
Platinum(IV) Prodrugs Incorporating an Indole-Based Derivative, 5-Benzyloxyindole-3-Acetic Acid in the Axial Position Exhibit Prominent Anticancer Activity

 , , , , , and
, , , , , and 
Abstract
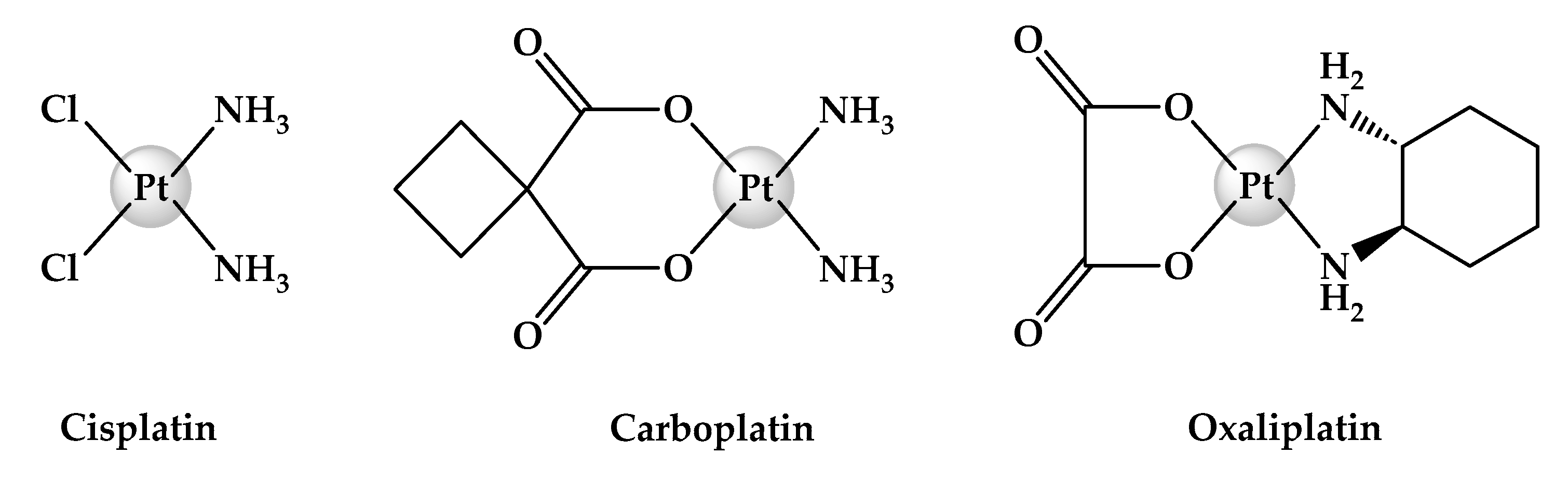
1. Introduction
2. Results and Discussion
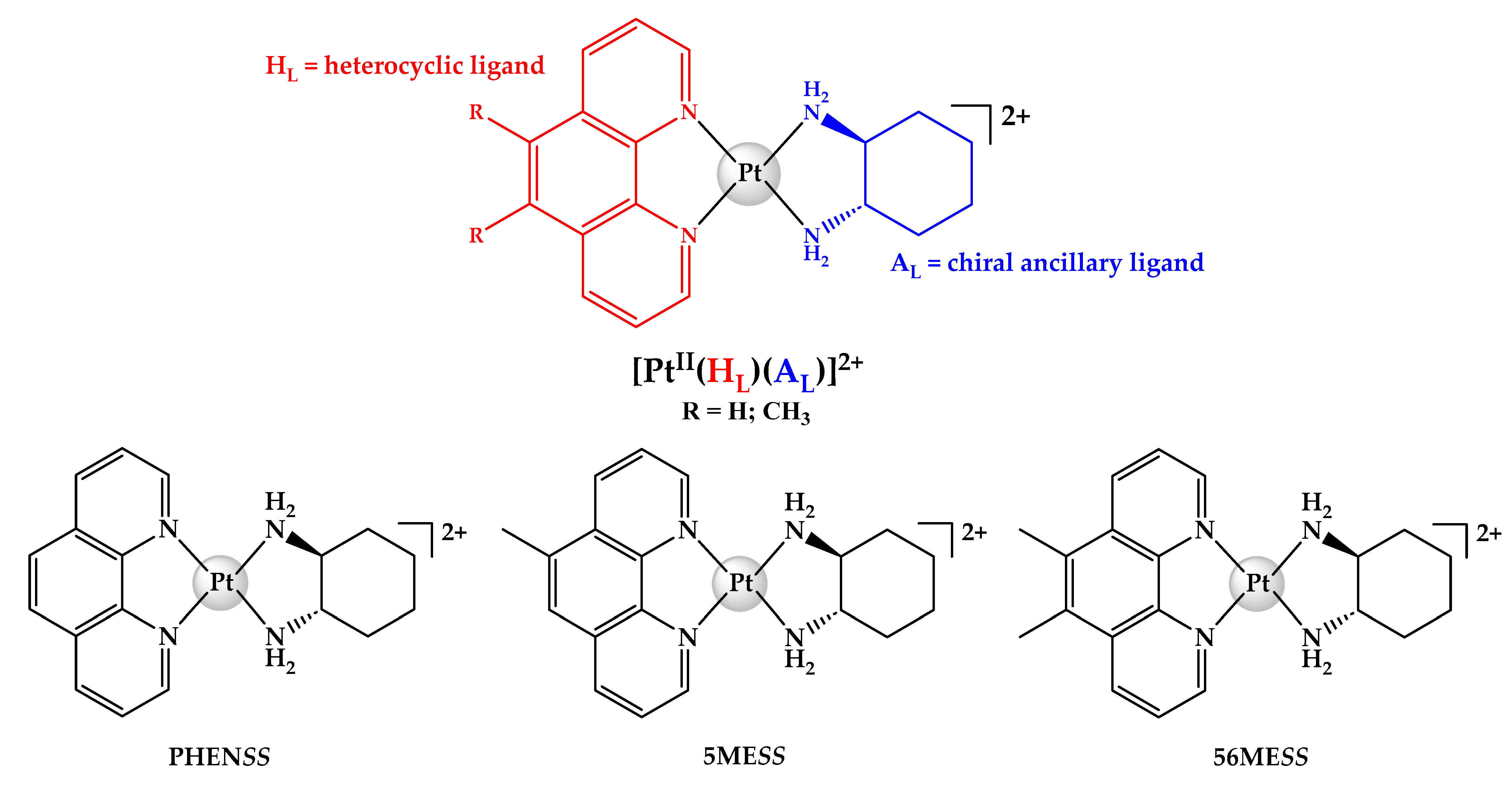
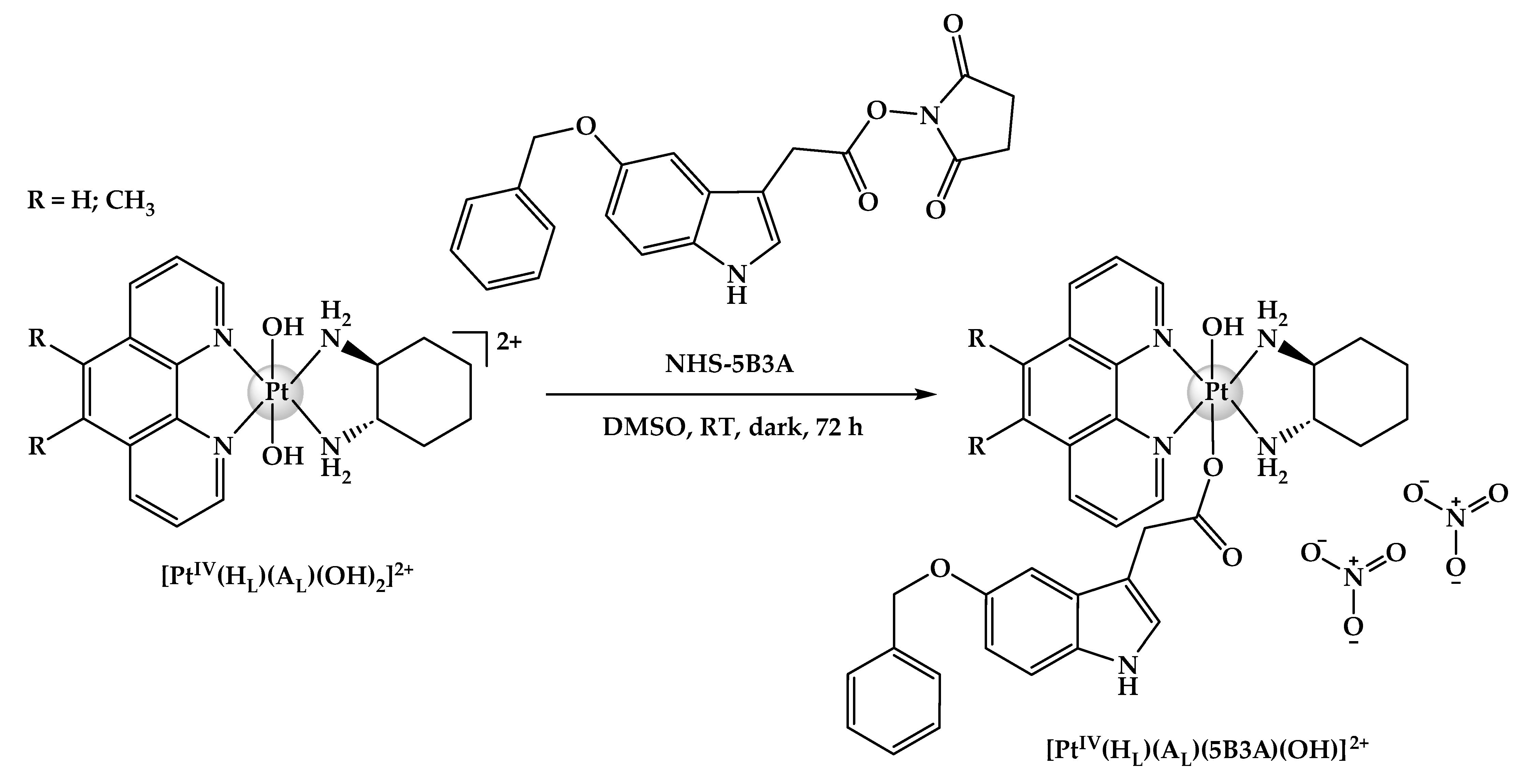
2.1. Syntheses and Characterisation
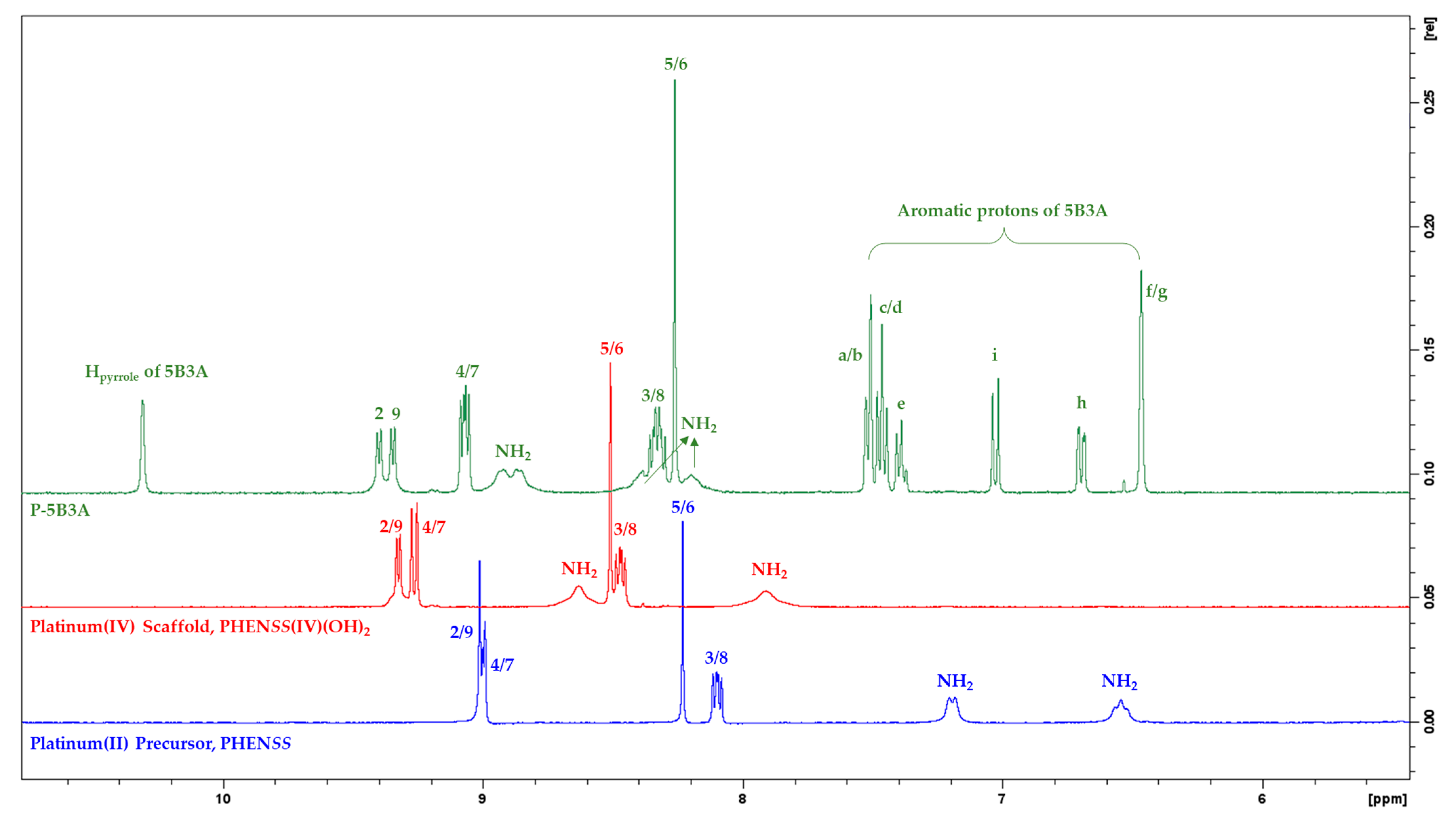
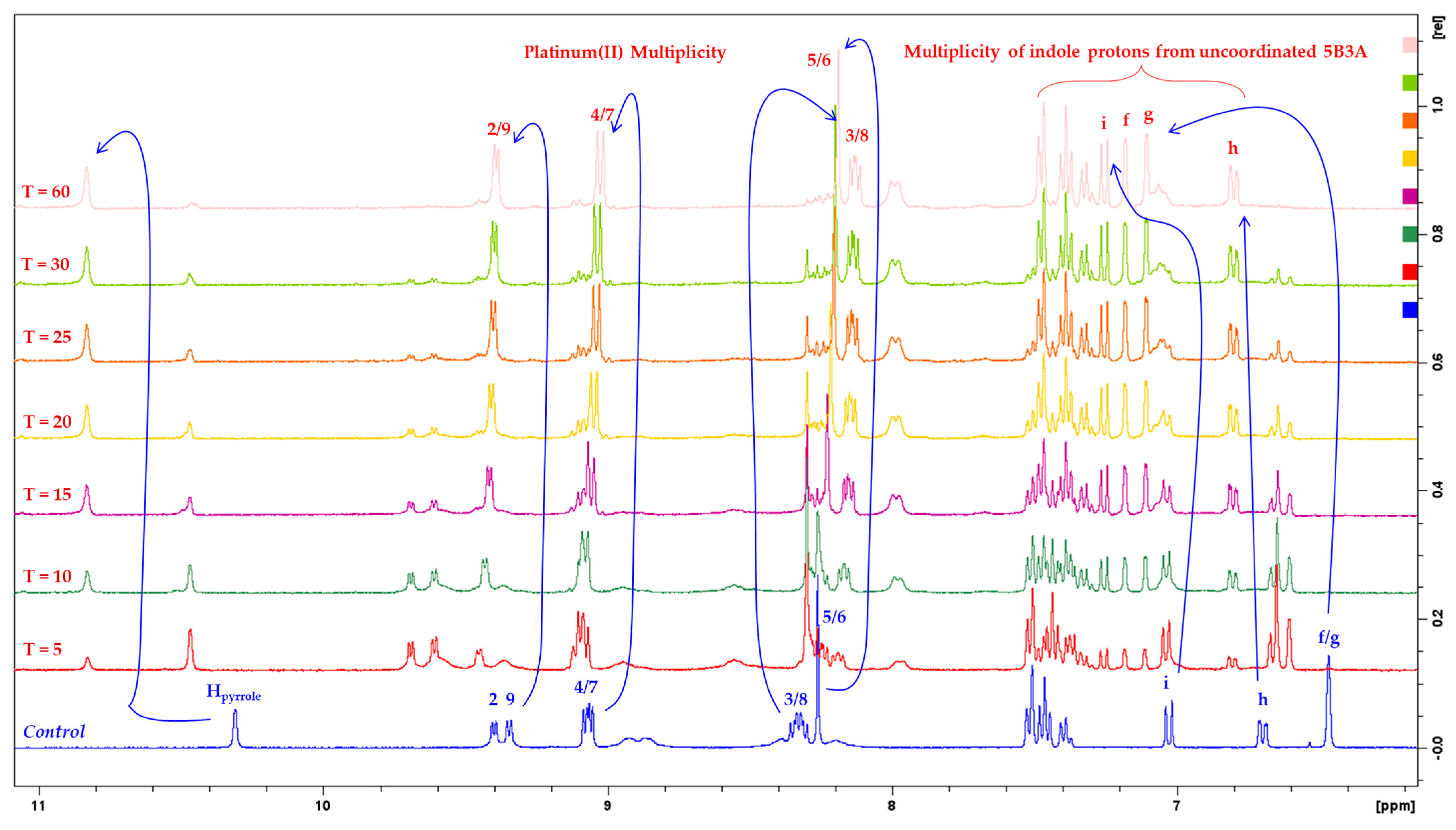
2.1.1. NMR Assignments
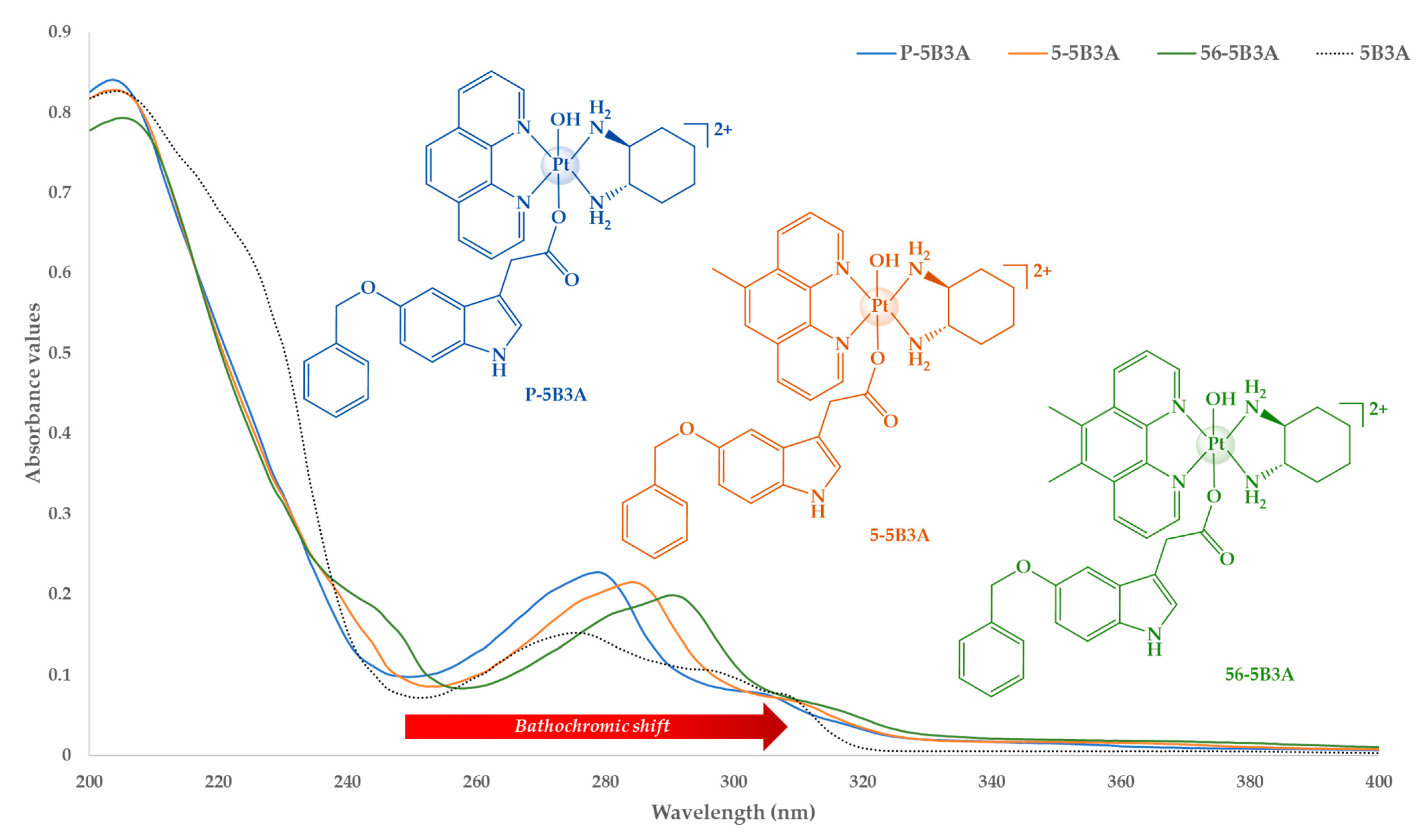
2.1.2. UV and CD Spectral Analysis
2.2. Physicochemical Investigations
2.2.1. Lipophilicity
2.2.2. Preliminary Reduction Measurements
2.2.3. Stability Measurements
2.3. Biological Investigations
2.3.1. In Vitro Anticancer Activity
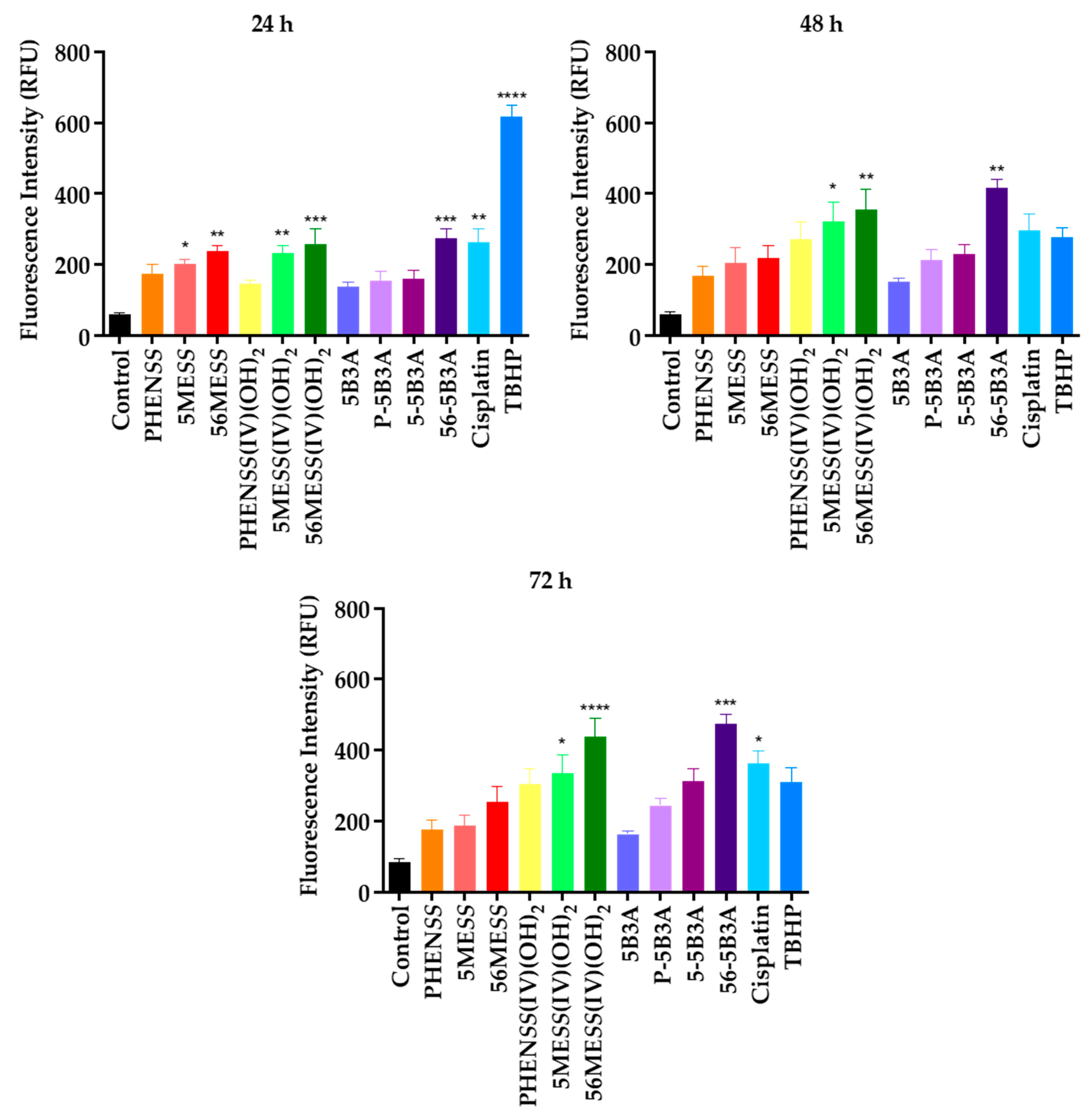
2.3.2. Drug-Induced ROS Production as a Treatment Strategy
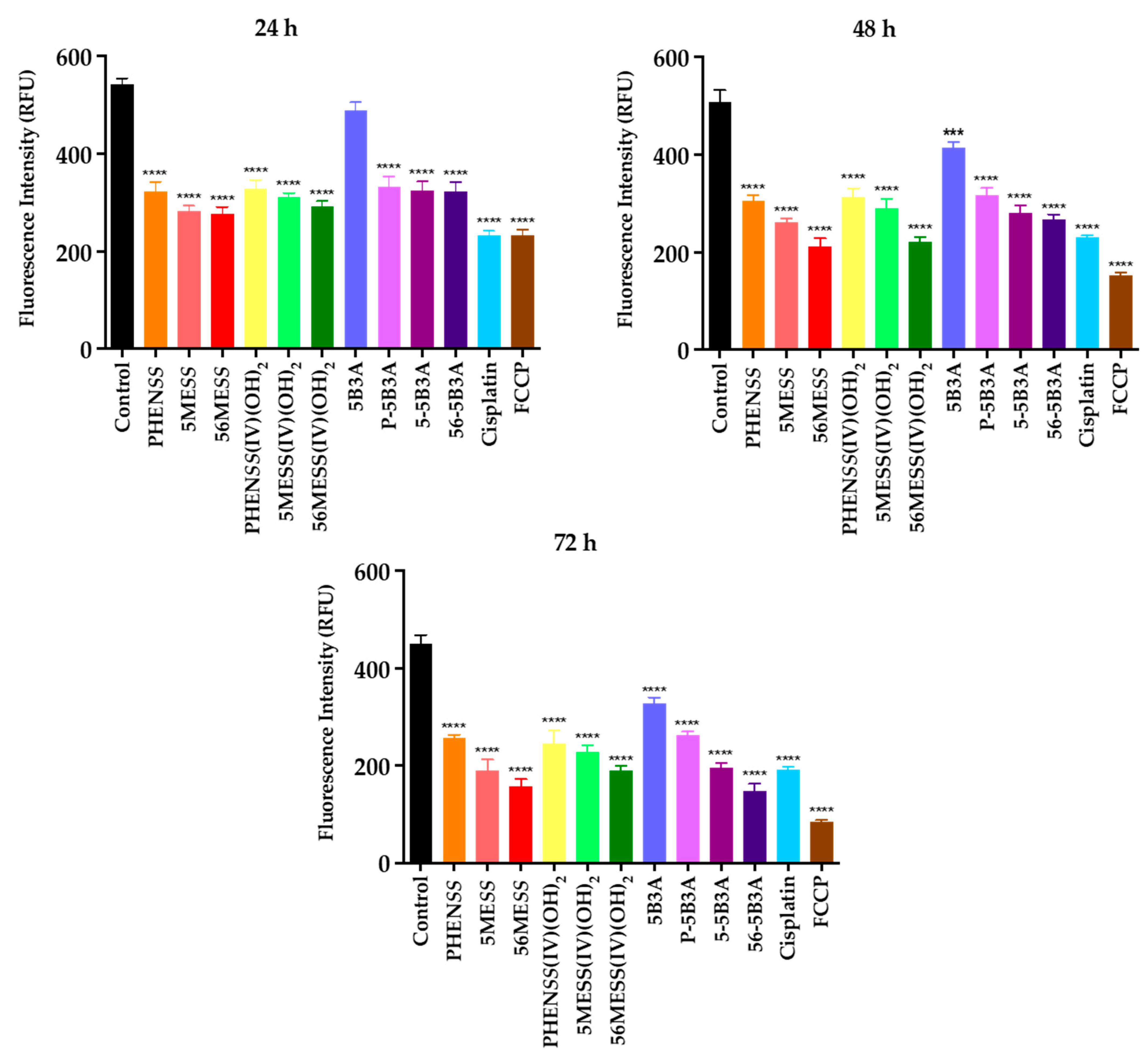
2.3.3. Drug-Induced Mitochondrial Dysfunction as a Treatment Strategy
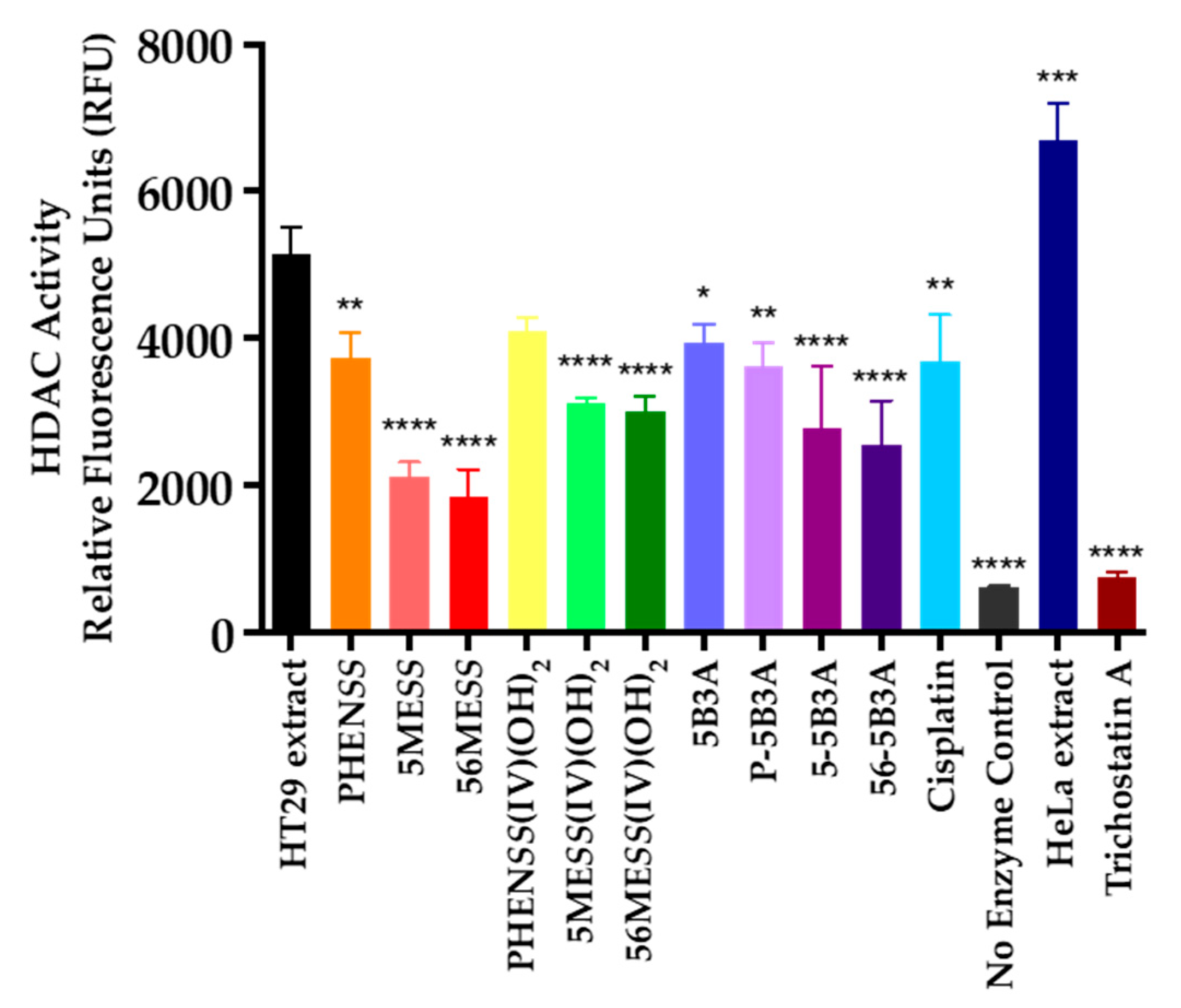
2.3.4. Histone Deacetylase (HDAC) Inhibition Studies
3. Materials and Methods
3.1. Materials
3.2. Chemistry
3.2.1. Synthesis Route of NHS-5B3A
3.2.2. Syntheses of Platinum(II) Precursors and Platinum(IV) Scaffolds
3.2.3. Syntheses of Platinum(IV) Derivatives Incorporating 5B3A (P-5B3A, 5-5B3A and 56-5B3A)
3.3. Laboratory Instrumentation
3.3.1. Flash Chromatography
3.3.2. High-Performance Liquid Chromatography (HPLC)
3.3.3. Nuclear Magnetic Resonance (NMR) Spectroscopy
3.3.4. Ultraviolet–Visible (UV) Spectroscopy
3.3.5. Circular Dichroism (CD) Spectroscopy
3.3.6. Elemental Microanalysis
3.4. Physicochemical Measurements
3.4.1. Solubility
3.4.2. Stability
3.4.3. Lipophilicity
3.4.4. Reduction
3.5. Biological Measurements
3.5.1. Cell Viability Assays
3.5.2. Drug-Induced Reactive Oxygen Species (ROS) Production
3.5.3. Mitochondrial Membrane Potential (MtMP) Changes
3.5.4. Histone Deacetylase (HDAC) Inhibition Measurements
4. Conclusions
5. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rosenberg, B.; Van Camp, L.; Krigas, T. Inhibition of cell division in Escherichia coli by electrolysis products from a platinum electrode. Nature 1965, 205, 698–699. [Google Scholar] [CrossRef]
- Kelland, L.R.; Sharp, S.Y.; O’Neill, C.F.; Raynaud, F.I.; Beale, P.J.; Judson, I.R. Mini-review: Discovery and development of platinum complexes designed to circumvent cisplatin resistance. J. Inorg. Biochem. 1999, 77, 111–115. [Google Scholar] [CrossRef] [PubMed]
- Galanski, M.; Jakupec, M.A.; Keppler, B.K. Update of the preclinical situation of anticancer platinum complexes: Novel design strategies and innovative analytical approaches. Curr. Med. Chem. 2005, 12, 2075–2094. [Google Scholar] [CrossRef] [PubMed]
- Lokich, J.; Anderson, N. Carboplatin versus cisplatin in solid tumours: An analysis of the literature. Ann. Oncol. 1998, 9, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Taamma, A.; Cvitkovic, E.; Raymond, E.; Chaney, S.G. Oxaliplatin: A review of preclinical and clinical studies. Ann. Oncol. 1998, 9, 1053–1071. [Google Scholar]
- Schoch, S.; Gajewski, S.; Rothfuß, J.; Hartwig, A.; Köberle, B. Comparative study of the mode of action of clinically approved platinum-based chemotherapeutics. Int. J. Mol. Sci. 2020, 21, 6928. [Google Scholar] [CrossRef] [PubMed]
- Rocha, C.R.R.; Silva, M.M.; Quinet, A.; Cabral-Neto, J.B.; Menck, C.F.M. DNA repair pathways and cisplatin resistance: An intimate relationship. Clinics 2018, 73 (Suppl. S1), e478s. [Google Scholar] [CrossRef]
- Mezencev, R. Interactions of cisplatin with non-DNA targets and their influence on anticancer activity and drug toxicity: The complex world of the platinum complex. Curr. Cancer Drug Targets 2015, 14, 794–816. [Google Scholar] [CrossRef]
- Wang, J.; Tao, J.; Jia, S.; Wang, M.; Jiang, H.; Du, Z. The protein-binding behavior of platinum anticancer drugs in blood revealed by mass spectrometry. Pharmaceuticals 2021, 14, 104. [Google Scholar] [CrossRef]
- Alassadi, S.; Pisani, M.J.; Wheate, N.J. A chemical perspective on the clinical use of platinum-based anticancer drugs. Dalton Trans. 2022, 51, 10835–10846. [Google Scholar] [CrossRef]
- Goldstein, R.S.; Mayor, G.H. The nephrotoxicity of cisplatin. Life Sci. 1983, 32, 685–690. [Google Scholar] [CrossRef] [PubMed]
- Kidera, Y.; Kawakami, H.; Sakiyama, T.; Okamoto, K.; Tanaka, K.; Takeda, M.; Kaneda, H.; Nishina, S.; Tsurutani, J.; Fujiwara, K.; et al. Risk factors for cisplatin-induced nephrotoxicity and potential of magnesium supplementation for renal protection. PLoS ONE 2014, 9, e101902. [Google Scholar] [CrossRef] [PubMed]
- McSweeney, K.R.; Gadanec, L.K.; Qaradakhi, T.; Ali, B.A.; Zulli, A.; Apostolopoulos, V. Mechanisms of cisplatin-induced acute kidney injury: Pathological mechanisms, pharmacological interventions, and genetic mitigations. Cancers 2021, 13, 1572. [Google Scholar] [CrossRef] [PubMed]
- Rogers, B.B.; Cuddahy, T.; Briscella, C.; Ross, N.; Olszanski, A.J.; Denlinger, C.S. Oxaliplatin. Clin. J. Oncol. Nurs. 2019, 23, 68–75. [Google Scholar] [PubMed]
- Fotopoulou, C. Limitations to the use of carboplatin-based therapy in advanced ovarian cancer. Eur. J. Cancer Suppl. 2014, 12, 13–16. [Google Scholar] [CrossRef] [PubMed]
- Coffetti, G.; Moraschi, M.; Facchetti, G.; Rimoldi, I. The challenging treatment of cisplatin-resistant tumours: State of the art and future perspectives. Molecules 2023, 28, 3407. [Google Scholar] [CrossRef] [PubMed]
