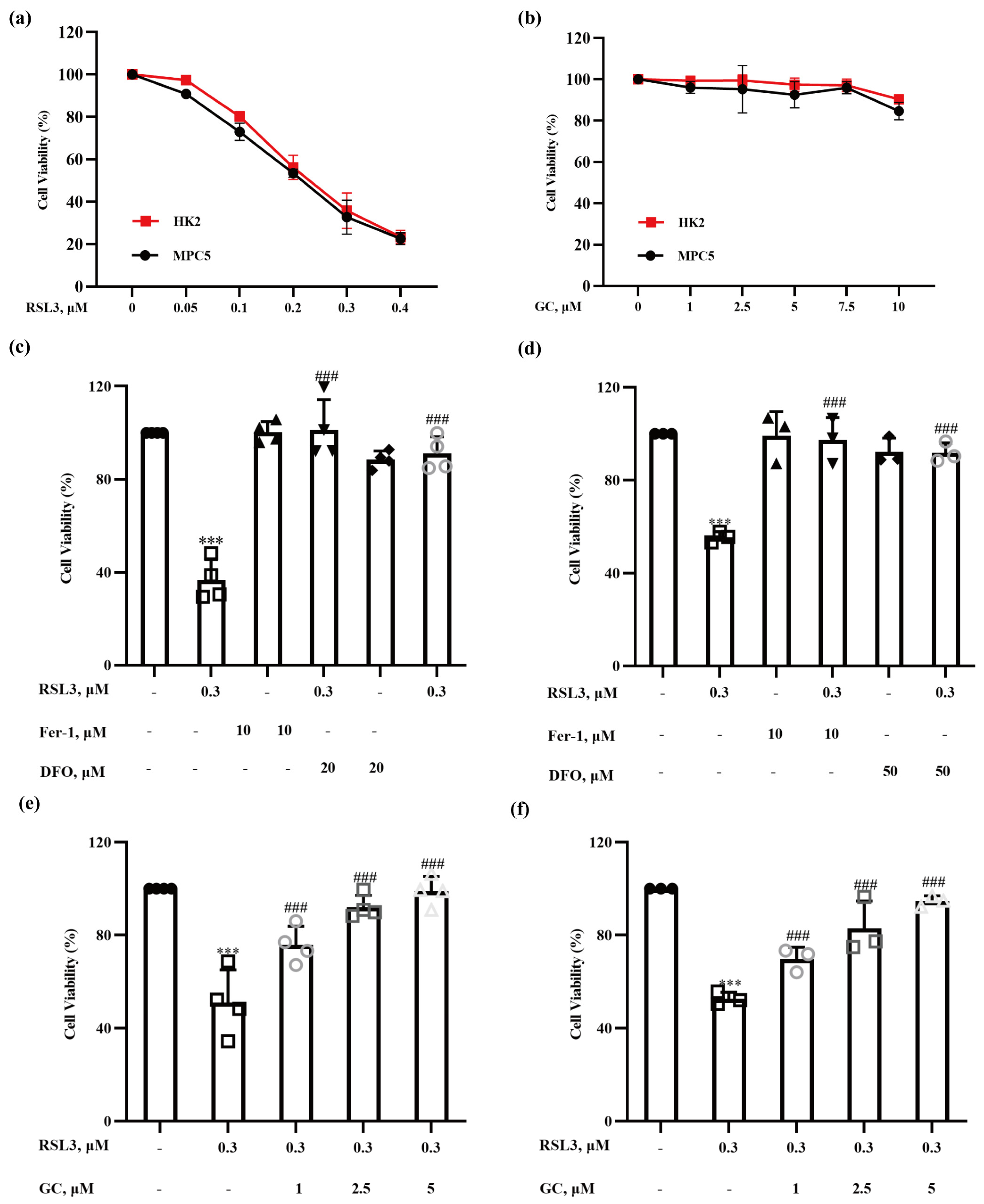
2.1. Glycyrol (GC) Exerts a Protective Effect on Nephrocyte Ferroptosis In Vitro
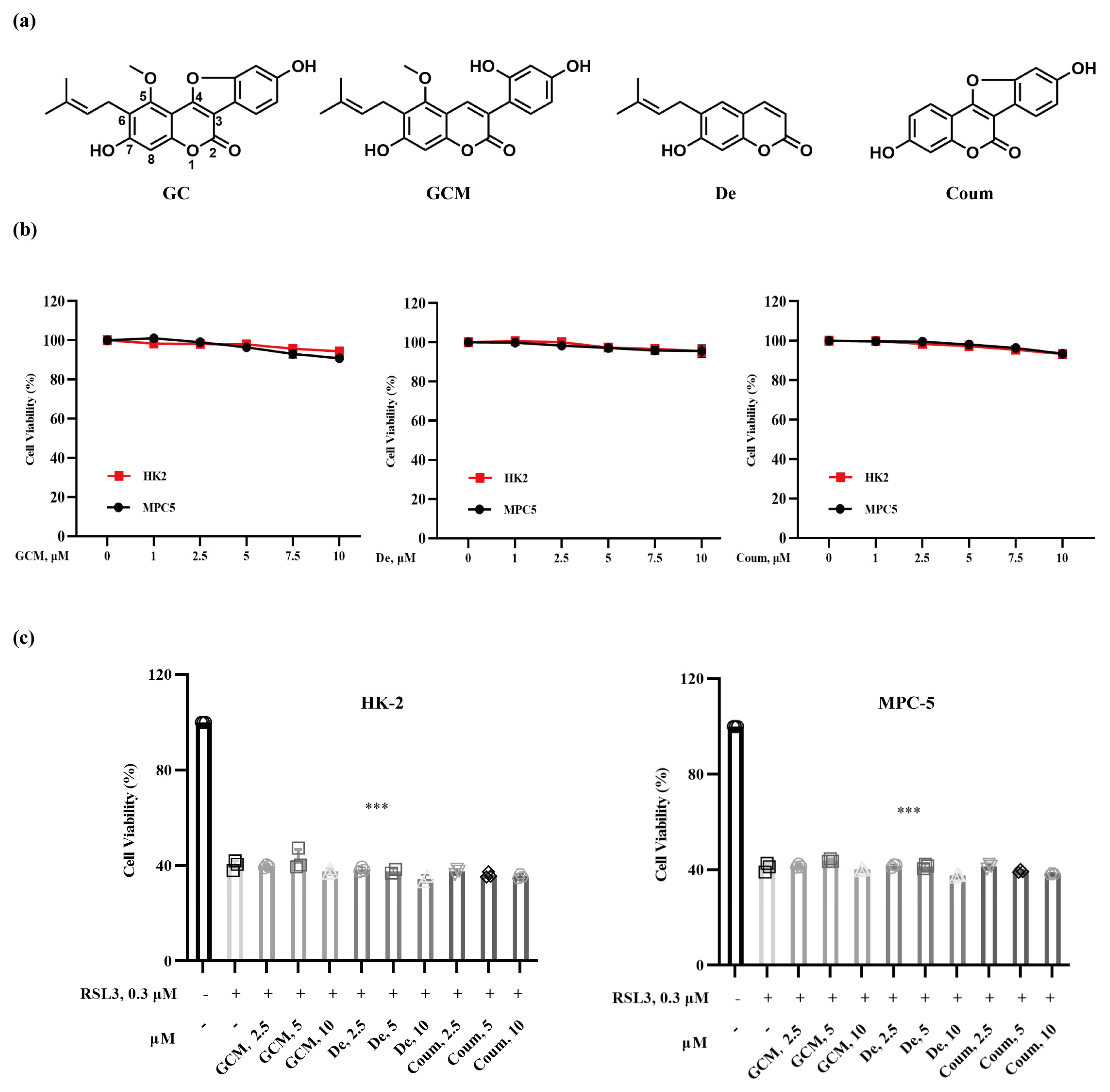
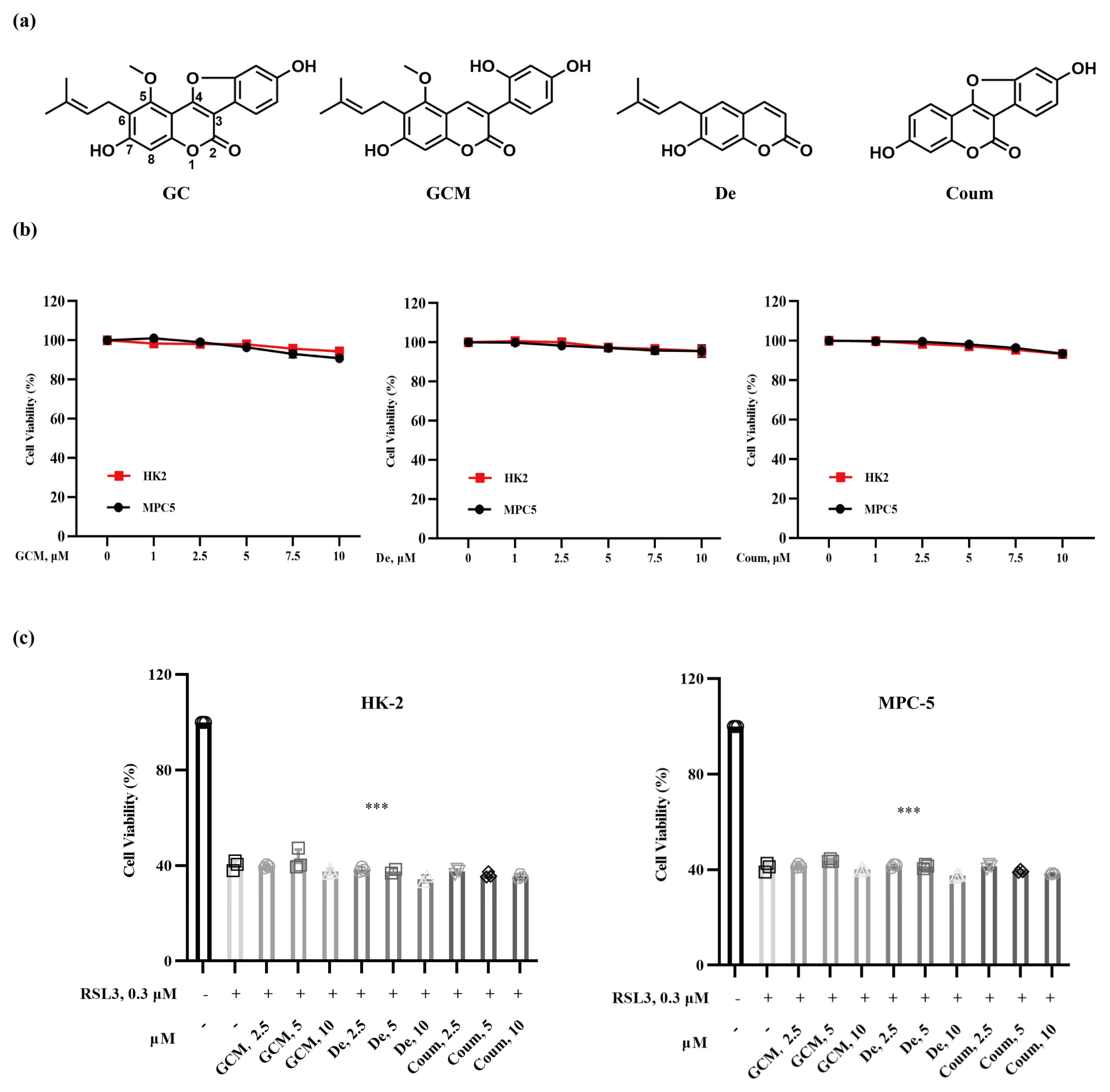
2.2. The Protective Effect of GC on Nephrocyte Ferroptosis Is Associated with Its Specific Structural Feature
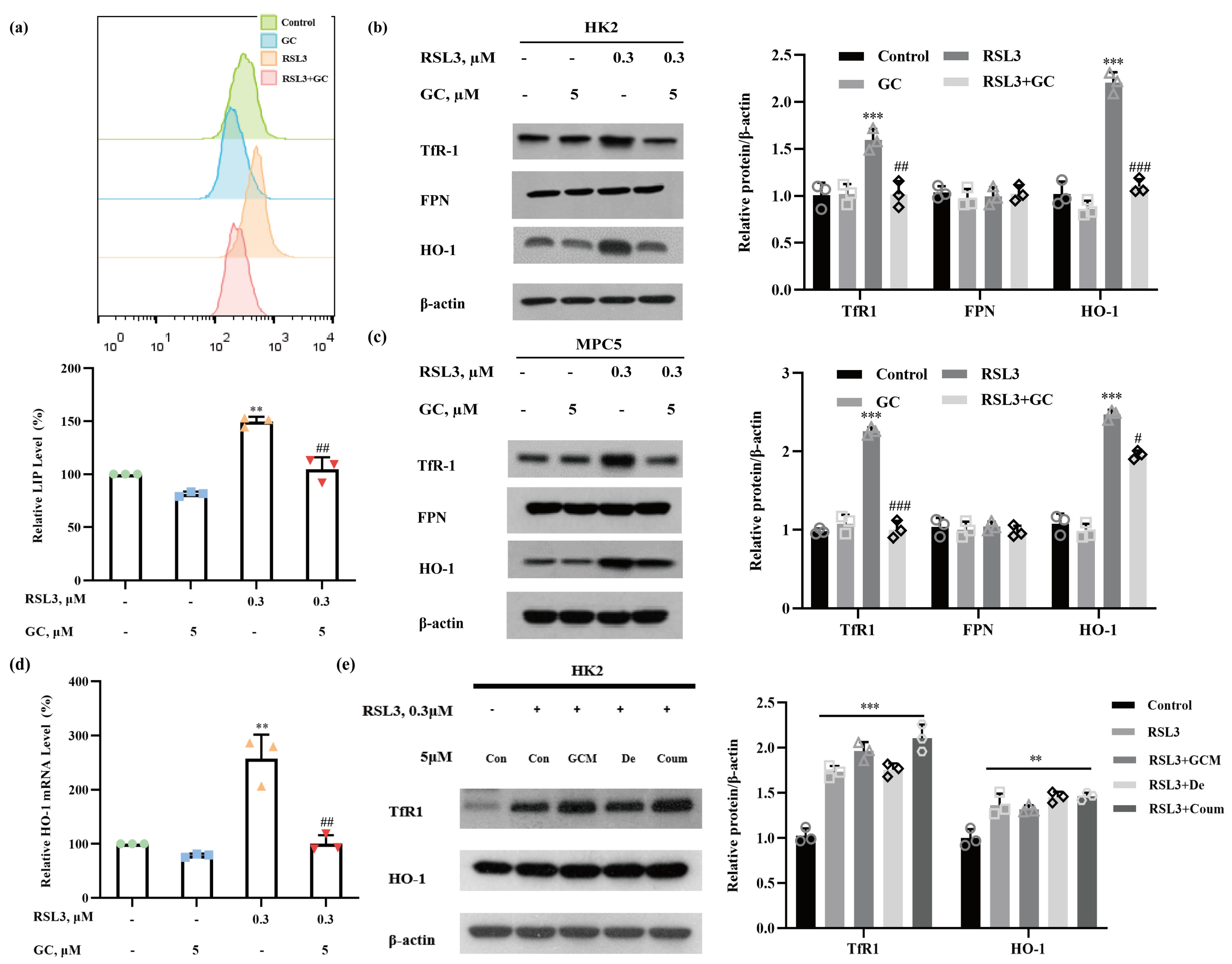
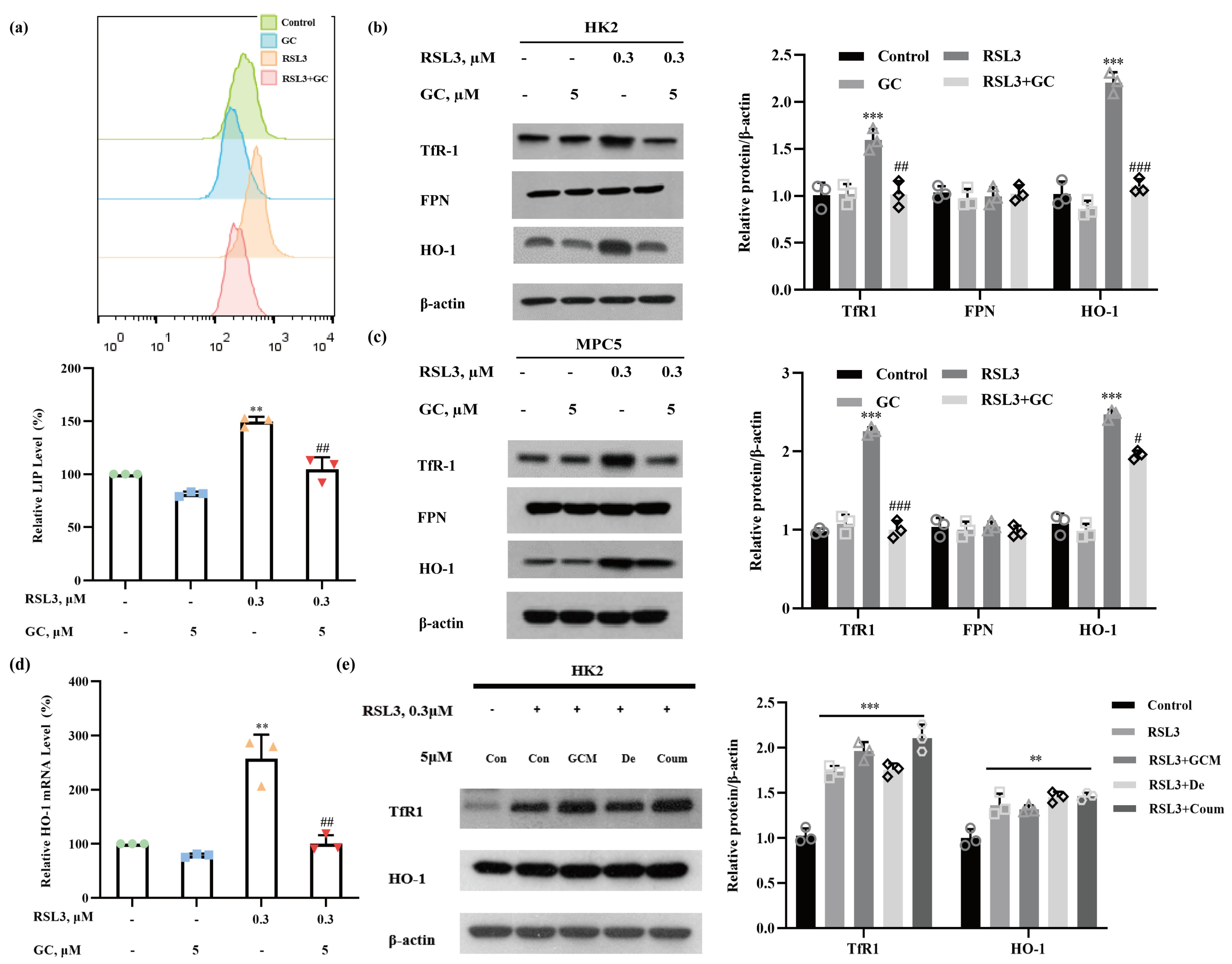
2.3. GC Prevents RSL3-Induced Iron Dysmetabolism in HK2 and MPC5 Cells
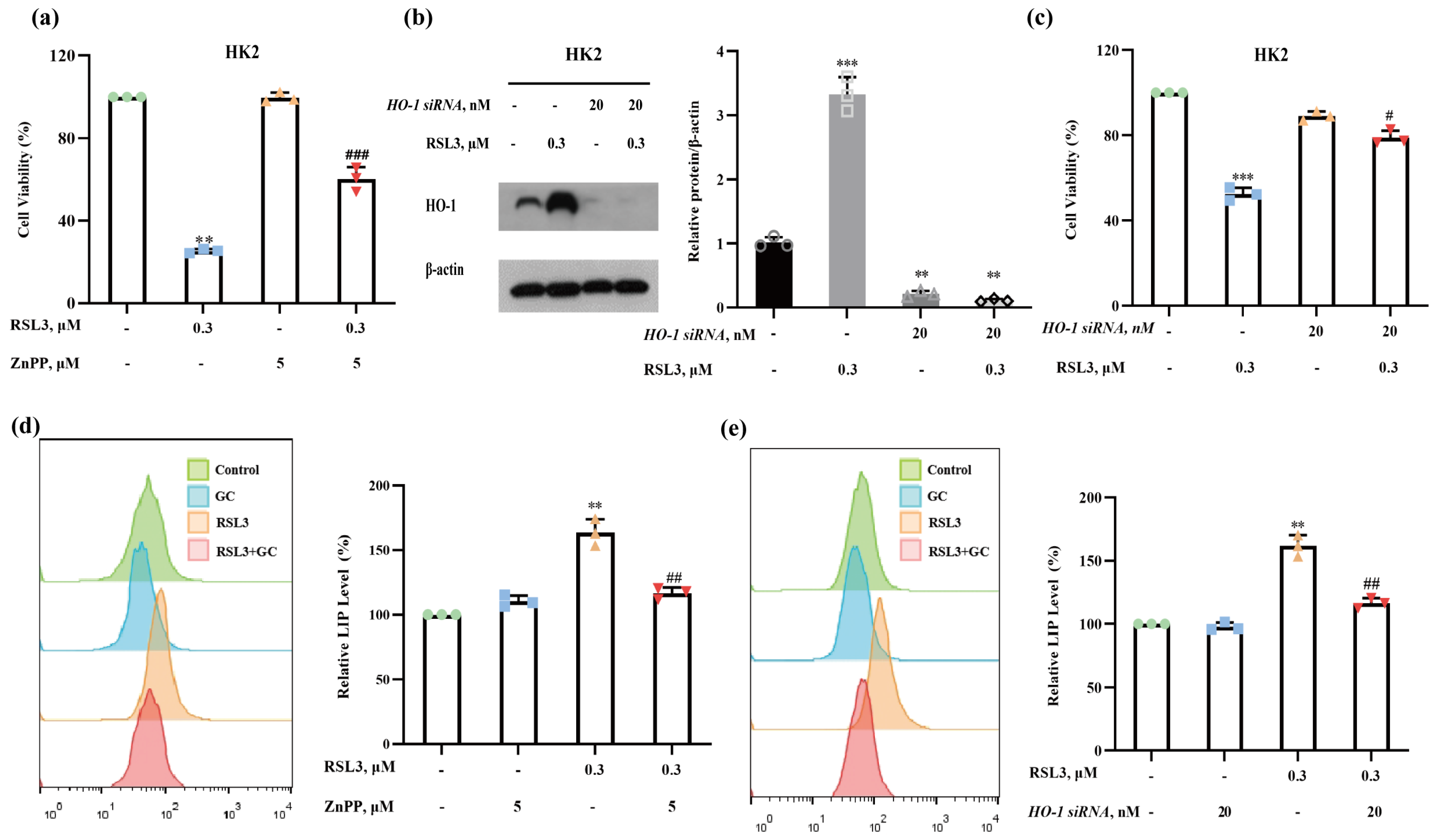
2.4. Induction of HO-1 Promotes RSL3-Induced Ferroptosis in HK2 Cells
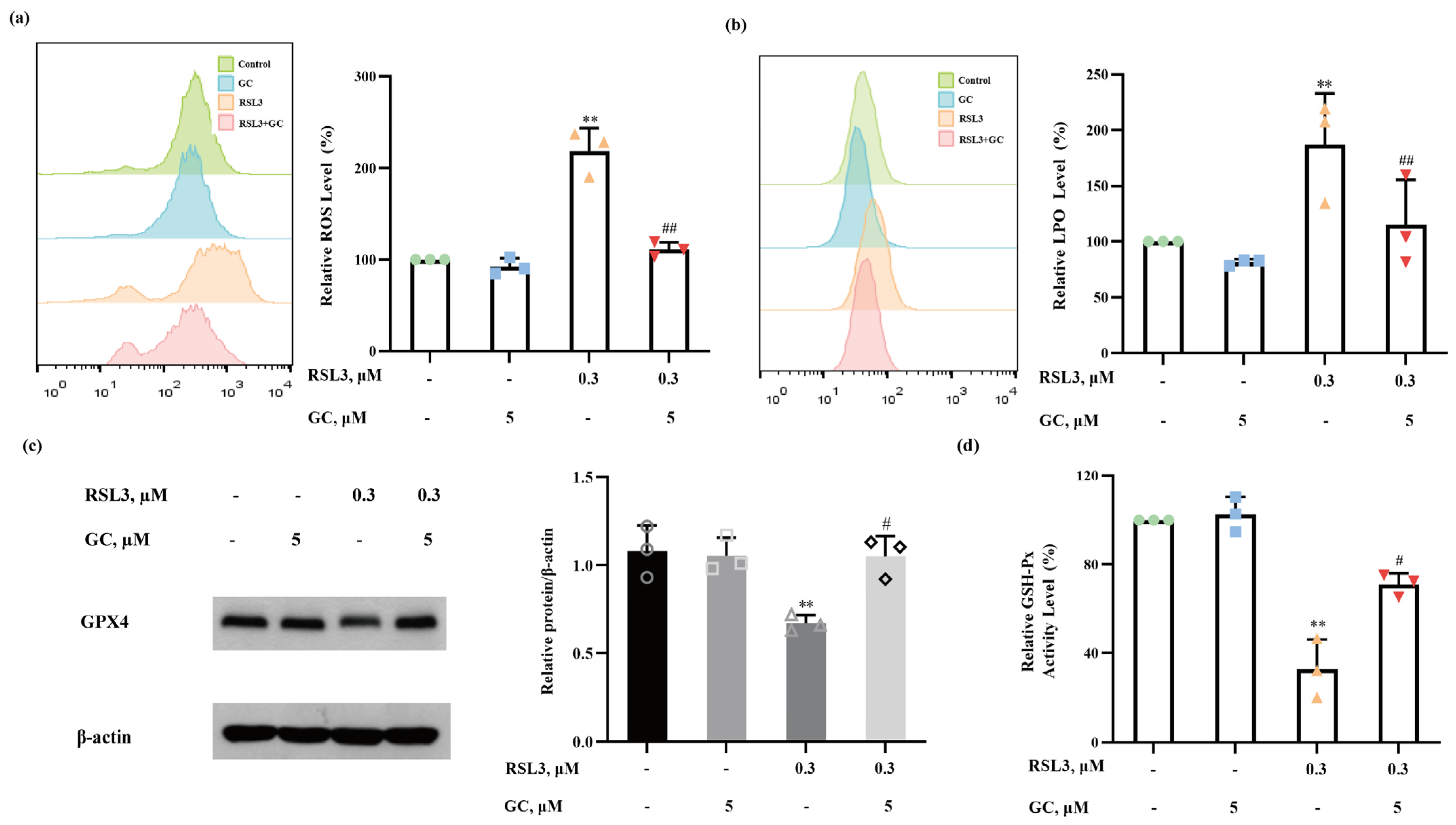
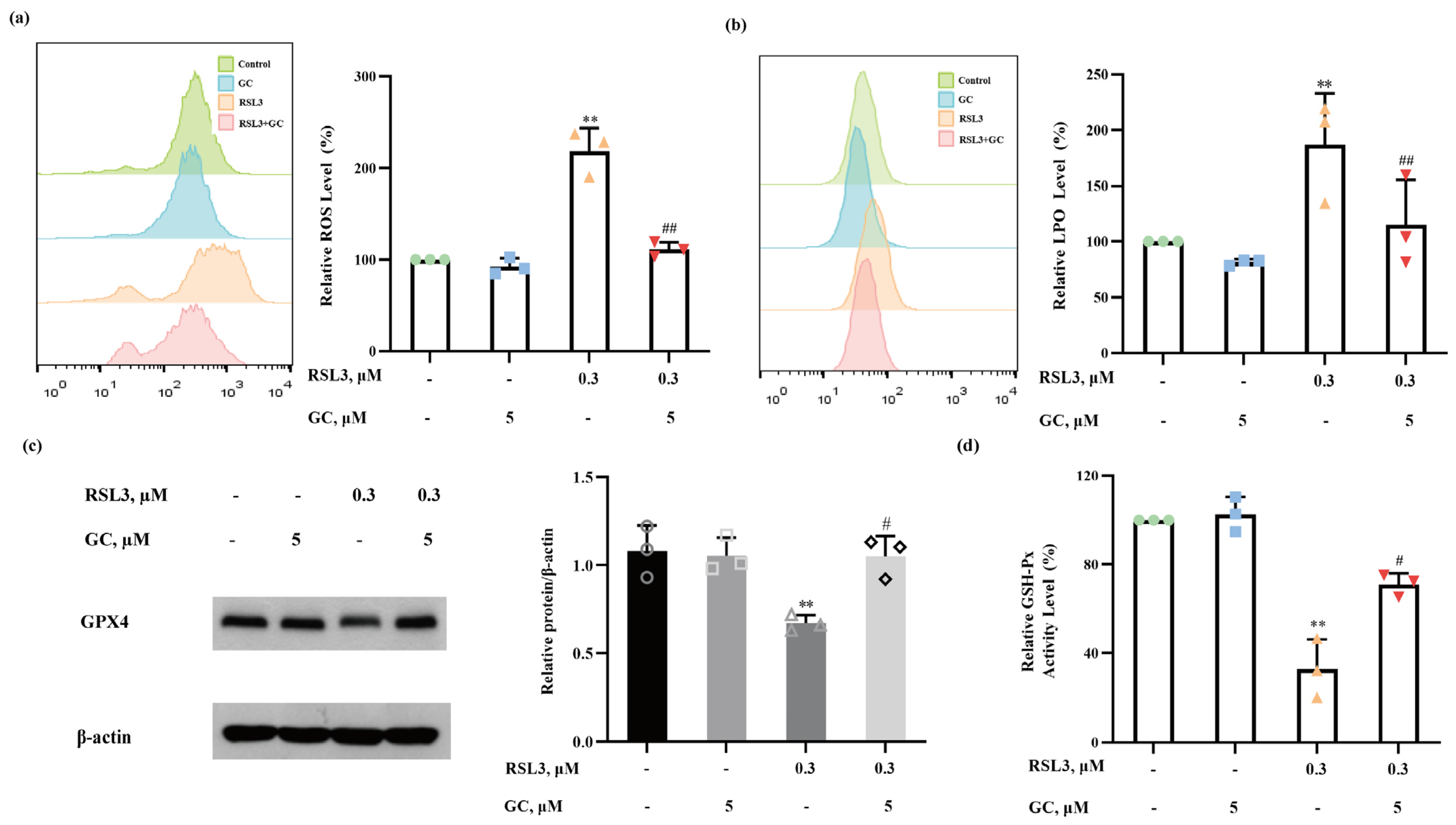
2.5. GC Suppresses ROS Generation and Lipid Peroxidation
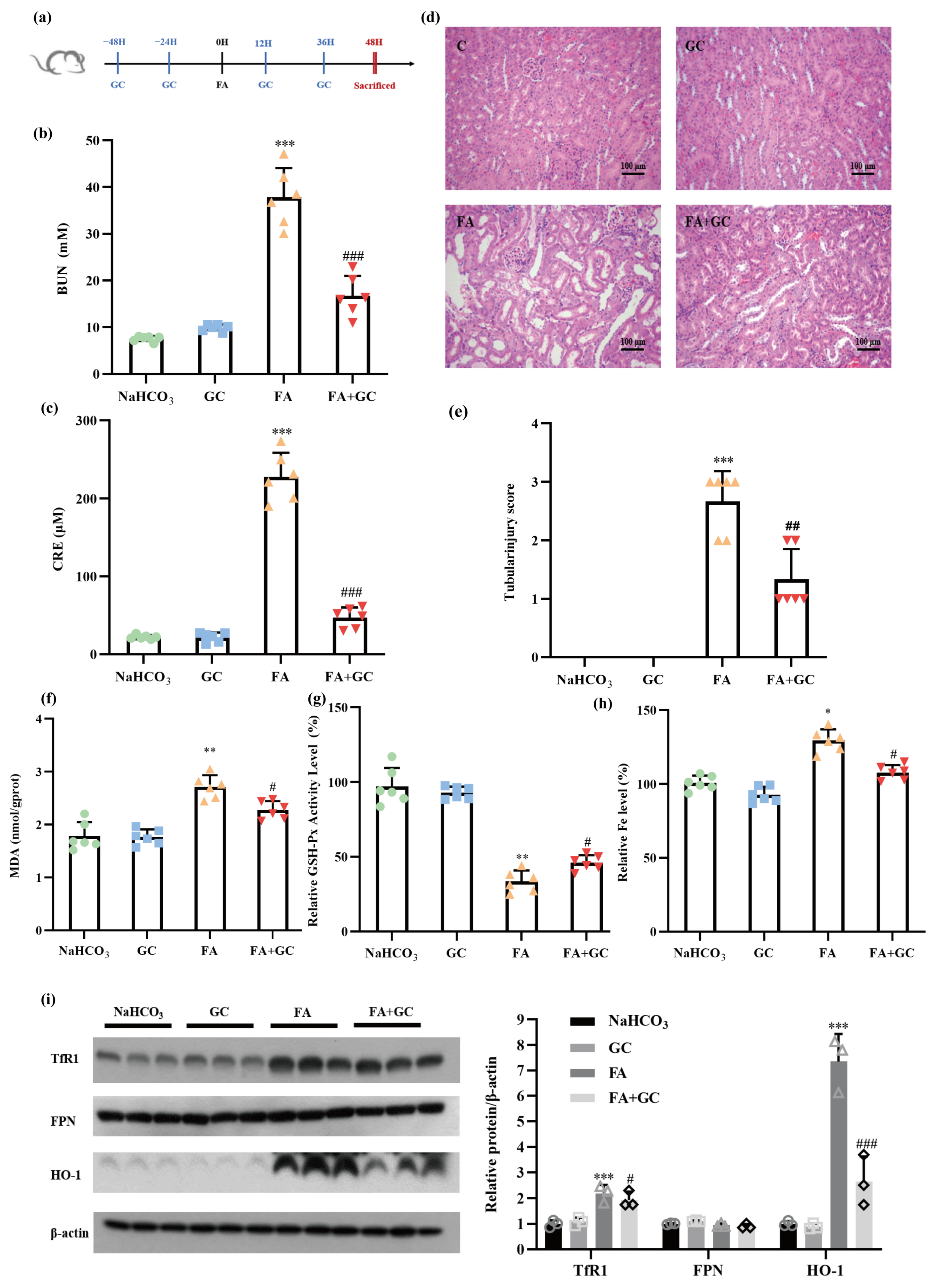
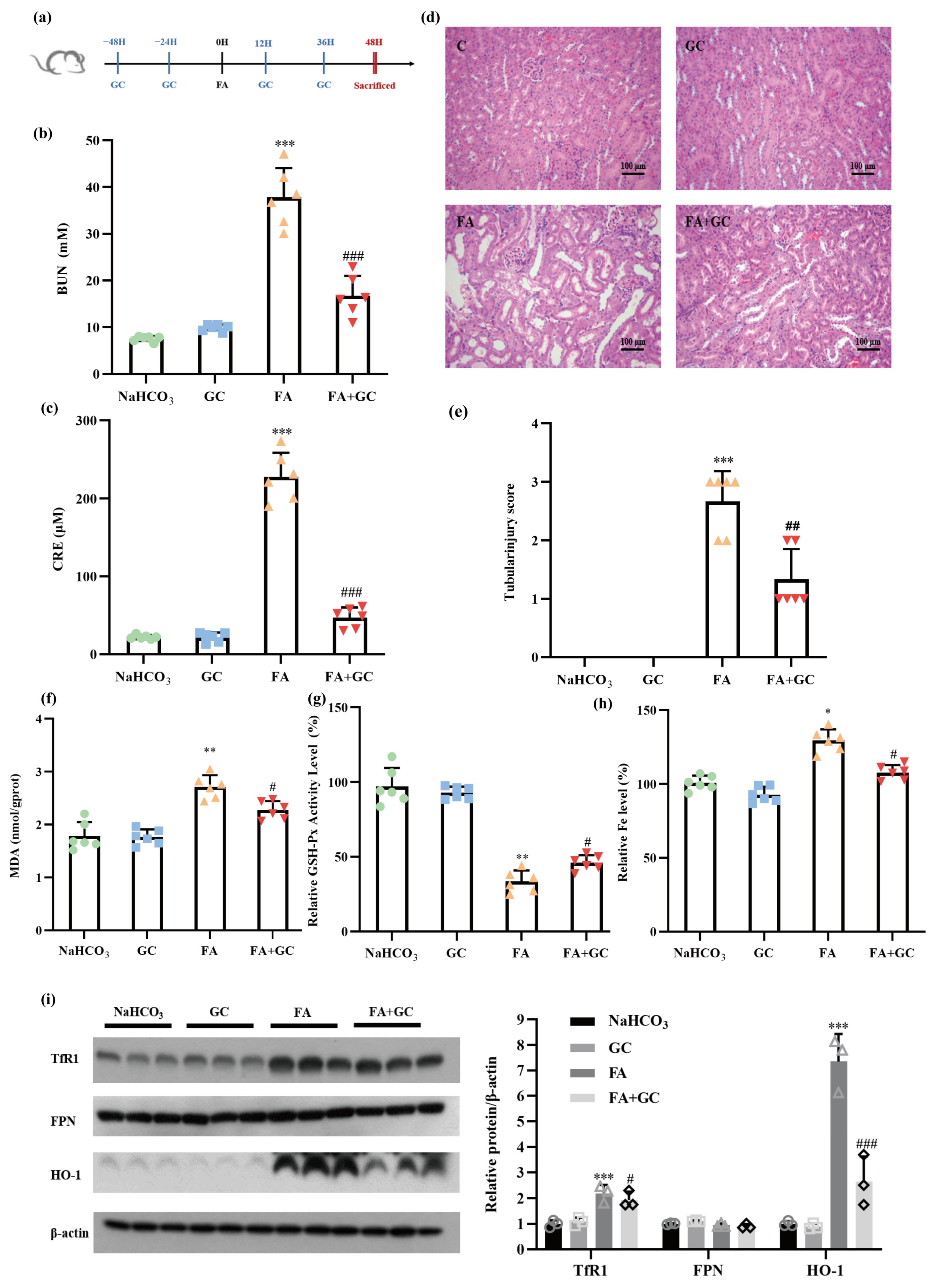
2.6. GC Protects against Folic Acid-Induced Acute Kidney Injury In Vivo
3. Discussion
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Cell Culture and Treatments
4.3. Crystal Violet Staining
4.4. Labile-Iron-Pool Assay
4.5. Measurement of Reactive Oxygen Species (ROS)
4.6. Detection of Lipid Peroxide
4.7. RNA Interference
