
Evaluation of Antitumor Activity of Xanthones Conjugated with Amino Acids

 ,
, 
 , ,
, ,  ,
,  ,
,  ,
,  ,
,  ,
,  and
and 
Abstract
1. Introduction
2. Results and Discussion
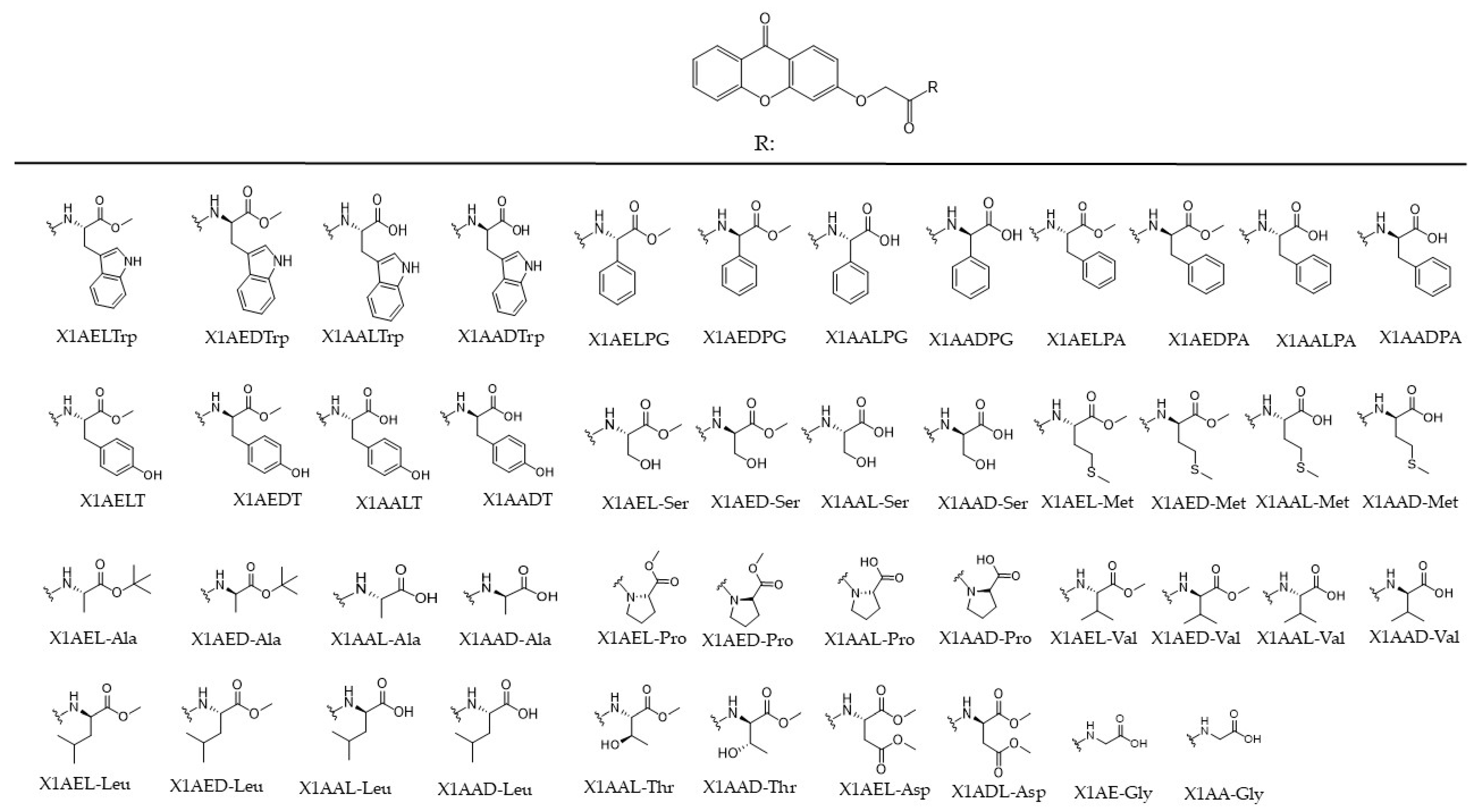
2.1. Effect of Chiral Derivatives of Xanthones on the Growth of Human Tumor and Non-Tumor Cell Lines
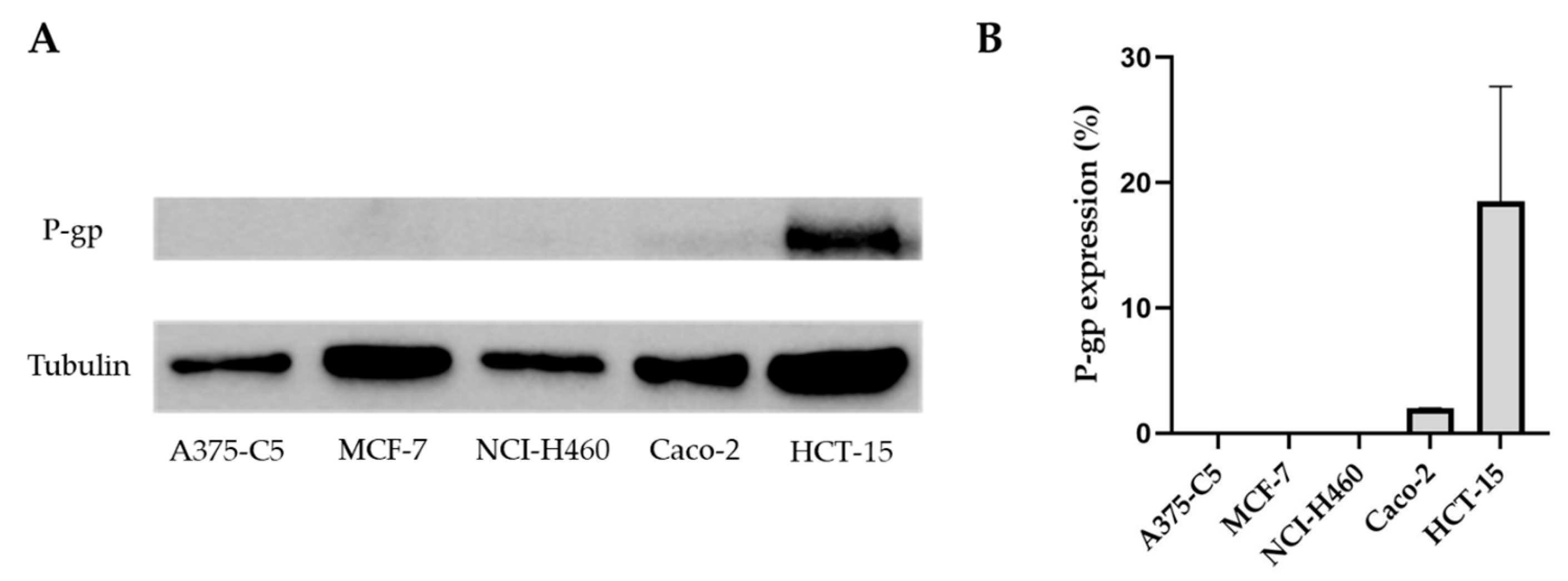
2.2. Pgp Expression in the Tumor Cell Lines A375-C5, MCF-7, NCI-H460, Caco-2, and HCT-15
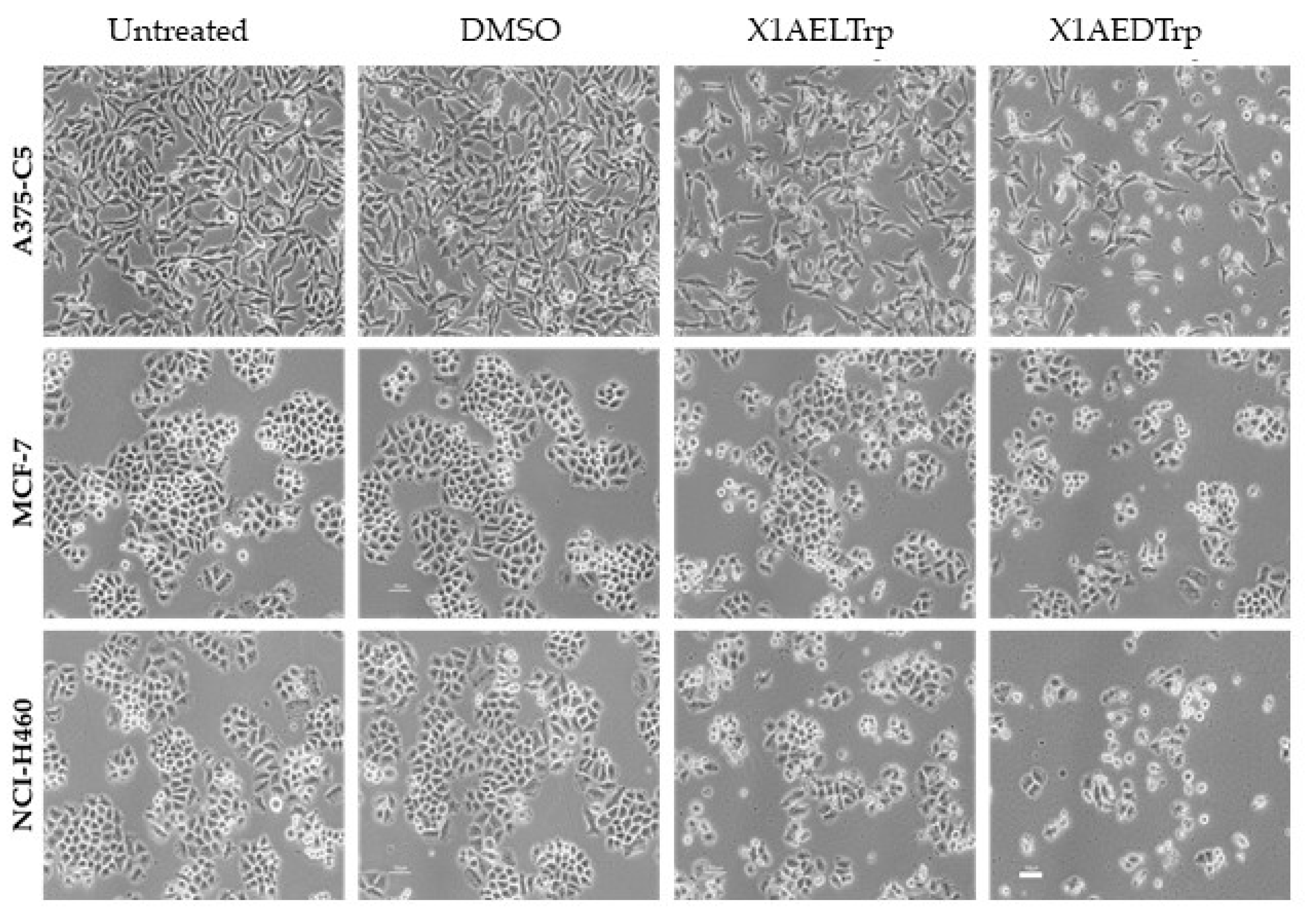
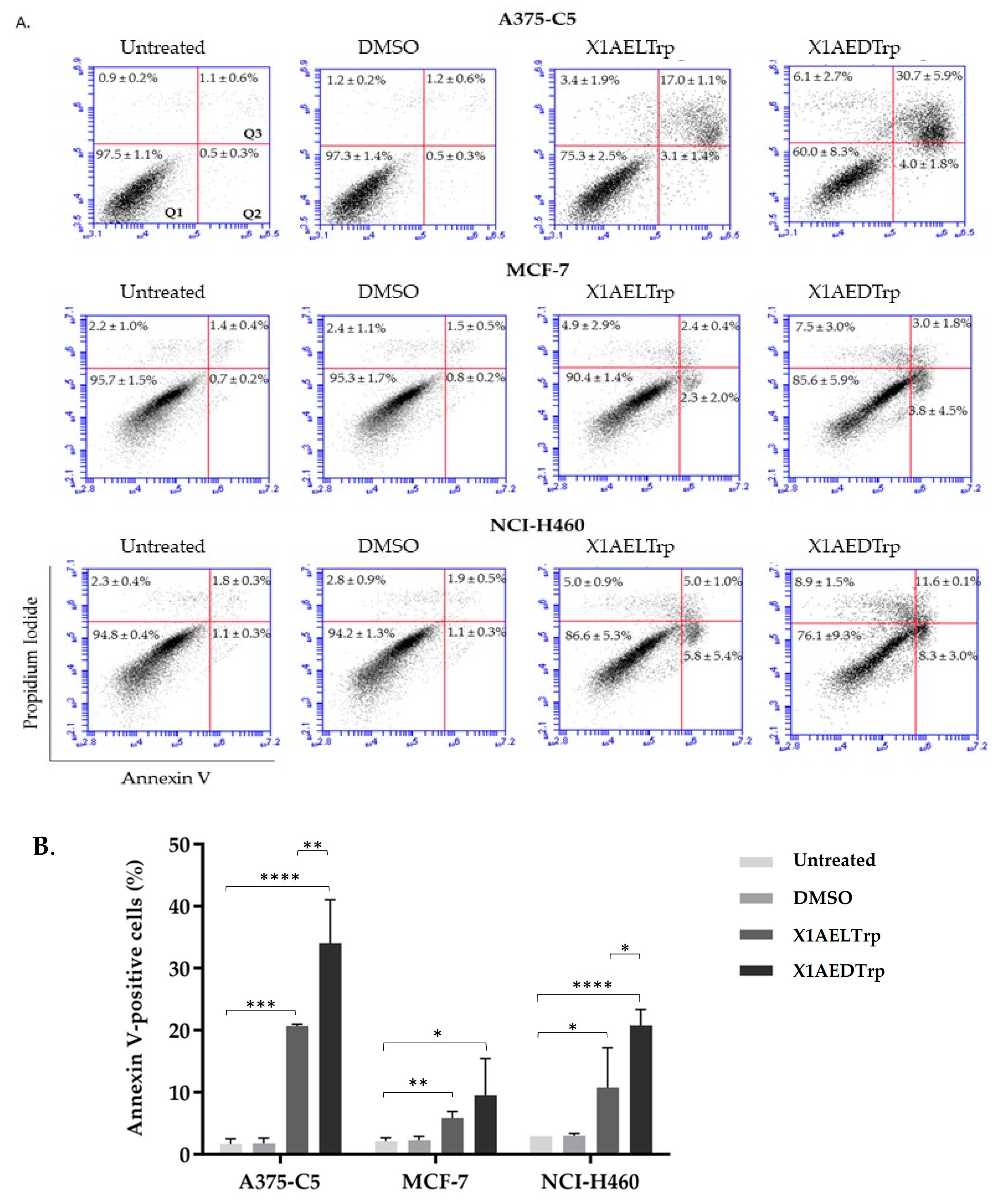
2.3. Mechanism of Cell Death, Induced by the Most Promising CDXs, X1AELTrp and X1AEDTrp
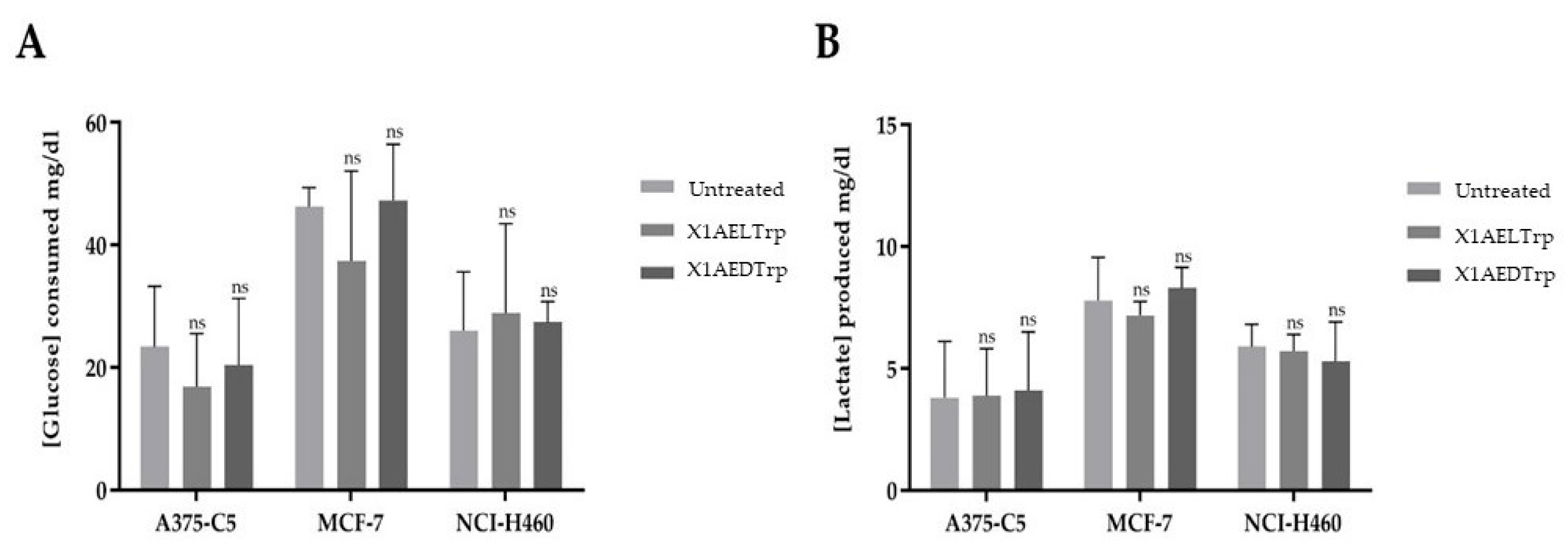
2.4. Alterations in Cancer Cell Metabolism, Induced by the Most Promising CDXs, X1AELTrp and X1AEDTrp
2.5. Evaluation of the Functionality of the Multidrug Resistance Protein Pgp Based on a Yeast Strain Expressing the Human MDR1 Gene
2.6. Phenotypic Evaluation of the Growth of the Saccharomyces Cerevisiae Yeast Strains AD1-8MDR and AD1-8GPD in the Presence of Different Concentrations of the CDXs X1AELTrp and X1AEDTrp
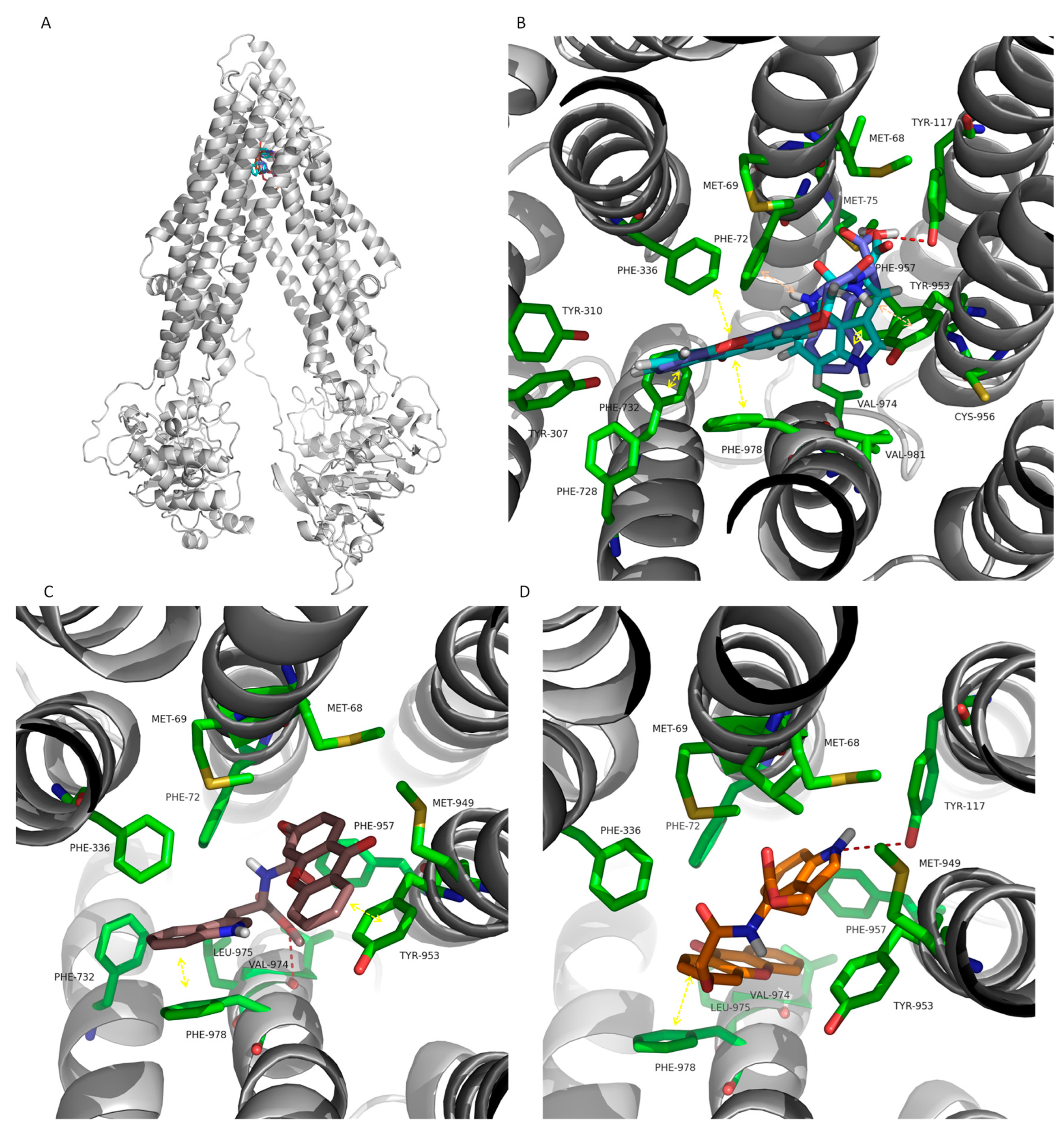
2.7. Docking Scores of the CDXs and Positive Controls Based on the Drug-Binding Pocket of the Human Inward Pgp Model
3. Materials and Methods
3.1. Chemicals
3.2. Cell Culture
3.3. Cell Viability Assays
3.4. Annexin V/Propidium Iodide Staining
3.5. Image Processing
3.6. Pgp Expression Evaluation
3.7. Metabolic Assays: Determination of Extracellular Levels of Glucose and Lactate
3.8. Yeast Strains and Media
3.9. Evaluation of CDXs Effect on the Growth of S. cerevisiae Strains, Expressing or Not Expressing Pgp
3.10. Statistical Analysis
3.11. Docking Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Anand, U.; Dey, A.; Chandel, A.K.S.; Sanyal, R.; Mishra, A.; Pandey, D.K.; De Falco, V.; Upadhyay, A.; Kandimalla, R.; Chaudhary, A.; et al. Cancer chemotherapy and beyond: Current status, drug candidates, associated risks and progress in targeted therapeutics. Genes Dis. 2023, 10, 1367–1401. [Google Scholar] [CrossRef]
- Barreca, M.; Spanò, V.; Rocca, R.; Bivacqua, R.; Gualtieri, G.; Raimondi, M.V.; Gaudio, E.; Bortolozzi, R.; Manfreda, L.; Bai, R.; et al. Identification of pyrrolo[3′,4′:3,4]cyclohepta[1,2-d][1,2]oxazoles as promising new candidates for the treatment of lymphomas. Eur. J. Med. Chem. 2023, 254, 115372. [Google Scholar] [CrossRef]
