

A Novel Drosophila Model of Alzheimer’s Disease to Study Aβ Proteotoxicity in the Digestive Tract


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
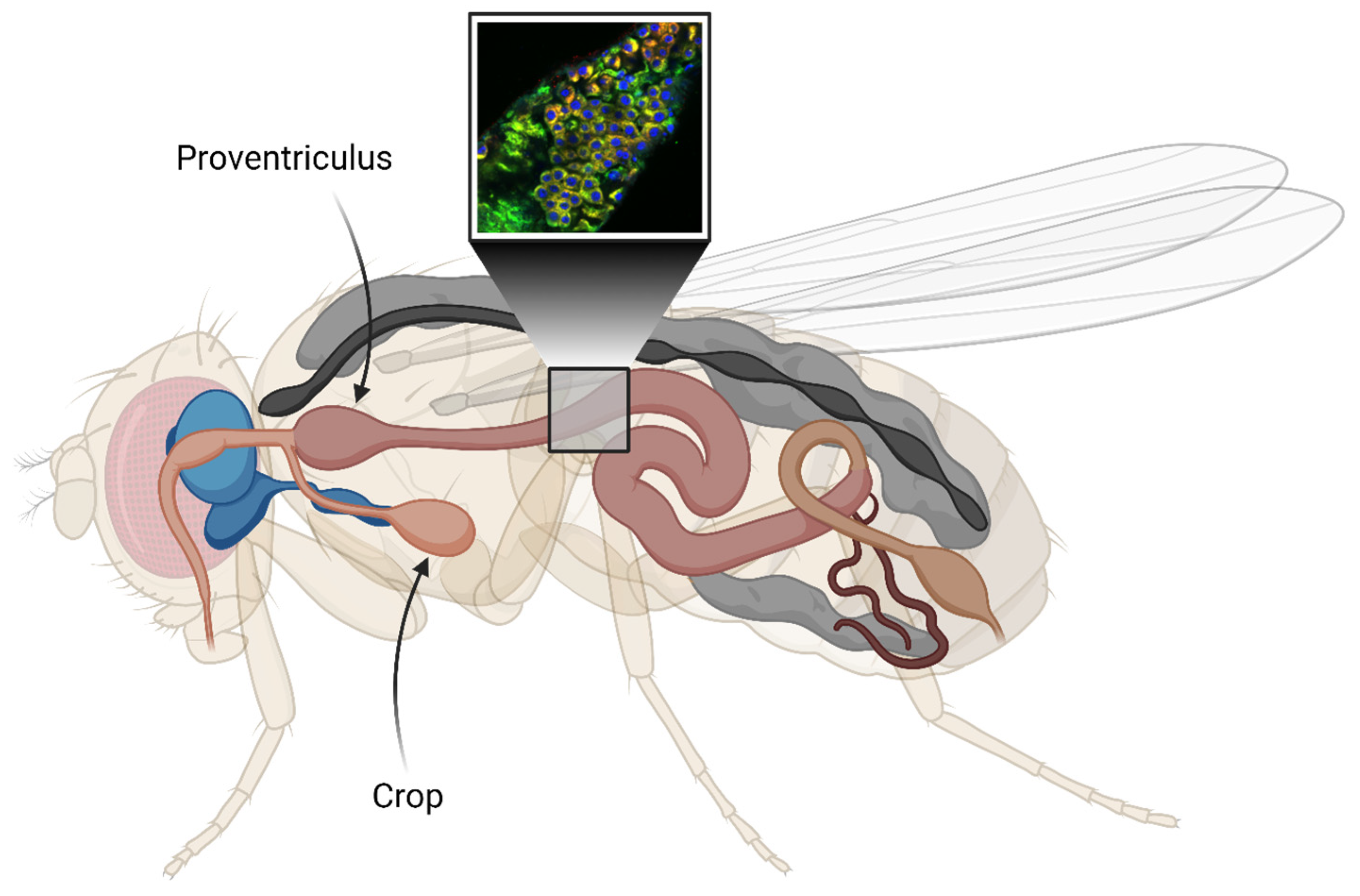
2. Results
2.1. Shortened Lifespan in Aβ-Expressing Flies
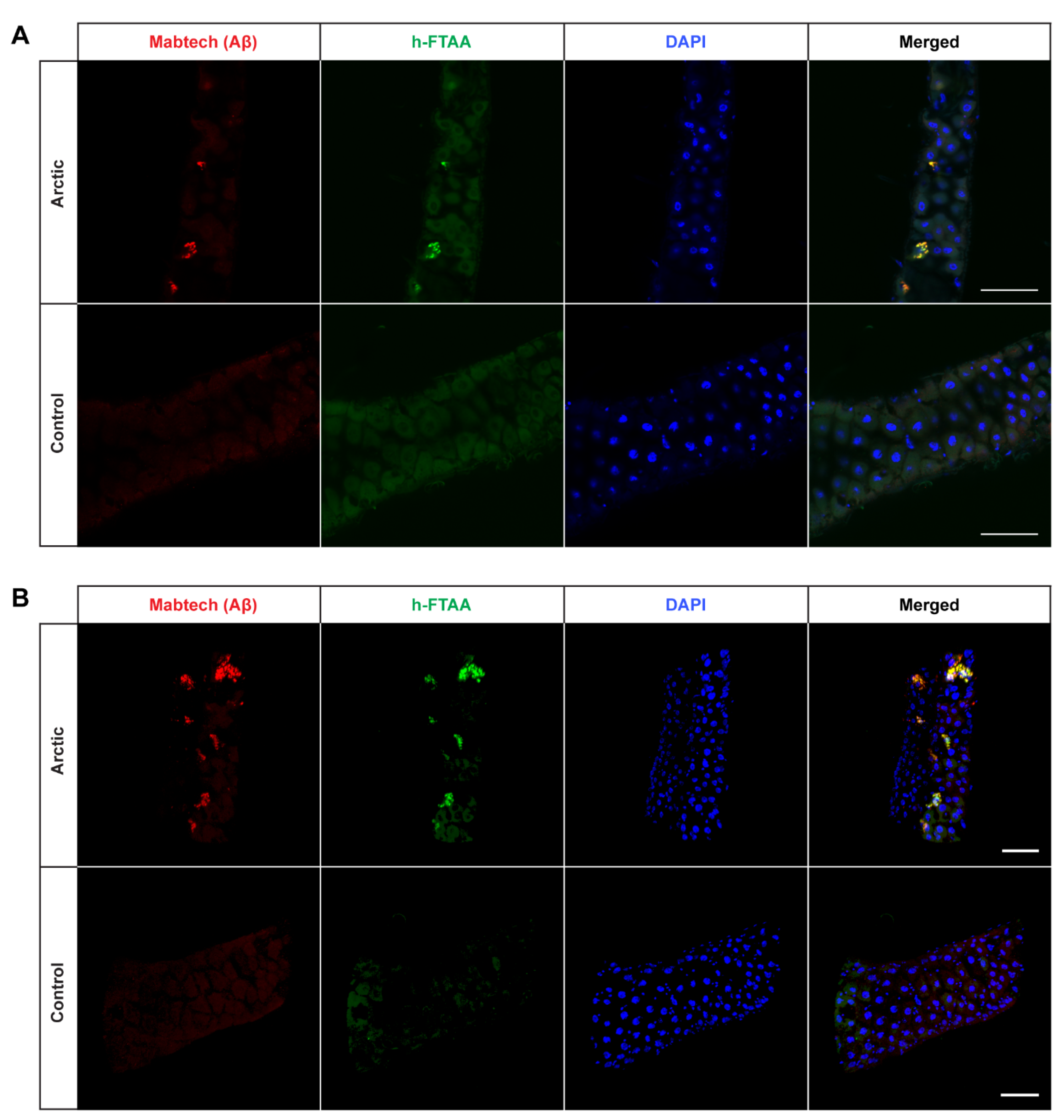
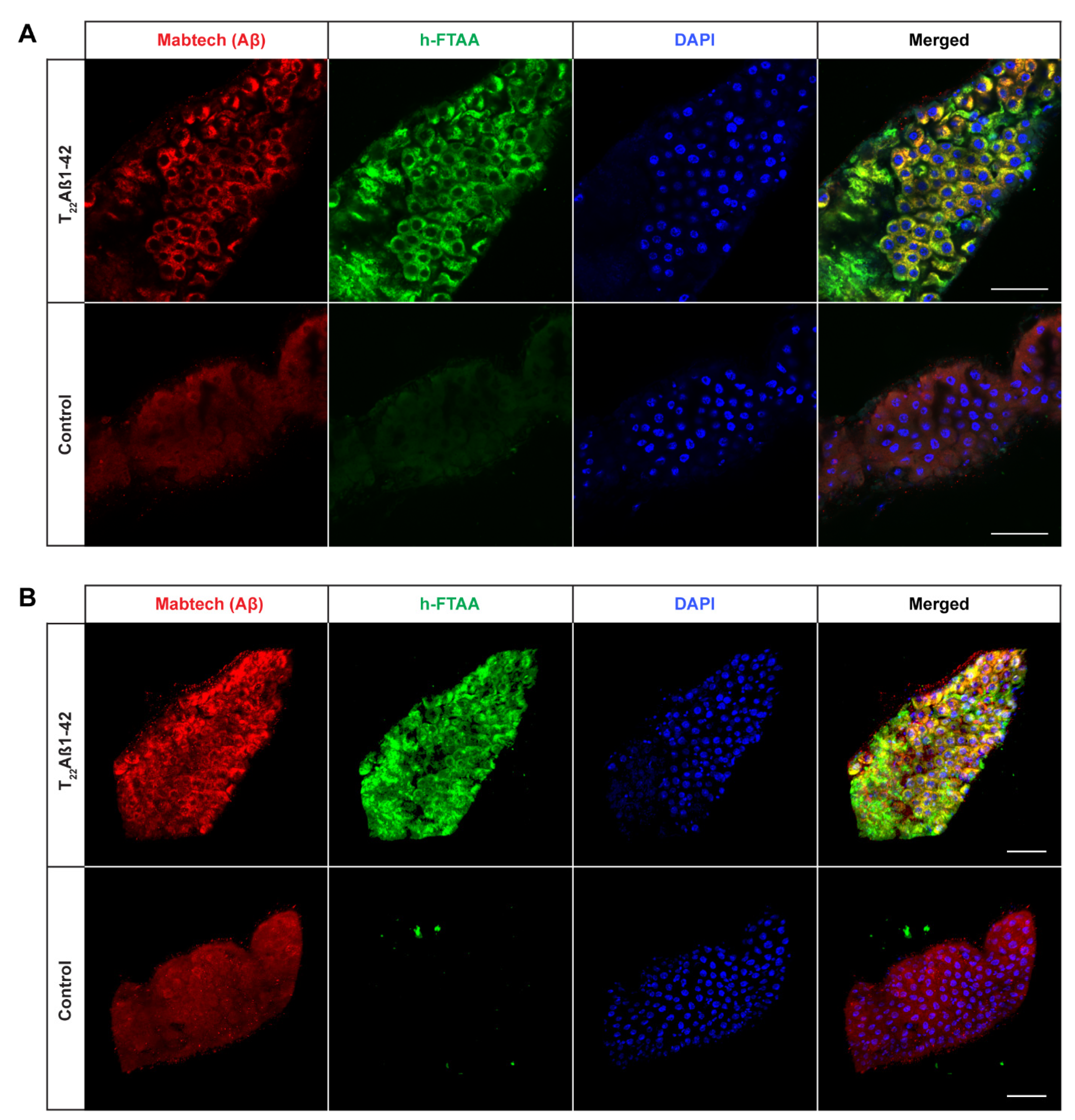
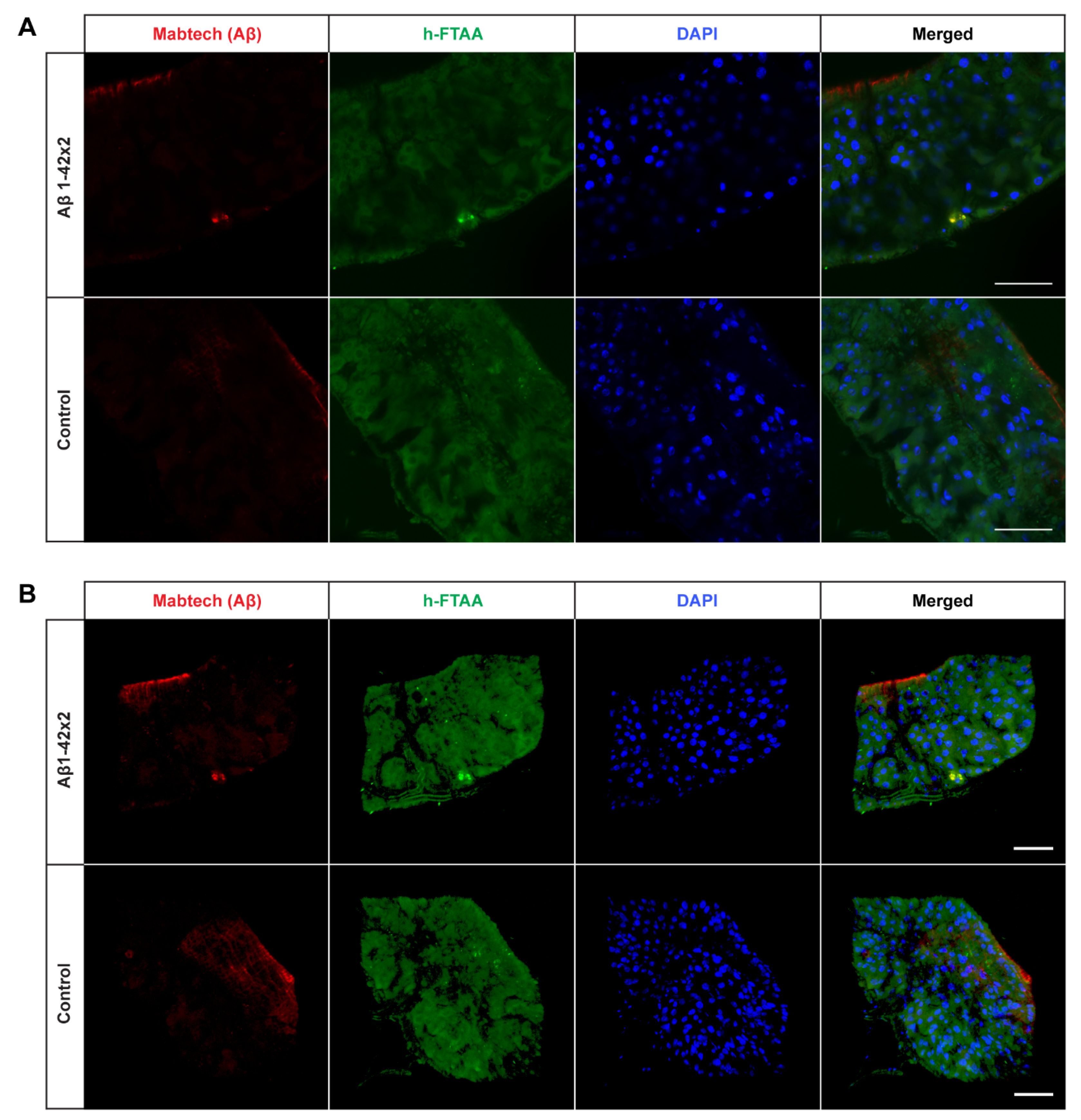
2.2. Detection of Aggregates in Aβ-Expressing Flies
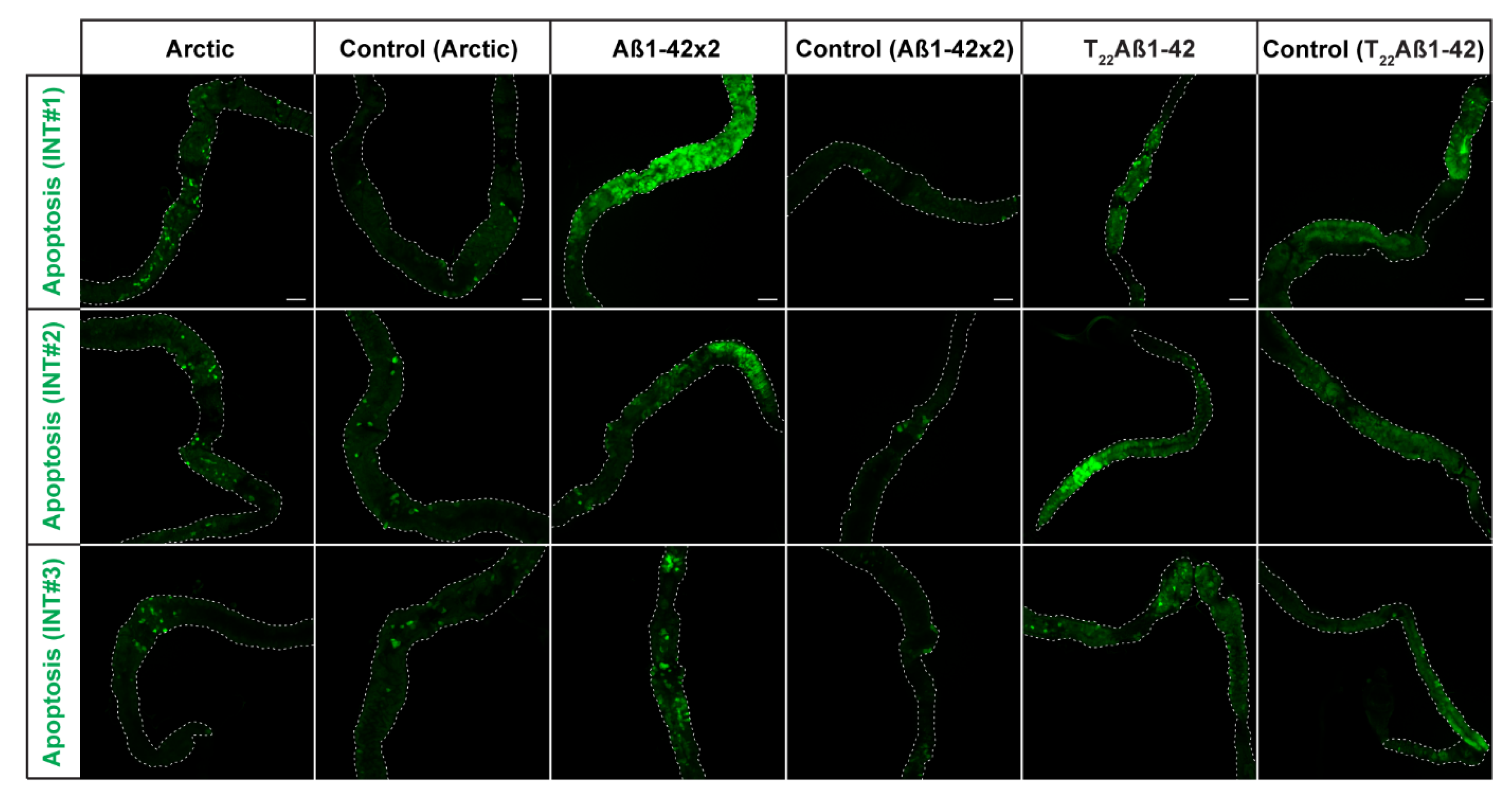
2.3. Increased Number of Apoptotic Cells in Aβ-Expressing Flies
3. Discussion
4. Materials and Methods
4.1. Drosophila Stocks
4.2. Longevity Assay
4.3. Dissection
4.4. Antibody and Ligand Double Staining
4.5. Apoptotic Assay
4.6. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Uysal, G.; Ozturk, M. Hippocampal Atrophy Based Alzheimer’s Disease Diagnosis via Machine Learning Methods. J. Neurosci. Methods 2020, 337, 108669. [Google Scholar] [CrossRef] [PubMed]
- Alzheimer’s Association. 2016 Alzheimer’s Disease Facts and Figures. Alzheimers Dement. J. Alzheimers Assoc. 2016, 12, 459–509. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, R.H.; Nagao, T.; Gouras, G.K. Plaque Formation and the Intraneuronal Accumulation of β-Amyloid in Alzheimer’s Disease. Pathol. Int. 2017, 67, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Thal, D.R.; Walter, J.; Saido, T.C.; Fändrich, M. Neuropathology and Biochemistry of Aβ and Its Aggregates in Alzheimer’s Disease. Acta Neuropathol. 2015, 129, 167–182. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.A.; Higgins, G.A. Alzheimer’s Disease: The Amyloid Cascade Hypothesis. Science 1992, 256, 184–185. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J. The Molecular Pathology of Alzheimer’s Disease. Neuron 1991, 6, 487–498. [Google Scholar] [CrossRef] [PubMed]