- Amable, L. Cisplatin resistance and opportunities for precision medicine. Pharmacol. Res. 2016, 106, 27–36. [Google Scholar] [CrossRef]
- Bayat Mokhtari, R.; Homayouni, T.S.; Baluch, N.; Morgatskaya, E.; Kumar, S.; Das, B.; Yeger, H. Combination therapy in combating cancer. Oncotarget 2017, 8, 38022–38043. [Google Scholar] [CrossRef]
- Tolcher, A.W.; Mayer, L.D. Improving combination cancer therapy: The CombiPlex® development platform. Future Oncol. 2018, 14, 1317–1332. [Google Scholar] [CrossRef]
- Boshuizen, J.; Peeper, D.S. Rational cancer treatment combinations: An urgent clinical need. Mol. Cell 2020, 78, 1002–1018. [Google Scholar] [CrossRef]
- Wheate, N.J.; Walker, S.; Craig, G.E.; Oun, R. The status of platinum anticancer drugs in the clinic and in clinical trials. Dalton Trans. 2010, 39, 8113–8127. [Google Scholar] [CrossRef] [PubMed]
- Johnstone, T.C.; Park, G.Y.; Lippard, S.J. Understanding and improving platinum anticancer drugs—Phenanthriplatin. Anticancer Res. 2014, 34, 471–476. [Google Scholar] [PubMed]
- Johnstone, T.C.; Suntharalingam, K.; Lippard, S.J. The The next generation of platinum drugs: Targeted Pt(ii) agents, nanoparticle delivery, and Pt(iv) prodrugs. Chem. Rev. 2016, 116, 3436–3486. [Google Scholar] [CrossRef] [PubMed]
- Ndagi, U.; Mhlongo, N.; Soliman, M.E. Metal complexes in cancer therapy—An update from drug design perspective. Drug Des. Devel. Ther. 2017, 11, 599–616. [Google Scholar] [CrossRef] [PubMed]
- Markman, M.; DeMarco, L.C.; Birkhofer, M.; Budman, D.; Hakes, T.; Reichman, B.; Rubin, S.; Jones, W.; Barakat, R.; Curtin, J.; et al. Phase II trial of zeniplatin (CL 286 558), a new patinum compound, in patients with advanced ovarian cancer previously treated with organoplatinum-based therapy. J. Cancer Res. Clin. Oncol. 1993, 119, 234–236. [Google Scholar] [CrossRef] [PubMed]
- Tanis, B.C.; Vermorken, J.B.; ten Bokkel Huinink, W.W.; van Oosterom, A.T.; Splinter, T.A.; Vendrik, K.J.; Sleijfer, D.T.; van der Burg, M.E.; van der Putten, E.; Pinedo, H.M. Multicenter phase II study of spiroplatin. Oncology 1992, 49, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Kudelka, A.P.; Siddik, Z.H.; Tresukosol, D.; Edwards, C.L.; Freedman, R.S.; Madden, T.L.; Rastogl, R.; Hord, M.; Kim, E.E.; Tornos, C.; et al. A phase II and pharmacokinetic study of enloplatin in patients with platinum refractory advanced ovarian carcinoma. Anti-Cancer Drugs 1997, 8, 649–656. [Google Scholar] [CrossRef]
- Dragovich, T.; Mendelson, D.; Hoos, A.; Lewis, J.; Kurtin, S.; Richardson, K.; Von Hoff, D. 268 A phase II trial of aroplatin (L-NDDP), a liposomal DACH platinum, in patients with metastatic colorectal cancer (CRC)—A preliminary report. Eur. J. Cancer Suppl. 2003, 1, S82–S83. [Google Scholar] [CrossRef]
- Lebwohl, D.; Canetta, R. Clinical development of platinum complexes in cancer therapy: An historical perspective and an update. Eur. J. Cancer 1998, 34, 1522–1534. [Google Scholar] [CrossRef]
- Brodie, C.R.; Collins, J.G.; Aldrich-Wright, J.R. DNA binding and biological activity of some platinum(ii) intercalating compounds containing methyl-substituted 1,10-phenanthrolines. Dalton Trans. 2004, 8, 1145–1152. [Google Scholar] [CrossRef]
- Wheate, N.J.; Taleb, R.I.; Krause-Heuer, A.M.; Cook, R.L.; Wang, S.; Higgins, V.J.; Aldrich-Wright, J.R. Novel platinum(II)-based anticancer complexes and molecular hosts as their drug delivery vehicles. Dalton Trans. 2007, 43, 5055–5064. [Google Scholar] [CrossRef] [PubMed]
- Fisher, D.M.; Fenton, R.; Aldrich-Wright, J. In vivo studies of a platinum(II) metallointercalator. Chem. Comm. 2008, 43, 5613–5615. [Google Scholar] [CrossRef] [PubMed]
- Kemp, S.; Wheate, N.J.; Buck, D.P.; Nikac, M.; Collins, J.G.; Aldrich-Wright, J.R. The effect of ancillary ligand chirality and phenanthroline functional group substitution on the cytotoxicity of platinum(II)-based metallointercalators. J. Inorg. Biochem. 2007, 101, 1049–1058. [Google Scholar] [CrossRef] [PubMed]
- Fisher, D.M.; Bednarski, P.J.; Grünert, R.; Turner, P.; Fenton, R.R.; Aldrich-Wright, J.R. Chiral platinum(ii) metallointercalators with potent in vitro cytotoxic activity. Chem. Med. Chem. 2007, 2, 488–495. [Google Scholar] [CrossRef]
- Krause-Heuer, A.M.; Grunert, R.; Kuhne, S.; Buczkowska, M.; Wheate, N.J.; Le Pevelen, D.D.; Boag, L.R.; Fisher, D.M.; Kasparkova, J.; Malina, J.; et al. Studies of the mechanism of action of platinum(II) complexes with potent cytotoxicity in human cancer cells. J. Med. Chem. 2009, 52, 5474–5484. [Google Scholar] [CrossRef] [PubMed]
- Kostrhunova, H.; Zajac, J.; Novohradsky, V.; Kasparkova, J.; Malina, J.; Aldrich-Wright, J.R.; Petruzzella, E.; Sirota, R.; Gibson, D.; Brabec, V. A subset of new platinum antitumour agents kills cells by a multimodal mechanism of action also involving changes in the organization of the microtubule cytoskeleton. J. Med. Chem. 2019, 62, 5176–5190. [Google Scholar] [CrossRef] [PubMed]
- Garbutcheon-Singh, K.B.; Myers, S.; Harper, B.W.; Ng, N.S.; Dong, Q.; Xie, C.; Aldrich-Wright, J.R. The effects of 56MESS on mitochondrial and cytoskeletal proteins and the cell cycle in MDCK cells. Metallomics 2013, 5, 1061–1067. [Google Scholar] [CrossRef] [PubMed]