- Rogova, A.; Gorbunova, I.A.; Karpov, T.E.; Sidorov, R.Y.; Rubtsov, A.E.; Shipilovskikh, D.A.; Muslimov, A.R.; Zyuzin, M.V.; Timin, A.S.; Shipilovskikh, S.A. Synthesis of thieno[3,2-e]pyrrolo[1,2-a]pyrimidine derivatives and their precursors containing 2-aminothiophenes fragments as anticancer agents for therapy of pulmonary metastatic melanoma. Eur. J. Med. Chem. 2023, 254, 115325. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Chao, T.; Liu, Y.; Gong, C.; Zhang, Y.; Xiong, H. Heterocyclic Molecular Targeted Drugs and Nanomedicines for Cancer: Recent Advances and Challenges. Pharmaceutics 2023, 15, 1706. [Google Scholar] [CrossRef]
- Zhang, Y.; Wu, C.; Zhang, N.; Fan, R.; Ye, Y.; Xu, J. Recent Advances in the Development of Pyrazole Derivatives as Anticancer Agents. Int. J. Mol. Sci. 2023, 24, 12724. [Google Scholar] [CrossRef]
- Hossain, M.; Habib, I.; Singha, K.; Kumar, A. FDA-approved heterocyclic molecules for cancer treatment: Synthesis, dosage, mechanism of action and their adverse effect. Heliyon 2024, 10, e23172. [Google Scholar] [CrossRef] [PubMed]
- Valentová, J.; Lintnerová, L.; Miklášová, N.; Oboňová, B.; Habala, L. Analogues of Anticancer Natural Products: Chiral Aspects. Int. J. Mol. Sci. 2023, 24, 5679. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, C.; Palmeira, A.; Ramos, I.I.; Carneiro, C.; Afonso, C.; Tiritan, M.E.; Cidade, H.; Pinto, P.; Saraiva, M.; Reis, S.; et al. Chiral Derivatives of Xanthones: Investigation of the Effect of Enantioselectivity on Inhibition of Cyclooxygenases (COX-1 and COX-2) and Binding Interaction with Human Serum Albumin. Pharmaceuticals 2017, 10, 50. [Google Scholar] [CrossRef] [PubMed]
- Araújo, J.; Fernandes, C.; Pinto, M.; Tiritan, M. Chiral Derivatives of Xanthones with Antimicrobial Activity. Molecules 2019, 24, 314–343. [Google Scholar] [CrossRef]
- Durães, F.; Cravo, S.; Freitas-Silva, J.; Szemerédi, N.; Martins-Da-costa, P.; Pinto, E.; Tiritan, M.E.; Spengler, G.; Fernandes, C.; Sousa, E.; et al. Enantioselectivity of chiral derivatives of xanthones in virulence effects of resistant bacteria. Pharmaceuticals 2021, 14, 1141. [Google Scholar] [CrossRef]
- Pinto, M.M.M.; Palmeira, A.; Fernandes, C.; Resende, D.I.S.P.; Sousa, E.; Cidade, H.; Tiritan, M.E.; Correia-Da-silva, M.; Cravo, S. From natural products to new synthetic small molecules: A journey through the world of xanthones. Molecules 2021, 26, 431. [Google Scholar] [CrossRef]