- Karran, E.; Mercken, M.; De Strooper, B. The Amyloid Cascade Hypothesis for Alzheimer’s Disease: An Appraisal for the Development of Therapeutics. Nat. Rev. Drug Discov. 2011, 10, 698–712. [Google Scholar] [CrossRef]
- Citron, M.; Teplow, D.B.; Selkoe, D.J. Generation of Amyloid β Protein from Its Precursor Is Sequence Specific. Neuron 1995, 14, 661–670. [Google Scholar] [CrossRef]
- Arbor, S.C.; LaFontaine, M.; Cumbay, M. Amyloid-Beta Alzheimer Targets—Protein Processing, Lipid Rafts, and Amyloid-Beta Pores. Yale J. Biol. Med. 2016, 89, 5–21. [Google Scholar]
- Dahlgren, K.N.; Manelli, A.M.; Stine, W.B.; Baker, L.K.; Krafft, G.A.; LaDu, M.J. Oligomeric and Fibrillar Species of Amyloid-β Peptides Differentially Affect Neuronal Viability. J. Biol. Chem. 2002, 277, 32046–32053. [Google Scholar] [CrossRef]
- Iijima, K.; Liu, H.-P.; Chiang, A.-S.; Hearn, S.A.; Konsolaki, M.; Zhong, Y. Dissecting the Pathological Effects of Human Aβ40 and Aβ42 in Drosophila: A Potential Model for Alzheimer’s Disease. Proc. Natl. Acad. Sci. USA 2004, 101, 6623–6628. [Google Scholar] [CrossRef]
- Nilsberth, C.; Westlind-Danielsson, A.; Eckman, C.B.; Condron, M.M.; Axelman, K.; Forsell, C.; Stenh, C.; Luthman, J.; Teplow, D.B.; Younkin, S.G.; et al. The “Arctic” APP Mutation (E693G) Causes Alzheimer’s Disease by Enhanced Aβ Protofibril Formation. Nat. Neurosci. 2001, 4, 887–893. [Google Scholar] [CrossRef] [PubMed]
- Iversen, L.L.; Mortishire-Smith, R.J.; Pollack, S.J.; Shearman, M.S. The Toxicity in Vitro of Beta-Amyloid Protein. Biochem. J. 1995, 311, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Törnquist, M.; Michaels, T.C.T.; Sanagavarapu, K.; Yang, X.; Meisl, G.; Cohen, S.I.A.; Knowles, T.P.J.; Linse, S. Secondary Nucleation in Amyloid Formation. Chem. Commun. Camb. Engl. 2018, 54, 8667–8684. [Google Scholar] [CrossRef] [PubMed]