4.8. Quantitative Polymerase Chain Reaction (PCR)
4.9. Animal Experiments
4.10. Biochemical Serum and Tissue Analysis
4.11. Haematoxylin and Eosin Staining
4.12. Western Blotting
4.13. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Dixon, S.J.S.J.; Lemberg, K.M.K.M.; Lamprecht, M.R.M.R.; Skouta, R.R.; Zaitsev, E.M.E.M.; Gleason, C.E.C.E.; Patel, D.N.D.N.; Bauer, A.J.A.J.; Cantley, A.M.A.M.; Yang, W.S.W.S.; et al. Ferroptosis: An Iron-Dependent Form of Non-Apoptotic Cell Death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [PubMed]
- Stockwell, B.R. Ferroptosis turns 10: Emerging mechanisms, physiological functions, and therapeutic applications. Cell 2022, 185, 2401–2421. [Google Scholar] [CrossRef] [PubMed]
- GBD Chronic Kidney Disease Collaboration. Global, regional, and national burden of chronic kidney disease, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2020, 395, 709–733. [Google Scholar] [CrossRef] [PubMed]
- Kellum, J.A.; Romagnani, P.; Ashuntantang, G.; Ronco, C.; Zarbock, A.; Anders, H.J. Acute kidney injury. Nat. Rev. Dis. Primers 2021, 7, 52. [Google Scholar] [CrossRef]