- Gogoi, U.; Pathak, K.; Saikia, R.; Pathak, M.P.; Paul, T.; Khan, S.A.; Das, A. Recent Advances on Natural and Non-Natural Xanthones as Potential Anticancer Agents: A Review. Med. Chem. 2023, 19, 757–784. [Google Scholar] [CrossRef]
- Fernandes, C.; Masawang, K.; Tiritan, M.E.; Sousa, E.; de Lima, V.; Afonso, C.; Bousbaa, H.; Sudprasert, W.; Pedro, M.; Pinto, M. New chiral derivatives of xanthones: Synthesis and investigation of enantioselectivity as inhibitors of growth of human tumor cell lines. Bioorg. Med. Chem. 2014, 22, 1049–1062. [Google Scholar] [CrossRef]
- Vieira, S.F.; Araújo, J.; Gonçalves, V.M.F.; Fernandes, C.; Pinto, M.; Ferreira, H.; Neves, N.M.; Tiritan, M.E. Synthesis and Anti-Inflammatory Evaluation of a Library of Chiral Derivatives of Xanthones Conjugated with Proteinogenic Amino Acids. Int. J. Mol. Sci. 2023, 24, 10357. [Google Scholar] [CrossRef]
- Alam, A.; Kowal, J.; Broude, E.; Roninson, I.; Locher, K.P. Structural insight into substrate and inhibitor discrimination by human P-glycoprotein. Science 2019, 363, 753–756. [Google Scholar] [CrossRef] [PubMed]
- Hancu, G.; Modroiu, A. Chiral Switch: Between Therapeutical Benefit and Marketing Strategy. Pharmaceuticals 2022, 15, 240. [Google Scholar] [CrossRef] [PubMed]
- Calcaterra, A.; D’Acquarica, I. The market of chiral drugs: Chiral switches versus de novo enantiomerically pure compounds. J. Pharm. Biomed. Anal. 2018, 147, 323–340. [Google Scholar] [CrossRef] [PubMed]
- Rossi, D.; Tarantino, M.; Rossino, G.; Rui, M.; Juza, M.; Collina, S. Approaches for multi-gram scale isolation of enantiomers for drug discovery. Expert Opin. Drug. Discov. 2017, 12, 1253–1269. [Google Scholar] [CrossRef] [PubMed]
- Carraro, M.L.; Marques, S.; Silva, A.S.; Freitas, B.; Silva, P.M.A.; Pedrosa, J.; De Marco, P.; Bousbaa, H.; Fernandes, C.; Tiritan, M.E.; et al. Synthesis of New Chiral Derivatives of Xanthones with Enantioselective Effect on Tumor Cell Growth and DNA Crosslinking. ChemistrySelect 2020, 5, 10285–10291. [Google Scholar] [CrossRef]
- Fernandes, C.; Carraro, M.L.; Ribeiro, J.; Araujo, J.; Tiritan, M.E.; Pinto, M.M.M. Synthetic Chiral Derivatives of Xanthones: Biological Activities and Enantioselectivity Studies. Molecules 2019, 24, 791. [Google Scholar] [CrossRef]
- Xu, Q.; Deng, H.; Li, X.; Quan, Z.S. Application of Amino Acids in the Structural Modification of Natural Products: A Review. Front. Chem. 2021, 9, 650569. [Google Scholar] [CrossRef]