- Walsh, D.M.; Selkoe, D.J. A Beta Oligomers—A Decade of Discovery. J. Neurochem. 2007, 101, 1172–1184. [Google Scholar] [CrossRef]
- Speretta, E.; Jahn, T.R.; Tartaglia, G.G.; Favrin, G.; Barros, T.P.; Imarisio, S.; Lomas, D.A.; Luheshi, L.M.; Crowther, D.C.; Dobson, C.M. Expression in Drosophila of Tandem Amyloid β Peptides Provides Insights into Links between Aggregation and Neurotoxicity. J. Biol. Chem. 2012, 287, 20748–20754. [Google Scholar] [CrossRef] [PubMed]
- Bolus, H.; Crocker, K.; Boekhoff-Falk, G.; Chtarbanova, S. Modeling Neurodegenerative Disorders in Drosophila melanogaster. Int. J. Mol. Sci. 2020, 21, 3055. [Google Scholar] [CrossRef]
- Elovsson, G.; Bergkvist, L.; Brorsson, A.-C. Exploring Aβ Proteotoxicity and Therapeutic Candidates Using Drosophila melanogaster. Int. J. Mol. Sci. 2021, 22, 10448. [Google Scholar] [CrossRef]
- Brand, A.H.; Perrimon, N. Targeted Gene Expression as a Means of Altering Cell Fates and Generating Dominant Phenotypes. Dev. Camb. Engl. 1993, 118, 401–415. [Google Scholar] [CrossRef]
- Berger, C.; Renner, S.; Lüer, K.; Technau, G.M. The Commonly Used Marker ELAV Is Transiently Expressed in Neuroblasts and Glial Cells in the Drosophila Embryonic CNS. Dev. Dyn. 2007, 236, 3562–3568. [Google Scholar] [CrossRef]
- Crowther, D.C.; Kinghorn, K.J.; Miranda, E.; Page, R.; Curry, J.A.; Duthie, F.A.I.; Gubb, D.C.; Lomas, D.A. Intraneuronal Aβ, non-amyloid Aggregates and neurodegeneration in a drosophila model of alzheimer’s disease. Neuroscience 2005, 132, 123–135. [Google Scholar] [CrossRef]