- Moretto, J.; Chauffert, B.; Ghiringhelli, F.; Aldrich-Wright, J.R.; Bouyer, F. Discrepancy between in vitro and in vivo antitumour effect of a new platinum(II) metallointercalator. Investig. N. Drugs 2011, 29, 1164–1176. [Google Scholar] [CrossRef]
- Harper, B.W.J.; Petruzzella, E.; Sirota, R.; Faccioli, F.F.; Aldrich-Wright, J.R.; Gandin, V.; Gibson, D. Synthesis, characterization and in vitro and in vivo anticancer activity of Pt(iv) derivatives of [Pt(1S,2S-DACH)(5,6-dimethyl-1,10-phenanthroline)]. Dalton Trans. 2017, 46, 7005–7019. [Google Scholar] [CrossRef]
- Macias, F.J.; Deo, K.M.; Pages, B.J.; Wormell, P.; Clegg, J.K.; Zhang, Y.; Li, F.; Zheng, G.; Sakoff, J.; Gilbert, J.; et al. Synthesis and analysis of the structure, diffusion and cytotoxicity of heterocyclic platinum(iv) complexes. Chemistry 2015, 21, 16990–17001. [Google Scholar] [CrossRef]
- Marotta, C.; Giorgi, E.; Binacchi, F.; Cirri, D.; Gabbiani, C.; Pratesi, A. An overview of recent advancements in anticancer Pt(IV) prodrugs: New smart drug combinations, activation and delivery strategies. Inorganica Chim. Acta 2023, 548, 121388. [Google Scholar] [CrossRef]
- Xu, Z.; Wang, Z.; Deng, Z.; Zhu, G. Recent advances in the synthesis, stability, and activation of platinum(IV) anticancer prodrugs. Coord. Chem. Rev. 2021, 442, 213991–214023. [Google Scholar] [CrossRef]
- Ponte, F.; Scoditti, S.; Mazzone, G.; Sicilia, E. The current status in computational exploration of Pt(iv) prodrug activation by reduction. Phys. Chem. Chem. Phys. 2023, 25, 15586–15599. [Google Scholar] [CrossRef] [PubMed]
- Wong, D.Y.Q.; Ang, W.H. Development of platinum(IV) complexes as anticancer prodrugs: The story so far. Cosmos 2012, 8, 121–135. [Google Scholar] [CrossRef]
- Zhong, Y.; Jia, C.; Zhang, X.; Liao, X.; Yang, B.; Cong, Y.; Pu, S.; Gao, C. Targeting drug delivery system for platinum(Ⅳ)-based antitumour complexes. Eur. J. Med. Chem. 2020, 194, 112229. [Google Scholar] [CrossRef] [PubMed]
- Hall, M.D.; Mellor, H.R.; Callaghan, R.; Hambley, T.W. Basis for design and development of platinum(IV) anticancer complexes. J. Med. Chem. 2007, 50, 3403–3411. [Google Scholar] [CrossRef] [PubMed]
- Ravera, M.; Gabano, E.; McGlinchey, M.J.; Osella, D. Pt(IV) antitumour prodrugs: Dogmas, paradigms, and realities. Dalton Trans. 2022, 51, 2121–2134. [Google Scholar] [CrossRef]
- Raveendran, R.; Braude, J.P.; Wexselblatt, E.; Novohradsky, V.; Stuchlikova, O.; Brabec, V.; Gandin, V.; Gibson, D. Pt(IV) derivatives of cisplatin and oxaliplatin with phenylbutyrate axial ligands are potent cytotoxic agents that act by several mechanisms of action. Chem. Sci. 2016, 7, 2381–2391. [Google Scholar] [CrossRef]
- Li, Z.; Wang, Q.; Li, L.; Chen, Y.; Cui, J.; Liu, M.; Zhang, N.; Liu, Z.; Han, J.; Wang, Z. Ketoprofen and loxoprofen platinum(iv) complexes displaying antimetastatic activities by inducing dna damage, inflammation suppression, and enhanced immune response. J. Med. Chem. 2021, 64, 17920–17935. [Google Scholar] [CrossRef]
- Han, W.; He, W.; Song, Y.; Zhao, J.; Song, Z.; Shan, Y.; Hua, W.; Sun, Y. Multifunctional platinum(iv) complex bearing HDAC inhibitor and biotin moiety exhibits prominent cytotoxicity and tumour-targeting ability. Dalton Trans. 2022, 51, 7343–7351. [Google Scholar] [CrossRef]
- Karmakar, S.; Kostrhunova, H.; Ctvrtlikova, T.; Novohradsky, V.; Gibson, D.; Brabec, V. Platinum(IV)-estramustine multiaction prodrugs are effective antiproliferative agents against prostate cancer cells. J. Med. Chem. 2020, 63, 13861–13877. [Google Scholar] [CrossRef] [PubMed]
- Teraiya, N.; Agrawal, K.; Patel, T.M.; Patel, A.; Patel, S.; Shah, U.; Shah, S.; Rathod, K.; Patel, K. A review of the therapeutic importance of indole scaffold in drug discovery. Curr. Drug Discov. Technol. 2023, 20, e050523216584. [Google Scholar] [CrossRef] [PubMed]
- Taliani, S.; Da Settimo, F.; Martini, C.; Laneri, S.; Novellino, E.; Greco, G. Exploiting the indole scaffold to design compounds binding to different pharmacological targets. Molecules 2020, 25, 2331. [Google Scholar] [CrossRef] [PubMed]
- Devi, N.; Kaur, K.; Biharee, A.; Jaitak, V. Recent development in indole derivatives as anticancer agent: A mechanistic approach. Anticancer Agents Med. Chem. 2021, 21, 1802–1824. [Google Scholar] [CrossRef] [PubMed]
- Dhuguru, J.; Skouta, R. Role of indole scaffolds as pharmacophores in the development of anti-lung cancer agents. Molecules 2020, 25, 1615. [Google Scholar] [CrossRef] [PubMed]
- Dhiman, A.; Sharma, R.; Singh, R.K. Target-based anticancer indole derivatives and insight into structure–Activity relationship: A mechanistic review update (2018–2021). Acta Pharm. Sin. B. 2022, 12, 3006–3027. [Google Scholar] [CrossRef] [PubMed]
- Oberhuber, N.; Ghosh, H.; Nitzsche, B.; Dandawate, P.; Höpfner, M.; Schobert, R.; Biersack, B. Synthesis and anticancer evaluation of new indole-based tyrphostin derivatives and their (p-cymene)dichloridoruthenium(ii) complexes. Int. J. Mol. Sci. 2023, 24, 854. [Google Scholar] [CrossRef]
- Bresciani, G.; Cervinka, J.; Kostrhunova, H.; Biancalana, L.; Bortoluzzi, M.; Pampaloni, G.; Novohradsky, V.; Brabec, V.; Marchetti, F.; Kasparkova, J. N-indolyl diiron vinyliminium complexes exhibit antiproliferative effects in cancer cells associated with disruption of mitochondrial homeostasis, ROS scavenging, and antioxidant activity. Chem. Biol. Interact. 2023, 385, 110742. [Google Scholar] [CrossRef]