- Arifian, H.; Maharani, R.; Megantara, S.; Gazzali, A.M.; Muchtaridi, M. Amino-Acid-Conjugated Natural Compounds: Aims, Designs and Results. Molecules 2022, 27, 7631. [Google Scholar] [CrossRef]
- Rakesh, K.P.; Darshini, N.; Manukumar, H.M.; Vivek, H.K.; Eissa, M.Y.H.; Prasanna, D.S.; Mallesha, N. Xanthone conjugated amino acids as potential anticancer and DNA binding agents: Molecular docking, cytotoxicity and SAR studies. Anti-Cancer Agents Med. Chem. 2018, 18, 2169–2177. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef]
- Karthika, C.; Sureshkumar, R.; Zehravi, M.; Akter, R.; Ali, F.; Ramproshad, S.; Mondal, B.; Tagde, P.; Ahmed, Z.; Khan, F.S.; et al. Multidrug Resistance of Cancer Cells and the Vital Role of P-Glycoprotein. Life 2022, 12, 897. [Google Scholar] [CrossRef] [PubMed]
- Ambudkar, S.V.; Kimchi-Sarfaty, C.; Sauna, Z.E.; Gottesman, M.M. P-glycoprotein: From genomics to mechanism. Oncogene 2003, 22, 7468–7485. [Google Scholar] [CrossRef] [PubMed]
- Jia, C.C.; Xue, J.J.; Gong, C.; Li, X.Y.; Li, D.H.; Li, Z.L.; Hua, H.M. Chiral resolution and anticancer effect of xanthones from Garcinia paucinervis. Fitoterapia 2018, 127, 220–225. [Google Scholar] [CrossRef]
- Lee, S.Y.; Mojulat, M.B.C.; Thangaperagasam, G.J.C.; Surugau, N.; Tan, S.A.; John, O.D. A Review on the Cytotoxic and Antimicrobial Properties of Xanthones from Cratoxylum cochinchinense. J. Trop. Life Sci. 2023, 13, 213–224. [Google Scholar] [CrossRef]
- Rech, J.; Sypniewski, D.; Żelaszczyk, D.; Szkaradek, N.; Rogóż, W.; Waszkielewicz, A.; Marona, H.; Bednarek, I. Novel xanthone derivatives impair growth and invasiveness of colon cancer cells in vitro. Biomolecules 2021, 11, 1480. [Google Scholar] [CrossRef]
- Lu, Y.; Guan, T.; Wang, S.; Zhou, C.; Wang, M.; Wang, X.; Zhang, K.; Han, X.; Lin, J.; Tang, Q.; et al. Novel xanthone antibacterials: Semi-synthesis, biological evaluation, and the action mechanisms. Bioorg. Med. Chem. 2023, 83, 117232. [Google Scholar] [CrossRef]
- Kalick, L.S.; Khan, H.A.; Maung, E.; Baez, Y.; Atkinson, A.N.; Wallace, C.E.; Day, F.; Delgadillo, B.E.; Mondal, A.; Watanapokasin, R.; et al. Mangosteen for malignancy prevention and intervention: Current evidence, molecular mechanisms, and future perspectives. Pharmacol. Res. 2023, 188, 106630. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Wang, S.; Ouyang, Y.; Tu, Y.; Liu, A.; Tian, Y.; He, M.; Pi, R. Garcinone D, a natural xanthone promotes C17.2 neural stem cell proliferation: Possible involvement of STAT3/Cyclin D1 pathway and Nrf2/HO-1 pathway. Neurosci. Lett. 2016, 626, 6–12. [Google Scholar] [CrossRef]