- Osterwalder, T.; Yoon, K.S.; White, B.H.; Keshishian, H. A Conditional Tissue-Specific Transgene Expression System Using Inducible GAL4. Proc. Natl. Acad. Sci. USA 2001, 98, 12596–12601. [Google Scholar] [CrossRef] [PubMed]
- Bergkvist, L.; Sandin, L.; Kågedal, K.; Brorsson, A.-C. AβPP Processing Results in Greater Toxicity per Amount of Aβ1-42 than Individually Expressed and Secreted Aβ1-42 in Drosophila Melanogaster. Biol. Open 2016, 5, 1030–1039. [Google Scholar] [CrossRef] [PubMed]
- Caesar, I.; Jonson, M.; Nilsson, K.P.R.; Thor, S.; Hammarström, P. Curcumin Promotes A-Beta Fibrillation and Reduces Neurotoxicity in Transgenic Drosophila. PLoS ONE 2012, 7, e31424. [Google Scholar] [CrossRef] [PubMed]
- Bergkvist, L.; Du, Z.; Elovsson, G.; Appelqvist, H.; Itzhaki, L.S.; Kumita, J.R.; Kågedal, K.; Brorsson, A. Mapping Pathogenic Processes Contributing to Neurodegeneration in Drosophila Models of Alzheimer’s Disease. FEBS Open Bio 2020, 10, 338–350. [Google Scholar] [CrossRef] [PubMed]
- Schott, S.; Ambrosini, A.; Barbaste, A.; Benassayag, C.; Gracia, M.; Proag, A.; Rayer, M.; Monier, B.; Suzanne, M. A Fluorescent Toolkit for Spatiotemporal Tracking of Apoptotic Cells in Living Drosophila Tissues. Dev. Camb. Engl. 2017, 144, 3840–3846. [Google Scholar] [CrossRef] [PubMed]
- Iijima, K.; Chiang, H.-C.; Hearn, S.A.; Hakker, I.; Gatt, A.; Shenton, C.; Granger, L.; Leung, A.; Iijima-Ando, K.; Zhong, Y. Aβ42 Mutants with Different Aggregation Profiles Induce Distinct Pathologies in Drosophila. PLoS ONE 2008, 3, e1703. [Google Scholar] [CrossRef]
- Jonson, M.; Pokrzywa, M.; Starkenberg, A.; Hammarstrom, P.; Thor, S. Systematic Aβ Analysis in Drosophila Reveals High Toxicity for the 1-42, 3-42 and 11-42 Peptides, and Emphasizes N- and C-Terminal Residues. PLoS ONE 2015, 10, e0133272. [Google Scholar] [CrossRef]