- Neyra, J.A.; Chawla, L.S. Acute Kidney Disease to Chronic Kidney Disease. Crit. Care Clin. 2021, 37, 453–474. [Google Scholar] [CrossRef]
- Feng, Q.; Yu, X.; Qiao, Y.; Pan, S.; Wang, R.; Zheng, B.; Wang, H.; Ren, K.D.; Liu, H.; Yang, Y. Ferroptosis and Acute Kidney Injury (AKI): Molecular Mechanisms and Therapeutic Potentials. Front. Pharmacol. 2022, 13, 858676. [Google Scholar] [CrossRef]
- Martin-Sanchez, D.; Ruiz-Andres, O.; Poveda, J.; Carrasco, S.; Cannata-Ortiz, P.; Sanchez-Niño, M.D.; Ruiz, O.M.; Egido, J.; Linkermann, A.; Ortiz, A.; et al. Ferroptosis, but Not Necroptosis, Is Important in Nephrotoxic Folic Acid-Induced AKI. J. Am. Soc. Nephrol. 2017, 28, 218–229. [Google Scholar] [CrossRef]
- Ni, L.; Yuan, C.; Wu, X. Targeting ferroptosis in acute kidney injury. Cell Death Dis. 2022, 13, 182. [Google Scholar] [CrossRef]
- Spinks, E.A.; Fenwick, G.R. The determination of glycyrrhizin in selected UK liquorice products. Food Addit. Contam. 1990, 7, 769–778. [Google Scholar] [CrossRef]
- Fu, Y.; Chen, J.; Li, Y.J.; Zheng, Y.F.; Li, P. Antioxidant and anti-inflammatory activities of six flavonoids separated from licorice. Food Chem. 2013, 141, 1063–1071. [Google Scholar] [CrossRef]