- Chang, J.; Hou, S.; Yan, X.; Li, W.; Xiao, J. Discovery of Novel STING Inhibitors Based on the Structure of the Mouse STING Agonist DMXAA. Molecules 2023, 28, 2906. [Google Scholar] [CrossRef] [PubMed]
- Gunter, N.V.; Teh, S.S.; Lim, Y.M.; Mah, S.H. Natural Xanthones and Skin Inflammatory Diseases: Multitargeting Mechanisms of Action and Potential Application. Front. Pharmacol. 2020, 11, 594202. [Google Scholar] [CrossRef]
- Kitaeva, K.V.; Rutland, C.S.; Rizvanov, A.A.; Solovyeva, V.V. Cell Culture Based in vitro Test Systems for Anticancer Drug Screening. Front. Bioeng. Biotechnol. 2020, 8, 322. [Google Scholar] [CrossRef]
- Novais, P.; Silva, P.M.A.; Moreira, J.; Palmeira, A.; Amorim, I.; Pinto, M.; Cidade, H.; Bousbaa, H. BP-M345, a new diarylpentanoid with promising antimitotic activity. Molecules 2021, 26, 7139. [Google Scholar] [CrossRef]
- Crowley, L.C.; Marfell, B.J.; Scott, A.P.; Waterhouse, N.J. Quantitation of apoptosis and necrosis by annexin V binding, propidium iodide uptake, and flow cytometry. Cold Spring Harbor Protoc. 2016, 2016, 953–957. [Google Scholar] [CrossRef] [PubMed]
- Costigan, A.; Hollville, E.; Martin, S.J. Discriminating Between Apoptosis, Necrosis, Necroptosis, and Ferroptosis by Microscopy and Flow Cytometry. Curr. Protoc. 2023, 3, e951. [Google Scholar] [CrossRef] [PubMed]
- Kotopka, B.J.; Smolke, C.D. Model-driven generation of artificial yeast promoters. Nat. Commun. 2020, 11, 2113. [Google Scholar] [CrossRef] [PubMed]
- Goffeau, A.; Barrell, G.; Bussey, H.; Davis, R.W.; Dujon, B.; Feldmann, H.; Galibert, F.; Hoheisel, J.D.; Jacq, C.; Johnston, M.; et al. Life with 6000 genes. Science 1996, 274, 546–567. [Google Scholar] [CrossRef] [PubMed]
- Pereira, C.; Coutinho, I.; Soares, J.; Bessa, C.; Leão, M.; Saraiva, L. New insights into cancer-related proteins provided by the yeast model. FEBS J. 2012, 279, 697–712. [Google Scholar] [CrossRef] [PubMed]
- Cannon, R.D.; Lamping, E.; Holmes, A.R.; Niimi, K.; Baret, P.V.; Keniya, M.V.; Tanabe, K.; Niimi, M.; Goffeau, A.; Monk, B.C. Efflux-mediated antifungal drug resistance. Clin. Microbiol. Rev. 2009, 22, 291–321. [Google Scholar] [CrossRef]