- Buchon, N.; Osman, D.; David, F.P.A.; Fang, H.Y.; Boquete, J.-P.; Deplancke, B.; Lemaitre, B. Morphological and Molecular Characterization of Adult Midgut Compartmentalization in Drosophila. Cell Rep. 2013, 3, 1725–1738. [Google Scholar] [CrossRef]
- Buchon, N.; Broderick, N.A.; Poidevin, M.; Pradervand, S.; Lemaitre, B. Drosophila Intestinal Response to Bacterial Infection: Activation of Host Defense and Stem Cell Proliferation. Cell Host Microbe 2009, 5, 200–211. [Google Scholar] [CrossRef]
- Chakrabarti, S.; Liehl, P.; Buchon, N.; Lemaitre, B. Infection-Induced Host Translational Blockage Inhibits Immune Responses and Epithelial Renewal in the Drosophila Gut. Cell Host Microbe 2012, 12, 60–70. [Google Scholar] [CrossRef]
- Sarkissian, T.; Timmons, A.; Arya, R.; Abdelwahid, E.; White, K. Detecting Apoptosis in Drosophila Tissues and Cells. Methods 2014, 68, 89–96. [Google Scholar] [CrossRef] [PubMed]
- Klingstedt, T.; Aslund, A.; Simon, R.A.; Johansson, L.B.G.; Mason, J.J.; Nyström, S.; Hammarström, P.; Nilsson, K.P.R. Synthesis of a Library of Oligothiophenes and Their Utilization as Fluorescent Ligands for Spectral Assignment of Protein Aggregates. Org. Biomol. Chem. 2011, 9, 8356–8370. [Google Scholar] [CrossRef] [PubMed]
- Nyström, S.; Psonka-Antonczyk, K.M.; Ellingsen, P.G.; Johansson, L.B.G.; Reitan, N.; Handrick, S.; Prokop, S.; Heppner, F.L.; Wegenast-Braun, B.M.; Jucker, M.; et al. Evidence for Age-Dependent in vivo Conformational Rearrangement within Aβ Amyloid Deposits. ACS Chem. Biol. 2013, 8, 1128–1133. [Google Scholar] [CrossRef] [PubMed]