- Zhang, Q.; Ye, M. Chemical analysis of the Chinese herbal medicine Gan-Cao (licorice). J. Chromatogr. A 2009, 1216, 1954–1969. [Google Scholar] [CrossRef]
- Ji, S.; Li, Z.; Song, W.; Wang, Y.; Liang, W.; Li, K.; Tang, S.; Wang, Q.; Qiao, X.; Zhou, D.; et al. Bioactive Constituents of Glycyrrhiza uralensis (Licorice): Discovery of the Effective Components of a Traditional Herbal Medicine. J. Nat. Prod. 2016, 79, 281–292. [Google Scholar] [CrossRef]
- Wang, Q.; Qiao, X.; Qian, Y.; Liu, C.F.; Yang, Y.F.; Ji, S.; Li, J.; Guo, D.A.; Ye, M. Metabolites identification of glycyrin and glycyrol, bioactive coumarins from licorice. J. Chromatogr. B 2015, 983, 39–46. [Google Scholar] [CrossRef]
- Lu, S.; Ye, L.; Yin, S.; Zhao, C.; Yan, M.; Liu, X.; Cui, J.; Hu, H. Glycyrol exerts potent therapeutic effect on lung cancer via directly inactivating T-LAK cell-originated protein kinase. Pharmacol. Res. 2019, 147, 104366. [Google Scholar] [CrossRef]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of Ferroptotic Cancer Cell Death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef] [PubMed]