- Decottignies, A.; Grant, A.M.; Nichols, J.W.; De Wet, H.; McIntosh, D.B.; Goffeau, A. ATPase and multidrug transport activities of the overexpressed yeast ABC protein Yor1p. J. Biol. Chem. 1998, 273, 12612–12622. [Google Scholar] [CrossRef]
- Rogers, B.; Decottignies, A.; Kolaczkowski, M.; Carvajal, E.; Balzi, E.; Goffeau, A. The pleitropic drug ABC transporters from Saccharomyces cerevisiae. J. Mol. Microbiol. Biotechnol. 2001, 3, 207–214. [Google Scholar]
- Kim, R.B. Drugs as P-glycoprotein substrates, inhibitors, and inducers. Drug Metab. Rev. 2002, 34, 47–54. [Google Scholar] [CrossRef]
- Zhou, S.F. Structure, function and regulation of P-glycoprotein and its clinical relevance in drug disposition. Xenobiotica 2008, 38, 802–832. [Google Scholar] [CrossRef]
- Kanaoka, S.; Kimura, Y.; Fujikawa, M.; Nakagawa, Y.; Ueda, K.; Akamatsu, M. Substrate recognition by P-glycoprotein efflux transporters: Structure-ATPase activity relationship of diverse chemicals and agrochemicals. J. Pestic. Sci. 2013, 38, 112–122. [Google Scholar] [CrossRef]
- De Lange, E.C.M. Multi Drug Resistance P Glycoprotein and other Transporters. In Encyclopedia of Stress; Elsevier Inc.: Amsterdam, The Netherlands, 2007; pp. 774–783. [Google Scholar]
- Seelig, A. P-Glycoprotein: One Mechanism, Many Tasks and the Consequences for Pharmacotherapy of Cancers. Front. Oncol. 2020, 10, 576559. [Google Scholar] [CrossRef]
- Ahmed Juvale, I.I.; Abdul Hamid, A.A.; Abd Halim, K.B.; Che Has, A.T. P-glycoprotein: New insights into structure, physiological function, regulation and alterations in disease. Heliyon 2022, 8, e09777. [Google Scholar] [CrossRef]
- Palmeira, A.; Sousa, E.; Vasconcelos, M.H.; Pinto, M.M. Three decades of P-gp Inhibitors: Skimming through several generations and scaffolds. Curr. Med. Chem. 2012, 19, 1946–2025. [Google Scholar] [CrossRef]
- Silva, V.; Gil-Martins, E.; Rocha-Pereira, C.; Lemos, A.; Palmeira, A.; Puthongking, P.; Sousa, E.; de Lourdes Bastos, M.; Remião, F.; Silva, R. Oxygenated xanthones as P-glycoprotein modulators at the intestinal barrier: In vitro and docking studies. Med. Chem. Res. 2020, 29, 1041–1057. [Google Scholar] [CrossRef]
- Silva, V.; Gil-Martins, E.; Silva, B.; Rocha-Pereira, C.; Sousa, M.E.; Remião, F.; Silva, R. Xanthones as P-glycoprotein modulators and their impact on drug bioavailability. Expert Opin. Drug Metab. Toxicol. 2021, 17, 441–482. [Google Scholar] [CrossRef] [PubMed]