- Sandin, L.; Bergkvist, L.; Nath, S.; Kielkopf, C.; Janefjord, C.; Helmfors, L.; Zetterberg, H.; Blennow, K.; Li, H.; Nilsberth, C.; et al. Beneficial Effects of Increased Lysozyme Levels in Alzheimer’s Disease Modelled in Drosophila Melanogaster. FEBS J. 2016, 283, 3508–3522. [Google Scholar] [CrossRef] [PubMed]
- Miguel-Aliaga, I.; Jasper, H.; Lemaitre, B. Anatomy and Physiology of the Digestive Tract of Drosophila Melanogaster. Genetics 2018, 210, 357–396. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, M.; Yoshida, H. Drosophila as a Model Organism. Adv. Exp. Med. Biol. 2018, 1076, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Murakami, K.; Irie, K.; Morimoto, A.; Ohigashi, H.; Shindo, M.; Nagao, M.; Shimizu, T.; Shirasawa, T. Synthesis, Aggregation, Neurotoxicity, and Secondary Structure of Various Aβ1-42 Mutants of Familial Alzheimer’s Disease at Positions 21–23. Biochem. Biophys. Res. Commun. 2002, 294, 5–10. [Google Scholar] [CrossRef]
- Lee, S.J.C.; Nam, E.; Lee, H.J.; Savelieff, M.G.; Lim, M.H. Towards an Understanding of Amyloid-β Oligomers: Characterization, Toxicity Mechanisms, and Inhibitors. Chem. Soc. Rev. 2017, 46, 310–323. [Google Scholar] [CrossRef]
- Hardy, J.; Selkoe, D.J. The Amyloid Hypothesis of Alzheimer’s Disease: Progress and Problems on the Road to Therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef]
- Luheshi, L.M.; Tartaglia, G.G.; Brorsson, A.-C.; Pawar, A.P.; Watson, I.E.; Chiti, F.; Vendruscolo, M.; Lomas, D.A.; Dobson, C.M.; Crowther, D.C. Systematic in vivo Analysis of the Intrinsic Determinants of Amyloid Beta Pathogenicity. PLoS Biol. 2007, 5, e290. [Google Scholar] [CrossRef] [PubMed]
- Nikoletopoulou, V.; Markaki, M.; Palikaras, K.; Tavernarakis, N. Crosstalk between Apoptosis, Necrosis and Autophagy. Biochim. Biophys. Acta 2013, 1833, 3448–3459. [Google Scholar] [CrossRef]