- Kawabata, H. Transferrin and transferrin receptors update. Free Radic. Biol. Med. 2019, 133, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zeng, X.; Lu, D.; Yin, M.; Shan, M.; Gao, Y. Erastin induces ferroptosis via ferroportin-mediated iron accumulation in endometriosis. Hum. Reprod. 2021, 36, 951–964. [Google Scholar] [CrossRef]
- Kerins, M.J.; Ooi, A. The Roles of NRF2 in Modulating Cellular Iron Homeostasis. Antioxid. Redox Signal. 2018, 29, 1756–1773. [Google Scholar] [CrossRef]
- Brigelius-Flohé, R.; Flohé, L. Regulatory Phenomena in the Glutathione Peroxidase Superfamily. Antioxid. Redox Signal. 2020, 33, 498–516. [Google Scholar] [CrossRef] [PubMed]
- Yan, L.J. Folic acid-induced animal model of kidney disease. Anim. Model. Exp. Med. 2021, 4, 329–342. [Google Scholar] [CrossRef]
- Wang, Y.; Quan, F.; Cao, Q.; Lin, Y.; Yue, C.; Bi, R.; Cui, X.; Yang, H.; Yang, Y.; Birnbaumer, L.; et al. Quercetin alleviates acute kidney injury by inhibiting ferroptosis. J. Adv. Res. 2021, 28, 231–243. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Liu, B.; Fan, Y.; Liu, M.; Han, B.; Meng, Y.; Xu, X.; Song, Z.; Liu, X.; Hao, Q.; et al. Nuciferine protects against folic acid-induced acute kidney injury by inhibiting ferroptosis. Br. J. Pharmacol. 2021, 178, 1182–1199. [Google Scholar] [CrossRef] [PubMed]
- Hassannia, B.; Vandenabeele, P.; Vanden Berghe, T. Targeting Ferroptosis to Iron Out Cancer. Cancer Cell 2019, 35, 830–849. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Wang, H.; Han, D.; Xie, E.; Yang, X.; Wei, J.; Gu, S.; Gao, F.; Zhu, N.; Yin, X.; et al. Ferroptosis as a target for protection against cardiomyopathy. Proc. Natl. Acad. Sci. USA 2019, 116, 2672–2680. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Ju, Y.; Dai, X.; Ni, N.; Liu, Y.; Zhang, D.; Gao, H.; Sun, H.; Zhang, J.; Gu, P. HO-1-mediated ferroptosis as a target for protection against retinal pigment epithelium degeneration. Redox Biol. 2021, 43, 101971. [Google Scholar] [CrossRef]
- Kwon, M.Y.; Park, E.; Lee, S.J.; Chung, S.W. Heme oxygenase-1 accelerates erastin-induced ferroptotic cell death. Oncotarget 2015, 6, 24393–24403. [Google Scholar] [CrossRef]