- Aputen, A.D.; Elias, M.G.; Gilbert, J.; Sakoff, J.A.; Gordon, C.P.; Scott, K.F.; Aldrich-Wright, J.R. Potent chlorambucil-platinum(IV) prodrugs. Int. J. Mol. Sci. 2022, 23, 10471–10499. [Google Scholar] [CrossRef]
- Barré, A.; Tintas, M.; Levacher, V.; Papamicael, C.; Vincent, G. An overview of the synthesis of highly versatile n-hydroxysuccinimide esters. Synthesis 2017, 49, 472–483. [Google Scholar]
- Aputen, A.D.; Elias, M.G.; Gilbert, J.; Sakoff, J.A.; Gordon, C.P.; Scott, K.F.; Aldrich-Wright, J.R. Versatile platinum(iv) prodrugs of naproxen and acemetacin as chemo-anti-inflammatory agents. Cancers 2023, 15, 2460. [Google Scholar] [CrossRef] [PubMed]
- Deo, K.M.; Sakoff, J.; Gilbert, J.; Zhang, Y.; Aldrich Wright, J.R. Synthesis, characterisation and potent cytotoxicity of unconventional platinum(iv) complexes with modified lipophilicity. Dalton Trans. 2019, 48, 17217–17227. [Google Scholar] [CrossRef] [PubMed]
- Khoury, A.; Elias, E.; Mehanna, S.; Shebaby, W.; Deo, K.M.; Mansour, N.; Khalil, C.; Sayyed, K.; Sakoff, J.A.; Gilbert, J.; et al. Novel platinum(ii) and platinum(iv) antitumour agents that exhibit potent cytotoxicity and selectivity. J. Med. Chem. 2022, 65, 16481–16493. [Google Scholar] [CrossRef] [PubMed]
- McGhie, B.S.; Sakoff, J.; Gilbert, J.; Aldrich-Wright, J.R. Synthesis and characterisation of platinum(IV) polypyridyl complexes with halide axial ligands. Inorganica Chim. Acta 2019, 495, 118964–118974. [Google Scholar] [CrossRef]
- Aputen, A.D.; Elias, M.G.; Gilbert, J.; Sakoff, J.A.; Gordon, C.P.; Scott, K.F.; Aldrich-Wright, J.R. Bioactive platinum(iv) complexes incorporating halogenated phenylacetates. Molecules 2022, 27, 7120. [Google Scholar] [CrossRef] [PubMed]
- Khoury, A.; Sakoff, J.A.; Gilbert, J.; Scott, K.F.; Karan, S.; Gordon, C.P.; Aldrich-Wright, J.R. Cyclooxygenase-inhibiting platinum(iv) prodrugs with potent anticancer activity. Pharmaceutics 2022, 14, 787–808. [Google Scholar] [CrossRef] [PubMed]
- Aputen, A. Novel platinum(IV) prodrugs. Master’s Thesis, Western Sydney University, Campbelltown, NSW, Australia, 2019. [Google Scholar]
- Garbutcheon-Singh, K.B.; Leverett, P.; Myers, S.; Aldrich-Wright, J.R. Cytotoxic platinum(II) intercalators that incorporate 1R,2R-diaminocyclopentane. Dalton Trans. 2013, 42, 918–926. [Google Scholar] [CrossRef]
- Khoury, A.; Sakoff, J.A.; Gilbert, J.; Karan, S.; Gordon, C.P.; Aldrich-Wright, J.R. Potent platinum(iv) prodrugs that incorporate a biotin moiety to selectively target cancer cells. Pharmaceutics 2022, 14, 2780. [Google Scholar] [CrossRef]
- Dasari, S.; Tchounwou, P.B. Cisplatin in cancer therapy: Molecular mechanisms of action. Eur. J. Pharmacol. 2014, 740, 364–378. [Google Scholar] [CrossRef]
- Chen, T.-Y.; Tsai, M.-J.; Chang, L.-C.; Wu, P.-C. Co-delivery of cisplatin and gemcitabine via viscous nanoemulsion for potential synergistic intravesical chemotherapy. Pharmaceutics 2020, 12, 949. [Google Scholar] [CrossRef]
- Tarleton, M.; Gilbert, J.; Robertson, M.J.; McCluskey, A.; Sakoff, J.A. Library synthesis and cytotoxicity of a family of 2-phenylacrylonitriles and discovery of an estrogen dependent breast cancer lead compound. Med. Chem. Comm. 2011, 2, 31–37. [Google Scholar] [CrossRef]
- Deo, K.M.; Sakoff, J.; Gilbert, J.; Zhang, Y.; Aldrich Wright, J.R. Synthesis, characterisation and influence of lipophilicity on cellular accumulation and cytotoxicity of unconventional platinum(iv) prodrugs as potent anticancer agents. Dalton Trans. 2019, 48, 17228–17240. [Google Scholar] [CrossRef] [PubMed]
- López-Lázaro, M. Two preclinical tests to evaluate anticancer activity and to help validate drug candidates for clinical trials. Oncoscience 2015, 2, 91–98. [Google Scholar] [CrossRef] [PubMed]
- López-Lázaro, M. A simple and reliable approach for assessing anticancer activity in vitro. Curr. Med. Chem. 2015, 22, 1324–1334. [Google Scholar] [CrossRef] [PubMed]
- Pilotto Heming, C.; Muriithi, W.; Wanjiku Macharia, L.; Niemeyer Filho, P.; Moura-Neto, V.; Aran, V. P-glycoprotein and cancer: What do we currently know? Heliyon 2022, 8, e11171. [Google Scholar] [CrossRef] [PubMed]
- Pizzino, G.; Irrera, N.; Cucinotta, M.; Pallio, G.; Mannino, F.; Arcoraci, V.; Squadrito, F.; Altavilla, D.; Bitto, A. Oxidative stress: Harms and benefits for human health. Oxid. Med. Cell. Longev. 2017, 2017, 8416763. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.Z.; Jiang, S.; Zhang, L.; Yu, Z.B. Mitochondrial electron transport chain, ROS generation and uncoupling (Review). Int. J. Mol. Med. 2019, 44, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Checa, J.; Aran, J.M. Reactive oxygen species: Drivers of physiological and pathological processes. J. Inflamm. Res. 2020, 13, 1057–1073. [Google Scholar] [CrossRef]
- Szechyńska-Hebda, M.; Ghalami, R.Z.; Kamran, M.; Van Breusegem, F.; Karpiński, S. To be or not to be? are reactive oxygen species, antioxidants, and stress signalling universal determinants of life or death? Cells 2022, 11, 4105. [Google Scholar] [CrossRef]
- Redza-Dutordoir, M.; Averill-Bates, D.A. Activation of apoptosis signalling pathways by reactive oxygen species. Biochim. Biophys. Acta Mol. Cell Res. 2016, 1863, 2977–2992. [Google Scholar] [CrossRef] [PubMed]