- D’Arcy, M.S. Cell Death: A Review of the Major Forms of Apoptosis, Necrosis and Autophagy. Cell Biol. Int. 2019, 43, 582–592. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Edgar, B.A. Intestinal Stem Cells in the Adult Drosophila Midgut. Exp. Cell Res. 2011, 317, 2780–2788. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Patel, P.H.; Kohlmaier, A.; Pavlovic, B.; Zhang, C.; Edgar, B.A. Intestinal Stem Cell Pool Regulation in Drosophila. Stem Cell Rep. 2017, 8, 1479–1487. [Google Scholar] [CrossRef] [PubMed]
- Luheshi, L.M.; Hoyer, W.; de Barros, T.P.; van Dijk Härd, I.; Brorsson, A.-C.; Macao, B.; Persson, C.; Crowther, D.C.; Lomas, D.A.; Ståhl, S.; et al. Sequestration of the Aβ Peptide Prevents Toxicity and Promotes Degradation in vivo. PLoS Biol. 2010, 8, e1000334. [Google Scholar] [CrossRef] [PubMed]
- Helmfors, L.; Boman, A.; Civitelli, L.; Nath, S.; Sandin, L.; Janefjord, C.; McCann, H.; Zetterberg, H.; Blennow, K.; Halliday, G.; et al. Protective Properties of Lysozyme on β-Amyloid Pathology: Implications for Alzheimer Disease. Neurobiol. Dis. 2015, 83, 122–133. [Google Scholar] [CrossRef]
- Pinsonneault, R.L.; Mayer, N.; Mayer, F.; Tegegn, N.; Bainton, R.J. Novel Models for Studying the Blood-Brain and Blood-Eye Barriers in Drosophila. Methods Mol. Biol. 2011, 686, 357–369. [Google Scholar] [CrossRef]
- Brankatschk, M.; Eaton, S. Lipoprotein Particles Cross the Blood-Brain Barrier in Drosophila. J. Neurosci. Off. J. Soc. Neurosci. 2010, 30, 10441–10447. [Google Scholar] [CrossRef]
- Schirmeier, S.; Klämbt, C. The Drosophila Blood-Brain Barrier as Interface between Neurons and Hemolymph. Mech. Dev. 2015, 138, 50–55. [Google Scholar] [CrossRef]