- Consoli, V.; Sorrenti, V.; Pittalà, V.; Greish, K.; D’Amico, A.G.; Romeo, G.; Intagliata, S.; Salerno, L.; Vanella, L. Heme Oxygenase Modulation Drives Ferroptosis in TNBC Cells. Int. J. Mol. Sci. 2022, 23, 5709. [Google Scholar] [CrossRef]
- Guerrero Hue, M.; García Caballero, C.; Palomino Antolín, A.; Rubio Navarro, A.; Vázquez Carballo, C.; Herencia, C.; Martín Sanchez, D.; Farré Alins, V.; Egea, J.; Cannata, P.; et al. Curcumin reduces renal damage associated with rhabdomyolysis by decreasing ferroptosis-mediated cell death. FASEB J. 2019, 33, 8961–8975. [Google Scholar] [CrossRef]
- Adedoyin, O.; Boddu, R.; Traylor, A.; Lever, J.M.; Bolisetty, S.; George, J.F.; Agarwal, A. Heme oxygenase-1 mitigates ferroptosis in renal proximal tubule cells. Am. J. Physiol.-Renal Physiol. 2018, 314, F702–F714. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Groups | Treatments |
|---|---|
| NaHCO3 | Intraperitoneal injection, equivalent volume of Tween-80: PBS (5:95, v/v) solution, equivalent volume of 0.3 M NaHCO3 aqueous solution |
| GC | Intraperitoneal injection, GC 10 mg/kg bw, equivalent volume of 0.3 M NaHCO3 aqueous solution |