- Villalpando-Rodriguez, G.E.; Gibson, S.B. Reactive oxygen species (ros) regulates different types of cell death by acting as a rheostat. Oxid. Med. Cell. Longev. 2021, 2021, 9912436. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, H.; Takada, K. Reactive oxygen species in cancer: Current findings and future directions. Cancer Sci. 2021, 112, 3945–3952. [Google Scholar] [CrossRef]
- Juan, C.A.; Pérez de la Lastra, J.M.; Plou, F.J.; Pérez-Lebeña, E. The chemistry of reactive oxygen species (ros) revisited: Outlining their role in biological macromolecules (DNA, lipids and proteins) and induced pathologies. Int. J. Mol. Sci. 2021, 22, 4642. [Google Scholar] [CrossRef]
- George, S.; Abrahamse, H. Redox potential of antioxidants in cancer progression and prevention. Antioxidants 2020, 9, 1156. [Google Scholar] [CrossRef] [PubMed]
- Galadari, S.; Rahman, A.; Pallichankandy, S.; Thayyullathil, F. Reactive oxygen species and cancer paradox: To promote or to suppress? Free Radic. Biol. Med. 2017, 104, 144–164. [Google Scholar] [CrossRef]
- Jia, P.; Dai, C.; Cao, P.; Sun, D.; Ouyang, R.; Miao, Y. The role of reactive oxygen species in tumour treatment. RSC Adv. 2020, 10, 7740–7750. [Google Scholar] [CrossRef]
- Aggarwal, V.; Tuli, H.S.; Varol, A.; Thakral, F.; Yerer, M.B.; Sak, K.; Varol, M.; Jain, A.; Khan, M.A.; Sethi, G. Role of reactive oxygen species in cancer progression: Molecular mechanisms and recent advancements. Biomolecules 2019, 9, 735. [Google Scholar] [CrossRef]
- Perillo, B.; Di Donato, M.; Pezone, A.; Di Zazzo, E.; Giovannelli, P.; Galasso, G.; Castoria, G.; Migliaccio, A. ROS in cancer therapy: The bright side of the moon. Exp. Mol. Med. 2020, 52, 192–203. [Google Scholar] [CrossRef]
- Estevão, M.S.; Carvalho, L.C.; Ribeiro, D.; Couto, D.; Freitas, M.; Gomes, A.; Ferreira, L.M.; Fernandes, E.; Marques, M.M.B. Antioxidant activity of unexplored indole derivatives: Synthesis and screening. Eur. J. Med. Chem. 2010, 45, 4869–4878. [Google Scholar] [CrossRef]
- Tolan, D.; Gandin, V.; Morrison, L.; El-Nahas, A.; Marzano, C.; Montagner, D.; Erxleben, A. Oxidative stress induced by Pt(IV) pro-drugs based on the cisplatin scaffold and indole carboxylic acids in axial position. Sci. Rep. 2016, 6, 29367–29379. [Google Scholar] [CrossRef] [PubMed]
- Javadov, S.; Kuznetsov, A.V. Mitochondria: The cell powerhouse and nexus of stress. Front. Physiol. 2013, 4, 207. [Google Scholar] [CrossRef]
- McBride, H.M.; Neuspiel, M.; Wasiak, S. Mitochondria: More than just a powerhouse. Curr. Biol. 2006, 16, R551–R560. [Google Scholar] [CrossRef] [PubMed]
- Pistritto, G.; Trisciuoglio, D.; Ceci, C.; Garufi, A.; D’Orazi, G. Apoptosis as anticancer mechanism: Function and dysfunction of its modulators and targeted therapeutic strategies. Aging 2016, 8, 603–619. [Google Scholar] [CrossRef] [PubMed]
- Shokolenko, I.; Venediktova, N.; Bochkareva, A.; Wilson, G.L.; Alexeyev, M.F. Oxidative stress induces degradation of mitochondrial DNA. Nucleic Acids Res. 2009, 37, 2539–2548. [Google Scholar] [CrossRef] [PubMed]
- Sahu, K.; Langeh, U.; Singh, C.; Singh, A. Crosstalk between anticancer drugs and mitochondrial functions. Curr. Res. Pharmacol. Drug Discov. 2021, 2, 100047. [Google Scholar] [CrossRef] [PubMed]
- Kuretu, A.; Arineitwe, C.; Mothibe, M.; Ngubane, P.; Khathi, A.; Sibiya, N. Drug-induced mitochondrial toxicity: Risks of developing glucose handling impairments. Front. Endocrinol. 2023, 14, 1123928. [Google Scholar] [CrossRef]
- Vieira, H.L.A.; Haouzi, D.; El Hamel, C.; Jacotot, E.; Belzacq, A.S.; Brenner, C.; Kroemer, G. Permeabilization of the mitochondrial inner membrane during apoptosis: Impact of the adenine nucleotide translocator. Cell Death Differ. 2000, 7, 1146–1154. [Google Scholar] [CrossRef]
- Chipuk, J.E.; Bouchier-Hayes, L.; Green, D.R. Mitochondrial outer membrane permeabilization during apoptosis: The innocent bystander scenario. Cell Death Differ. 2006, 13, 1396–1402. [Google Scholar] [CrossRef]
- Li, L.; Qi, R.; Zhang, L.; Yu, Y.; Hou, J.; Gu, Y.; Song, D.; Wang, X. Potential biomarkers and targets of mitochondrial dynamics. Clin. Transl. Med. 2021, 11, e529. [Google Scholar] [CrossRef]
- Zorova, L.D.; Popkov, V.A.; Plotnikov, E.J.; Silachev, D.N.; Pevzner, I.B.; Jankauskas, S.S.; Zorov, S.D.; Babenko, V.A.; Zorov, D.B. Functional significance of the mitochondrial membrane potential. Biochem. (Mosc.) Suppl. A Membr. 2018, 12, 20–26. [Google Scholar] [CrossRef]
- Vasan, K.; Clutter, M.; Fernandez Dunne, S.; George, M.D.; Luan, C.-H.; Chandel, N.S.; Martínez-Reyes, I. Genes involved in maintaining mitochondrial membrane potential upon electron transport chain disruption. Front. Cell Dev. Biol. 2022, 10, 781558. [Google Scholar] [CrossRef] [PubMed]
- Teodoro, J.S.; Palmeira, C.M.; Rolo, A.P. Mitochondrial Membrane Potential (ΔΨ) Fluctuations Associated with the Metabolic States of Mitochondria. In Mitochondrial Bioenergetics: Methods and Protocols; Palmeira, C.M., Moreno, A.J., Eds.; Springer New York: New York, NY, USA, 2018; pp. 109–119. [Google Scholar]
- Hüser, J.; Blatter, L.A. Fluctuations in mitochondrial membrane potential caused by repetitive gating of the permeability transition pore. Biochem. J. 1999, 343, 311–317. [Google Scholar] [CrossRef] [PubMed]
- Kuznetsov, A.V.; Margreiter, R.; Ausserlechner, M.J.; Hagenbuchner, J. The complex interplay between mitochondria, ros and entire cellular metabolism. Antioxidants 2022, 11, 1995. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P. Mitochondrial Dysfunction Indirectly Elevates ROS Production by the Endoplasmic Reticulum. Cell Metab. 2013, 18, 145–146. [Google Scholar] [CrossRef] [PubMed]