- Syed, S.B.; Arya, H.; Fu, I.H.; Yeh, T.K.; Periyasamy, L.; Hsieh, H.P.; Coumar, M.S. Targeting P-glycoprotein: Investigation of piperine analogs for overcoming drug resistance in cancer. Sci. Rep. 2017, 7, 7972. [Google Scholar] [CrossRef] [PubMed]
- Loo, T.W.; Clarke, D.M. Functional consequences of phenylalanine mutations in the predicted transmembrane domain of P-glycoprotein. J. Biol. Chem. 1993, 268, 19965–19972. [Google Scholar] [CrossRef] [PubMed]
- Gozalpour, E.; Wittgen, H.G.M.; van den Heuvel, J.J.M.W.; Greupink, R.; Russel, F.G.M.; Koenderink, J.B. Interaction of digitalis-like compounds with P-glycoprotein. Toxicol. Sci. 2013, 131, 502–511. [Google Scholar] [CrossRef] [PubMed]
- Lagares, L.M.; Pérez-castillo, Y.; Minovski, N.; Novič, M. Structure–function relationships in the human p-glycoprotein (Abcb1): Insights from molecular dynamics simulations. Int. J. Mol. Sci. 2022, 23, 362. [Google Scholar] [CrossRef] [PubMed]
- Tarcsay, A.; Keseru, G.M. Homology modeling and binding site assessment of the human P-glycoprotein. Future Med. Chem. 2011, 3, 297–307. [Google Scholar] [CrossRef]
- Nosol, K.; Romane, K.; Irobalieva, R.N.; Alam, A.; Kowal, J.; Fujita, N.; Locher, K.P. Cryo-EM structures reveal distinct mechanisms of inhibition of the human multidrug transporter ABCB1. Proc. Natl. Acad. Sci. USA 2020, 117, 26245–26253. [Google Scholar] [CrossRef]
- Mumberg, D.; Müller, R.; Funk, M. Yeast vectors for the controlled expression of heterologous proteins in different genetic backgrounds. Gene 1995, 156, 119–122. [Google Scholar] [CrossRef] [PubMed]
- Froimowitz, M. HyperChem(TM): A software package for computational chemistry and molecular modeling. Biotechniques 1993, 14, 1010–1013. [Google Scholar] [PubMed]
- Morris, G.M.; Huey, R.; Olson, A.J. UNIT using AutoDock for ligand-receptor docking. Curr. Protoc. Bioinform. 2008, 24, 8.14.1-8.14.40. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. Software news and update AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Martins, E.; Silva, V.; Lemos, A.; Palmeira, A.; Puthongking, P.; Sousa, E.; Rocha-Pereira, C.; Ghanem, C.I.; Carmo, H.; Remião, F.; et al. Newly synthesized oxygenated xanthones as potential P-glycoprotein activators: In vitro, ex vivo, and in silico studies. Molecules 2019, 24, 707. [Google Scholar] [CrossRef]