| FA | Intraperitoneal injection, equivalent volume of Tween-80: PBS (5:95, v/v) solution, FA 250 mg/kg bw |
| FA+GC | Intraperitoneal injection, GC 10 mg/kg bw, FA 250 mg/kg bw |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cao, L.; Han, K.; Fan, L.; Zhao, C.; Yin, S.; Hu, H. Glycyrol Alleviates Acute Kidney Injury by Inhibiting Ferroptsis. Int. J. Mol. Sci. 2024, 25, 2458. https://doi.org/10.3390/ijms25052458
Cao L, Han K, Fan L, Zhao C, Yin S, Hu H. Glycyrol Alleviates Acute Kidney Injury by Inhibiting Ferroptsis. International Journal of Molecular Sciences. 2024; 25(5):2458. https://doi.org/10.3390/ijms25052458
Chicago/Turabian StyleCao, Lixing, Kai Han, Lihong Fan, Chong Zhao, Shutao Yin, and Hongbo Hu. 2024. "Glycyrol Alleviates Acute Kidney Injury by Inhibiting Ferroptsis" International Journal of Molecular Sciences 25, no. 5: 2458. https://doi.org/10.3390/ijms25052458
APA StyleCao, L., Han, K., Fan, L., Zhao, C., Yin, S., & Hu, H. (2024). Glycyrol Alleviates Acute Kidney Injury by Inhibiting Ferroptsis. International Journal of Molecular Sciences, 25(5), 2458. https://doi.org/10.3390/ijms25052458