- Suski, J.; Lebiedzinska, M.; Bonora, M.; Pinton, P.; Duszynski, J.; Wieckowski, M.R. Relation Between Mitochondrial Membrane Potential and ROS Formation. In Mitochondrial Bioenergetics: Methods and Protocols; Palmeira, C.M., Moreno, A.J., Eds.; Springer: New York, NY, USA, 2018; pp. 357–381. [Google Scholar]
- Sullivan, L.B.; Chandel, N.S. Mitochondrial reactive oxygen species and cancer. Cancer Metab. 2014, 2, 17. [Google Scholar] [CrossRef] [PubMed]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.J.; Seto, E. HATs and HDACs: From structure, function and regulation to novel strategies for therapy and prevention. Oncogene 2007, 26, 5310–5318. [Google Scholar] [CrossRef]
- Prado, F.; Jimeno-González, S.; Reyes, J.C. Histone availability as a strategy to control gene expression. RNA Biol. 2017, 14, 281–286. [Google Scholar] [CrossRef]
- Marmorstein, R.; Roth, S.Y. Histone acetyltransferases: Function, structure, and catalysis. Curr. Opin. Genet. Dev. 2001, 11, 155–161. [Google Scholar] [CrossRef]
- Dueva, R.; Akopyan, K.; Pederiva, C.; Trevisan, D.; Dhanjal, S.; Lindqvist, A.; Farnebo, M. Neutralization of the positive charges on histone tails by rna promotes an open chromatin structure. Cell Chem. Biol. 2019, 26, 1436–1449. [Google Scholar] [CrossRef] [PubMed]
- Seto, E.; Yoshida, M. Erasers of histone acetylation: The histone deacetylase enzymes. Cold Spring Harb. Perspect. Biol. 2014, 6, a018713. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Salz, T.; Zajac-Kaye, M.; Liao, D.; Huang, S.; Qiu, Y. Overexpression of histone deacetylases in cancer cells is controlled by interplay of transcription factors and epigenetic modulators. FASEB J. 2014, 28, 4265–4279. [Google Scholar] [CrossRef] [PubMed]
- Ho, T.C.S.; Chan, A.H.Y.; Ganesan, A. Thirty years of HDAC inhibitors: 2020 insight and hindsight. J. Med. Chem. 2020, 63, 12460–12484. [Google Scholar] [CrossRef] [PubMed]
- Moran, B.; Davern, M.; Reynolds, J.V.; Donlon, N.E.; Lysaght, J. The impact of histone deacetylase inhibitors on immune cells and implications for cancer therapy. Cancer Lett. 2023, 559, 216121. [Google Scholar] [CrossRef] [PubMed]
- Uba, A.I.; Zengin, G. In the quest for histone deacetylase inhibitors: Current trends in the application of multilayered computational methods. Amino Acids 2023, 55, 1709–1726. [Google Scholar] [CrossRef] [PubMed]
- Bondarev, A.D.; Attwood, M.M.; Jonsson, J.; Chubarev, V.N.; Tarasov, V.V.; Schiöth, H.B. Recent developments of HDAC inhibitors: Emerging indications and novel molecules. Br. J. Clin. Pharmacol. 2021, 87, 4577–4597. [Google Scholar] [CrossRef] [PubMed]
- Göttlicher, M.; Minucci, S.; Zhu, P.; Krämer, O.H.; Schimpf, A.; Giavara, S.; Sleeman, J.P.; Lo Coco, F.; Nervi, C.; Pelicci, P.G.; et al. Valproic acid defines a novel class of HDAC inhibitors inducing differentiation of transformed cells. EMBO J. 2001, 20, 6969–6978. [Google Scholar] [CrossRef]
- Phiel, C.J.; Zhang, F.; Huang, E.Y.; Guenther, M.G.; Lazar, M.A.; Klein, P.S. Histone Deacetylase is a direct target of valproic acid, a potent anticonvulsant, mood stabilizer, and teratogen *. J. Biol. Chem. 2001, 276, 36734–36741. [Google Scholar] [CrossRef]
- He, B.; Moreau, R. Lipid-regulating properties of butyric acid and 4-phenylbutyric acid: Molecular mechanisms and therapeutic applications. Pharmacol. Res. 2019, 144, 116–131. [Google Scholar] [CrossRef]
- Moinul, M.; Amin, S.A.; Khatun, S.; Das, S.; Jha, T.; Gayen, S. A detail survey and analysis of selectivity criteria for indole-based histone deacetylase 8 (HDAC8) inhibitors. J. Mol. Struct. 2023, 1271, 133967. [Google Scholar] [CrossRef]
- Jiang, B.-E.; Hu, J.; Liu, H.; Liu, Z.; Wen, Y.; Liu, M.; Zhang, H.-K.; Pang, X.; Yu, L.-F. Design, synthesis, and biological evaluation of indole-based hydroxamic acid derivatives as histone deacetylase inhibitors. Eur. J. Med. Chem. 2022, 227, 113893. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Jin, X.; Yan, Y.; Shao, Y.; Pan, Y.; Roberts, L.R.; Zhang, J.; Huang, H.; Jiang, J. Inhibiting histone deacetylases suppresses glucose metabolism and hepatocellular carcinoma growth by restoring FBP1 expression. Sci. Rep. 2017, 7, 43864. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.T.T.; Zhang, Y.; Shang, E.; Shu, C.; Torrini, C.; Zhao, J.; Bianchetti, E.; Mela, A.; Humala, N.; Mahajan, A.; et al. HDAC inhibitors elicit metabolic reprogramming by targeting super-enhancers in glioblastoma models. J. Clin. Investig. 2020, 130, 3699–3716. [Google Scholar] [CrossRef] [PubMed]
- Shanmugam, G.; Rakshit, S.; Sarkar, K. HDAC inhibitors: Targets for tumour therapy, immune modulation and lung diseases. Transl. Oncol. 2022, 16, 101312. [Google Scholar] [CrossRef] [PubMed]
- Islam, S.; Espitia, C.M.; Persky, D.O.; Carew, J.S.; Nawrocki, S.T. Resistance to histone deacetylase inhibitors confers hypersensitivity to oncolytic reovirus therapy. Blood Adv. 2020, 4, 5297–5310. [Google Scholar] [CrossRef] [PubMed]
- Fantin, V.R.; Richon, V.M. Mechanisms of resistance to histone deacetylase inhibitors and their therapeutic implications. Clin. Cancer Res. 2007, 13, 7237–7242. [Google Scholar] [CrossRef]