- Rigsby, R.E.; Parker, A.B. Using the PyMOL application to reinforce visual understanding of protein structure. Biochem. Mol. Biol. Educ. 2016, 44, 433–437. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
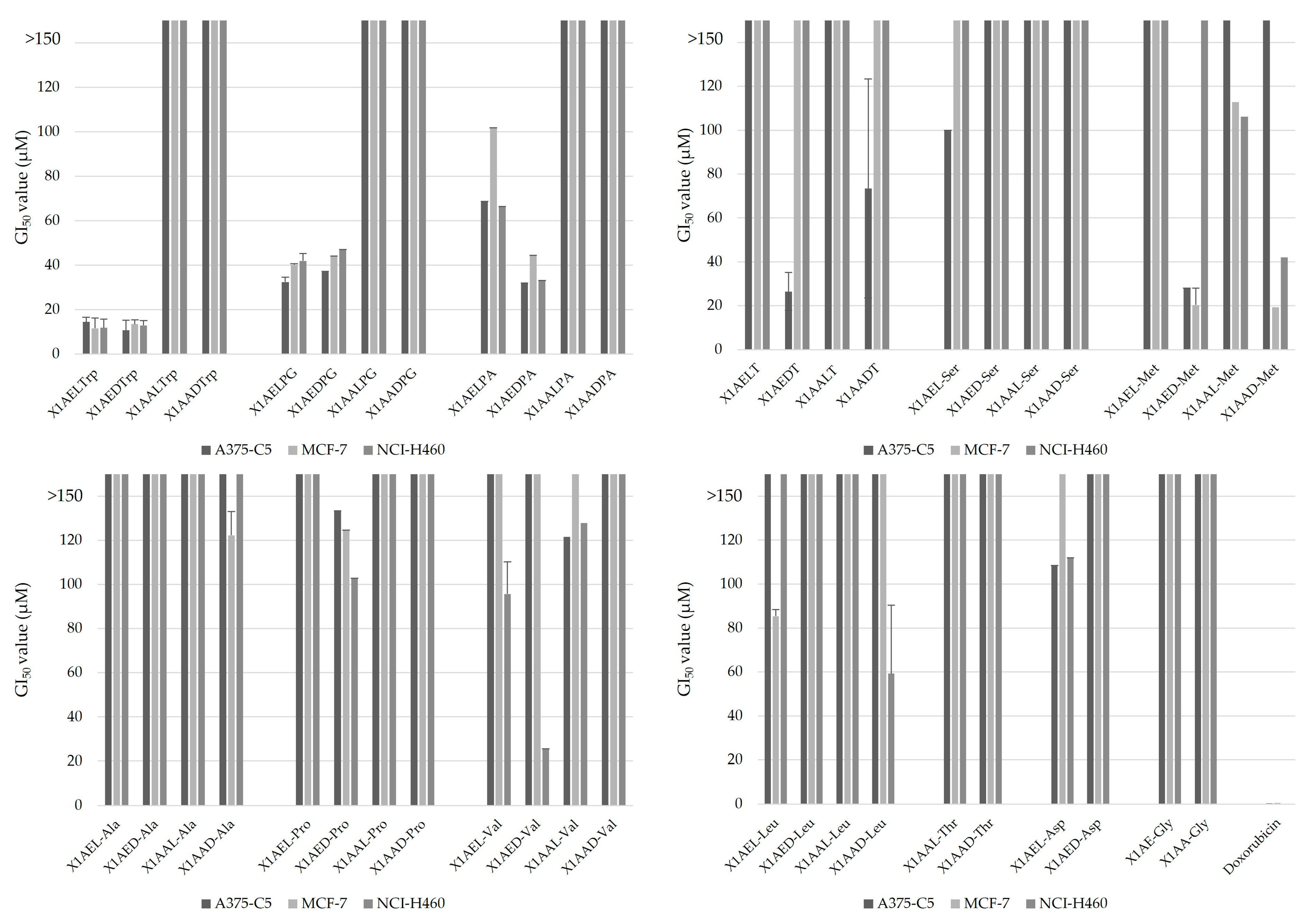
| GI50 (µM) | |||
|---|---|---|---|
| Compounds | A375-C5 | MCF-7 | NCI-H460 |
| X1AELTrp | 14.46 ± 2.10 | 11.61 ± 4.63 | 11.82 ± 3.97 |
| X1AEDTrp | 10.66 ± 4.32 | 13.52 ± 1.94 | 12.94 ± 2.12 |
| X1AELPG | 32.42 ± 0.25 | 40.08 ± 7.16 | 41.94 ± 1.04 |
| X1AEDPG | 37.16 ± 2.17 | 44.06 ± 0.56 | 46.95 ± 3.27 |
| X1AEDPA | 32.03 ± 5.64 | 44.43 ± 8.26 | 33.13 ± 6.72 |
| X1AEDT | 26.52 ± 8.71 | >150 | >150 |
| X1AED-Met | 28.05 ± 7.85 | 20.35 ± 4.6 | >150 |
| X1AAD-Met | >150 | 19.33 ± 7.75 | 42.1 ± 35.1 |
| X1AED-Val | >150 | >150 | 25.6 ± 7.19 |
| Doxorubicin | 0.41 ± 0.097 | 0.47 ± 0.22 | 0.35 ± 0.05 |
| GI50 (μM) 1 | Selectivity Index 2 | |||
|---|---|---|---|---|
| HPAepiC | A375-C5 | MCF-7 | NCI-H460 | |
| X1AELTrp | 40.90 ± 1.17 | 2.83 | 3.52 | 3.46 |
| X1AEDTrp | 43.58 ± 3.32 | 4.09 | 3.22 | 3.37 |
| Doxorubicin | 0.056 ± 0.013 | 0.14 | 0.12 | 0.16 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barbosa, F.; Araújo, J.; Gonçalves, V.M.F.; Palmeira, A.; Cunha, A.; Silva, P.M.A.; Fernandes, C.; Pinto, M.; Bousbaa, H.; Queirós, O.; et al. Evaluation of Antitumor Activity of Xanthones Conjugated with Amino Acids. Int. J. Mol. Sci. 2024, 25, 2121. https://doi.org/10.3390/ijms25042121
Barbosa F, Araújo J, Gonçalves VMF, Palmeira A, Cunha A, Silva PMA, Fernandes C, Pinto M, Bousbaa H, Queirós O, et al. Evaluation of Antitumor Activity of Xanthones Conjugated with Amino Acids. International Journal of Molecular Sciences. 2024; 25(4):2121. https://doi.org/10.3390/ijms25042121
Chicago/Turabian StyleBarbosa, Flávia, Joana Araújo, Virgínia M. F. Gonçalves, Andreia Palmeira, Andrea Cunha, Patrícia M. A. Silva, Carla Fernandes, Madalena Pinto, Hassan Bousbaa, Odília Queirós, and et al. 2024. "Evaluation of Antitumor Activity of Xanthones Conjugated with Amino Acids" International Journal of Molecular Sciences 25, no. 4: 2121. https://doi.org/10.3390/ijms25042121
APA StyleBarbosa, F., Araújo, J., Gonçalves, V. M. F., Palmeira, A., Cunha, A., Silva, P. M. A., Fernandes, C., Pinto, M., Bousbaa, H., Queirós, O., & Tiritan, M. E. (2024). Evaluation of Antitumor Activity of Xanthones Conjugated with Amino Acids. International Journal of Molecular Sciences, 25(4), 2121. https://doi.org/10.3390/ijms25042121