- Limmer, S.; Weiler, A.; Volkenhoff, A.; Babatz, F.; Klämbt, C. The Drosophila Blood-Brain Barrier: Development and Function of a Glial Endothelium. Front. Neurosci. 2014, 8, 365. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.L.; Yue, Z.; Arnold, D.M.; Artiushin, G.; Sehgal, A. A Circadian Clock in the Blood-Brain Barrier Regulates Xenobiotic Efflux. Cell 2018, 173, 130–139.e10. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, E.L.; Meier, P. Nonparametric estimation from incomplete observations. J. Am. Stat. Assoc. 1958, 53, 457. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Elovsson, G.; Klingstedt, T.; Brown, M.; Nilsson, K.P.R.; Brorsson, A.-C. A Novel Drosophila Model of Alzheimer’s Disease to Study Aβ Proteotoxicity in the Digestive Tract. Int. J. Mol. Sci. 2024, 25, 2105. https://doi.org/10.3390/ijms25042105
Elovsson G, Klingstedt T, Brown M, Nilsson KPR, Brorsson A-C. A Novel Drosophila Model of Alzheimer’s Disease to Study Aβ Proteotoxicity in the Digestive Tract. International Journal of Molecular Sciences. 2024; 25(4):2105. https://doi.org/10.3390/ijms25042105
Chicago/Turabian StyleElovsson, Greta, Therése Klingstedt, Mikaela Brown, K. Peter R. Nilsson, and Ann-Christin Brorsson. 2024. "A Novel Drosophila Model of Alzheimer’s Disease to Study Aβ Proteotoxicity in the Digestive Tract" International Journal of Molecular Sciences 25, no. 4: 2105. https://doi.org/10.3390/ijms25042105
APA StyleElovsson, G., Klingstedt, T., Brown, M., Nilsson, K. P. R., & Brorsson, A.-C. (2024). A Novel Drosophila Model of Alzheimer’s Disease to Study Aβ Proteotoxicity in the Digestive Tract. International Journal of Molecular Sciences, 25(4), 2105. https://doi.org/10.3390/ijms25042105