- Blaszczak, W.; Liu, G.; Zhu, H.; Barczak, W.; Shrestha, A.; Albayrak, G.; Zheng, S.; Kerr, D.; Samsonova, A.; La Thangue, N.B. Immune modulation underpins the anti-cancer activity of HDAC inhibitors. Mol. Oncol. 2021, 15, 3280–3298. [Google Scholar] [CrossRef]
- Suraweera, A.; O’Byrne, K.J.; Richard, D.J. Combination therapy with histone deacetylase inhibitors (hdaci) for the treatment of cancer: Achieving the full therapeutic potential of HDACi. Front. Oncol. 2018, 8, 92. [Google Scholar] [CrossRef]
- Vancurova, I.; Uddin, M.M.; Zou, Y.; Vancura, A. Combination therapies targeting HDAC and IKK in solid tumours. Trends Pharmacol. Sci. 2018, 39, 295–306. [Google Scholar] [CrossRef]
- Mendrina, T.; Poetsch, I.; Schueffl, H.; Baier, D.; Pirker, C.; Ries, A.; Keppler, B.K.; Kowol, C.R.; Gibson, D.; Grusch, M.; et al. Influence of the fatty acid metabolism on the mode of action of a cisplatin(iv) complex with phenylbutyrate as axial ligands. Pharmaceutics 2023, 15, 677. [Google Scholar] [CrossRef] [PubMed]
- Diyabalanage, H.V.; Granda, M.L.; Hooker, J.M. Combination therapy: Histone deacetylase inhibitors and platinum-based chemotherapeutics for cancer. Cancer Lett. 2013, 329, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Li, N.; Caron, C.; Matthias, G.; Hess, D.; Khochbin, S.; Matthias, P. HDAC-6 interacts with and deacetylates tubulin and microtubules in vivo. EMBO J. 2003, 22, 1168–1179. [Google Scholar] [CrossRef] [PubMed]
- Balmik, A.A.; Chinnathambi, S. Inter-relationship of histone deacetylase-6 with tau-cytoskeletal organization and remodeling. Eur. J. Cell Biol. 2022, 101, 151202. [Google Scholar] [CrossRef] [PubMed]
- Boyault, C.; Sadoul, K.; Pabion, M.; Khochbin, S. HDAC6, at the crossroads between cytoskeleton and cell signaling by acetylation and ubiquitination. Oncogene 2007, 26, 5468–5476. [Google Scholar] [CrossRef]
- Xu, G.; Niu, L.; Wang, Y.; Yang, G.; Zhu, X.; Yao, Y.; Zhao, G.; Wang, S.; Li, H. HDAC6-dependent deacetylation of TAK1 enhances sIL-6R release to promote macrophage M2 polarization in colon cancer. Cell Death Dis. 2022, 13, 888. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.L.; Zhu, H.Y.; Zhou, B.Y.; Chu, Y.; Huo, J.R.; Tan, Y.Y.; Liu, D.L. Histone deacetylase 6 is overexpressed and promotes tumour growth of colon cancer through regulation of the MAPK/ERK signal pathway. Onco Targets Ther. 2019, 12, 2409–2419. [Google Scholar] [CrossRef]
- Mehanna, S.; Mansour, N.; Daher, C.F.; Elias, M.G.; Dagher, C.; Khnayzer, R.S. Drug-free phototherapy of superficial tumours: White light at the end of the tunnel. J. Photochem. Photobiol. B 2021, 224, 112324. [Google Scholar] [CrossRef]
- Elias, M.G.; Mehanna, S.; Elias, E.; Khnayzer, R.S.; Daher, C.F. A photoactivatable chemotherapeutic Ru(II) complex bearing bathocuproine ligand efficiently induces cell death in human malignant melanoma cells through a multi-mechanistic pathway. Chem. Biol. Interact. 2021, 348, 109644. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Platinum(IV) Complexes | UV λmax nm (ε/M.cm−1 ± SD × 104) | CD λmax nm (Δε/M.cm−1 × 101) |
|---|---|---|
| P-5B3A | 304 (0.73 ± 3.15), 279 (2.16 ± 1.63), 204 (8.08 ± 1.70) | 202 (−463), 203 (−485), 205 (−551), 228 (−58.9), 239 (−99.6), 263 (+31), 269 (+5.15), 274 (+21.3), 285 (−16.5), 313 (−32.2) |
| 5-5B3A | 310 (1.01 ± 0.73), 286 (3.63 ± 0.33), 204 (13.5 ± 3.61) | 202 (−433), 203 (−460), 204 (−490), 230 (−35.1), 240 (−65.1), 257 (+16.9), 263 (+28.6), 270 (+30.7), 286 (−53.5) |
| 56-5B3A | 315 (0.59 ± 0.94), 291 (2.02 ± 1.78), 250 (1.31 ± 2.53), 206 (8.22 ± 1.75) | 202 (−376), 204 (−384), 208 (−425), 230 (−6.55), 235 (−25.4), 239 (+7.34), 248 (+73.4), 257 (+101), 275 (+80.7), 288 (+0.91) |
| Platinum(IV) Complexes | log kw Values |
|---|---|
| P-5B3A | 1.82 |
| 5-5B3A | 1.91 |
| 56-5B3A | 2.11 |
| Platinum(IV) Complexes | Approximate Reduction Time Points (min) | |
|---|---|---|
| T50% | T100% | |
| P-5B3A | 10–15 | 60 |
| 5-5B3A | 5–10 | 30 |
| 56-5B3 | 0–5 | 10 |
| GI50 (nM) | |||||||
|---|---|---|---|---|---|---|---|
| Cell Lines | 5B3A | P-5B3A | 5-5B3A | 56-5B3A | Cisplatin | Oxaliplatin | Carboplatin |
| HT29 | >50,000 *** | 200 ± 27 ** | 18 ± 0.67 ** | 4.4 ± 1.6 ** | 11,300 ± 1900 | 900 ± 200 | >50,000 |
| U87 | >50,000 *** | 1900 ± 150 * | 240 ± 23 | 88 ± 7 * | 3800 ± 1100 | 1800 ± 200 | >50,000 |
| H460 | >50,000 **** | 430 ± 50 * | 30 ± 3 | 7.8 ± 2.2 * | 900 ± 200 | 1600 ± 100 | 14,000 ± 1000 |
| A431 | >50,000 **** | 1200 ± 100 ** | 93 ± 8 ** | 33 ± 8.4 ** | 2400 ± 300 | 4100 ± 500 | 24,000 ± 2200 |
| Du145 | >50,000 **** | 190 ± 23 ** | 10 ± 2 ** | 1.2 ± 0.6 ** | 1200 ± 100 | 2900 ± 400 | 15,000 ± 1200 |
| BE2-C | >50,000 **** | 13,000 ± 5000 ** | 340 ± 40 | 150 ± 24 ** | 1900 ± 200 | 900 ± 200 | 19,000 ± 1200 |
| SJ-G2 | >50,000 **** | 1300 ± 200 | 200 ± 40 * | 110 ± 20 | 400 ± 100 | 3000 ± 1200 | 5700 ± 200 |
| MIA | >50,000 **** | 310 ± 26 ** | 31 ± 3 ** | 12 ± 3 ** | 7500 ± 1300 | 900 ± 200 | >50,000 |
| A2780 | >50,000 ** | 470 ± 18 ** | 42 ± 4 ** | 18 ± 6.1 ** | 1000 ± 100 | 160 ± 100 | 9200 ± 2900 |
| ADDP | >50,000 **** | 330 ± 8.8 **** | 23 ± 0.88 **** | 6 ± 3 **** | 28,000 ± 1600 | 800 ± 100 | >50,000 |
| MCF-7 | >50,000 ** | 5400 ± 2800 ** | 200 ± 21 | 68 ± 10 ** | 6500 ± 800 | 500 ± 100 | >50,000 |
| MCF10A | 44,000 ± 3000 ** | 280 ± 39 ** | 19 ± 1.5 ** | 3.3 ± 0.9 ** | 5200 ± 520 | Not determined | >50,000 |
| Mean GI50 | 49,500 ± 3000 | 2084 ± 703 | 104 ± 12 | 42 ± 7 | 5842 ± 610 | 1463 ± 320 | 32,242 ± 1450 |
| GI50 (nM) | ||||||
|---|---|---|---|---|---|---|
| Cell Lines | PHENSS | 5MESS | 56MESS | PHENSS(IV)(OH)2 | 5MESS(IV)(OH)2 | 56MESS(IV)(OH)2 |
| HT29 | 160 ± 45 ** | 33 ± 4 ** | 10 ± 1.6 ** | 710 ± 300 ** | 60 ± 6 ** | 36 ± 7 ** |
| U87 | 980 ± 270 * | 320 ± 26 * | 35 ± 6.4 * | 4900 ± 610 | 900 ± 58 | 190 ± 23 * |
| H460 | 360 ± 35 * | 41 ± 5 * | 21 ± 2 * | 1700 ± 200 | 60 ± 5 * | 190 ± 150 * |
| A431 | 480 ± 170 ** | 120 ± 25 ** | 29 ± 1 ** | 4300 ± 530 * | 360 ± 58 ** | 120 ± 22 ** |
| Du145 | 100 ± 38 ** | 22 ± 3 ** | 4.6 ± 0.4 ** | 310 ± 92 ** | 41 ± 5 ** | 15 ± 2.6 ** |
| BE2-C | 380 ± 46 ** | 270 ± 38 ** | 59 ± 4 ** | 3000 ± 530 | 1400 ± 300 | 240 ± 22 ** |
| SJ-G2 | 330 ± 66 | 220 ± 10 | 66 ± 22 ** | 1700 ± 350 ** | 640 ± 70 | 210 ± 45 * |
| MIA | 200 ± 57 ** | 48 ± 2 ** | 13 ± 2 ** | 3400 ± 2200 | 160 ± 29 ** | 43 ± 2.5 ** |
| A2780 | 230 ± 30 ** | 61 ± 10 ** | 76 ± 57 ** | 800 ± 84 | 240 ± 9 ** | 59 ± 7 ** |
| ADDP | 190 ± 47 **** | 34 ± 2 **** | 13 ± 2 **** | 1300 ± 350 **** | 130 ± 22 **** | 170 ± 120 **** |
| MCF-7 | 1500 ± 500 ** | 200 ± 12 ** | 93 ± 44 ** | 16,000 ± 4500 | 1200 ± 390 ** | 480 ± 140 ** |
| MCF10A | 300 ± 58 ** | 30 ± 2 ** | 16 ± 1 ** | 1700 ± 200 | 130 ± 19 ** | 61 ± 7 ** |
| Mean GI50 | 434 ± 110 | 117 ± 12 | 36 ± 10 | 3318 ± 880 | 443 ± 81 | 151 ± 50 |
| Cell Lines | Platinum Complexes | |||
|---|---|---|---|---|
| P-5B3A | 5-5B3A | 56-5B3A | Cisplatin | |
| HT29 | 1.40 | 1.06 | 0.75 | 0.46 |
| U87 | 0.15 | 0.08 | 0.04 | 1.37 |
| MCF-7 | 0.05 | 0.10 | 0.05 | 0.80 |
| H460 | 0.65 | 0.63 | 0.42 | 5.78 |
| A431 | 0.23 | 0.20 | 0.10 | 2.17 |
| Du145 | 1.47 | 1.90 | 2.75 | 4.33 |
| BE2-C | 0.02 | 0.06 | 0.02 | 2.74 |
| SJ-G2 | 0.22 | 0.10 | 0.03 | 13.00 |
| MIA | 0.90 | 0.61 | 0.28 | 0.69 |
| A2780 | 0.60 | 0.45 | 0.18 | 5.20 |
| ADDP | 0.85 | 0.83 | 0.55 | 0.19 |
| Compounds and Platinum Complexes | ROS Production * at Different Time Intervals (RFU) | ||
|---|---|---|---|
| 24 h | 48 h | 72 h | |
| Control | 60 ± 3 | 63 ± 4 | 85 ± 6 |
| Cisplatin | 265 ± 19 | 299 ± 22 | 364 ± 18 |
| (Tert-butyl hydroperoxide) TBHP | 614 ± 21 | 477 ± 23 | 311 ± 18 |
| PHENSS | 174 ± 2 | 172 ± 9 | 176 ± 7 |
| 5MESS | 204 ± 4 | 205 ± 3 | 188 ± 3 |
| 56MESS | 240 ± 5 | 218 ± 3 | 255 ± 4 |
| PHENSS(IV)(OH)2 | 144 ± 5 | 273 ± 4 | 303 ± 1 |
| 5MESS(IV)(OH)2 | 234 ± 1 | 323 ± 9 | 335 ± 2 |
| 56MESS(IV)(OH)2 | 259 ± 3 | 356 ± 11 | 438 ± 7 |
| 5B3A | 138 ± 8 | 153 ± 6 | 164 ± 4 |
| P-5B3A | 156 ± 16 | 214 ± 12 | 246 ± 11 |
| 5-5B3A | 160 ± 11 | 230 ± 9 | 315 ± 14 |
| 56-5B3A | 274 ± 9 | 418 ± 15 | 474 ± 10 |
| Compounds and Platinum Complexes | MtMP Changes * at Different Time Intervals (RFU) | ||
|---|---|---|---|
| 24 h | 48 h | 72 h | |
| Control | 542 ± 4 | 507 ± 11 | 449 ± 10 |
| Cisplatin | 232 ± 4 | 230 ± 4 | 192 ± 7 |
| Carbonyl cyanide-p-trifluoromethoxyphenylhydrazone (FCCP) | 234 ± 3 | 151 ± 7 | 84 ± 5 |
| PHENSS | 321 ± 7 | 305 ± 5 | 257 ± 6 |
| 5MESS | 283 ± 4 | 261 ± 9 | 189 ± 11 |
| 56MESS | 277 ± 5 | 211 ± 7 | 151 ± 7 |
| PHENSS(IV)(OH)2 | 328 ± 7 | 314 ± 6 | 234 ± 11 |
| 5MESS(IV)(OH)2 | 311 ± 3 | 291 ± 9 | 228 ± 7 |
| 56MESS(IV)(OH)2 | 291 ± 9 | 221 ± 5 | 188 ± 5 |
| 5B3A | 489 ± 6 | 415 ± 5 | 327 ± 9 |
| P-5B3A | 332 ± 7 | 317 ± 7 | 263 ± 5 |
| 5-5B3A | 324 ± 9 | 280 ± 12 | 184 ± 4 |
| 56-5B3A | 321 ± 11 | 267 ± 8 | 148 ± 7 |
| Compounds and Complexes | HDAC Inhibition * at 72 h |
|---|---|
| HT29 extract | 5154 ± 40 |
| PHENSS | 3733 ± 37 |
| 5MESS | 2124 ± 21 |
| 56MESS | 1847 ± 42 |
| PHENSS(IV)(OH)2 | 4107 ± 19 |
| 5MESS(IV)(OH)2 | 3119 ± 9 |
| 56MESS(IV)(OH)2 | 3011 ± 23 |
| 5B3A | 3942 ± 28 |
| P-5B3A | 3620 ± 35 |
| 5-5B3A | 2791 ± 72 |
| 56-5B3A | 2550 ± 57 |
| Cisplatin | 3698 ± 59 |
| No Enzyme Control | 617 ± 3 |
| HeLa Nuclear Extract | 6697 ± 55 |
| Trichostatin A (TSA) | 749 ± 8 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aputen, A.D.; Elias, M.G.; Gilbert, J.; Sakoff, J.A.; Gordon, C.P.; Scott, K.F.; Aldrich-Wright, J.R. Platinum(IV) Prodrugs Incorporating an Indole-Based Derivative, 5-Benzyloxyindole-3-Acetic Acid in the Axial Position Exhibit Prominent Anticancer Activity. Int. J. Mol. Sci. 2024, 25, 2181. https://doi.org/10.3390/ijms25042181
Aputen AD, Elias MG, Gilbert J, Sakoff JA, Gordon CP, Scott KF, Aldrich-Wright JR. Platinum(IV) Prodrugs Incorporating an Indole-Based Derivative, 5-Benzyloxyindole-3-Acetic Acid in the Axial Position Exhibit Prominent Anticancer Activity. International Journal of Molecular Sciences. 2024; 25(4):2181. https://doi.org/10.3390/ijms25042181
Chicago/Turabian StyleAputen, Angelico D., Maria George Elias, Jayne Gilbert, Jennette A. Sakoff, Christopher P. Gordon, Kieran F. Scott, and Janice R. Aldrich-Wright. 2024. "Platinum(IV) Prodrugs Incorporating an Indole-Based Derivative, 5-Benzyloxyindole-3-Acetic Acid in the Axial Position Exhibit Prominent Anticancer Activity" International Journal of Molecular Sciences 25, no. 4: 2181. https://doi.org/10.3390/ijms25042181
APA StyleAputen, A. D., Elias, M. G., Gilbert, J., Sakoff, J. A., Gordon, C. P., Scott, K. F., & Aldrich-Wright, J. R. (2024). Platinum(IV) Prodrugs Incorporating an Indole-Based Derivative, 5-Benzyloxyindole-3-Acetic Acid in the Axial Position Exhibit Prominent Anticancer Activity. International Journal of Molecular Sciences, 25(4), 2181. https://doi.org/10.3390/ijms25042181