
Genomic and Comparative Transcriptomic Analyses Reveal Key Genes Associated with the Biosynthesis Regulation of Okaramine B in Penicillium daleae NBP-49626

 , , and
, , and
Abstract
1. Introduction
2. Results
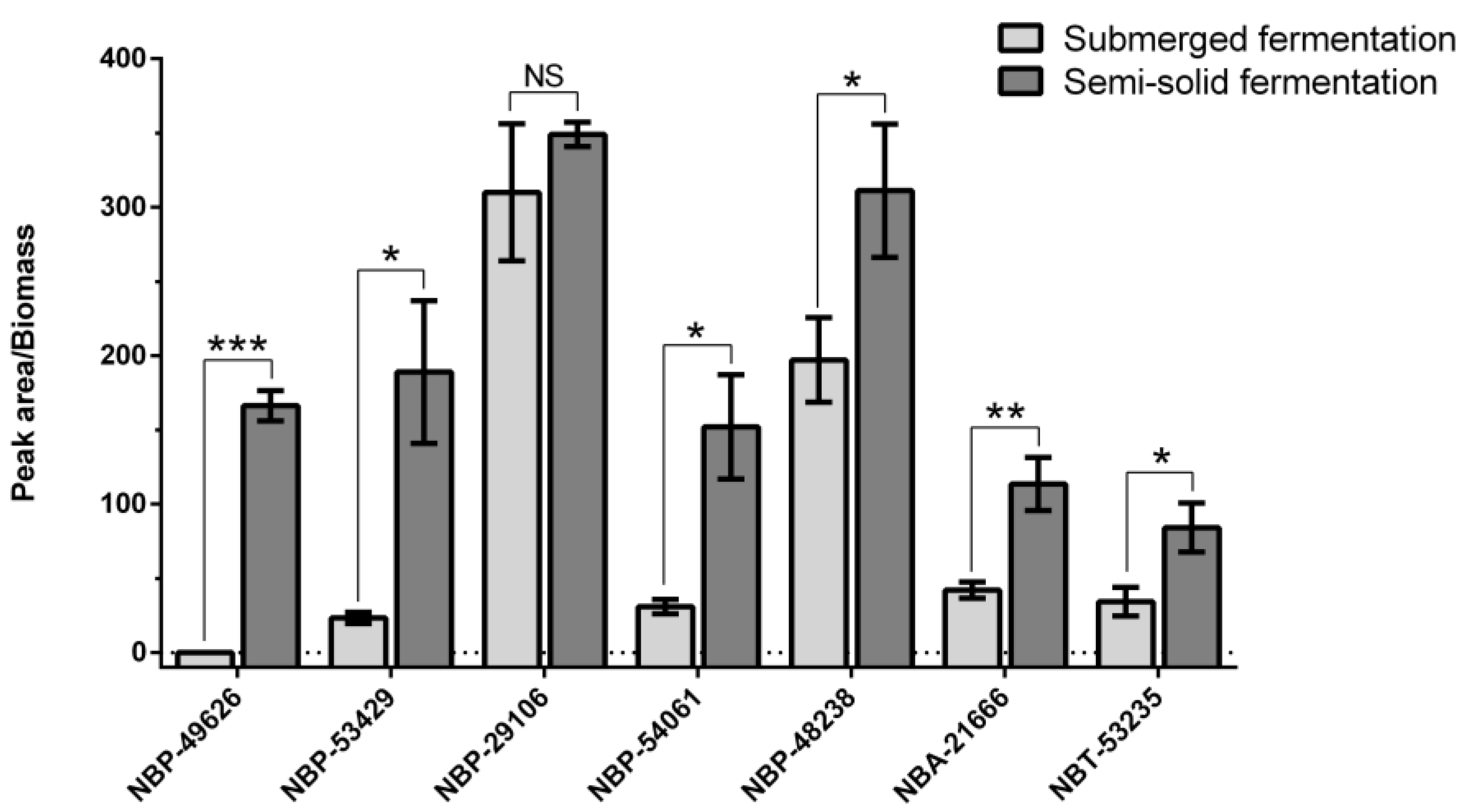
2.1. The Yield Differences of Okaramine B
2.2. Genomic Data Evaluation
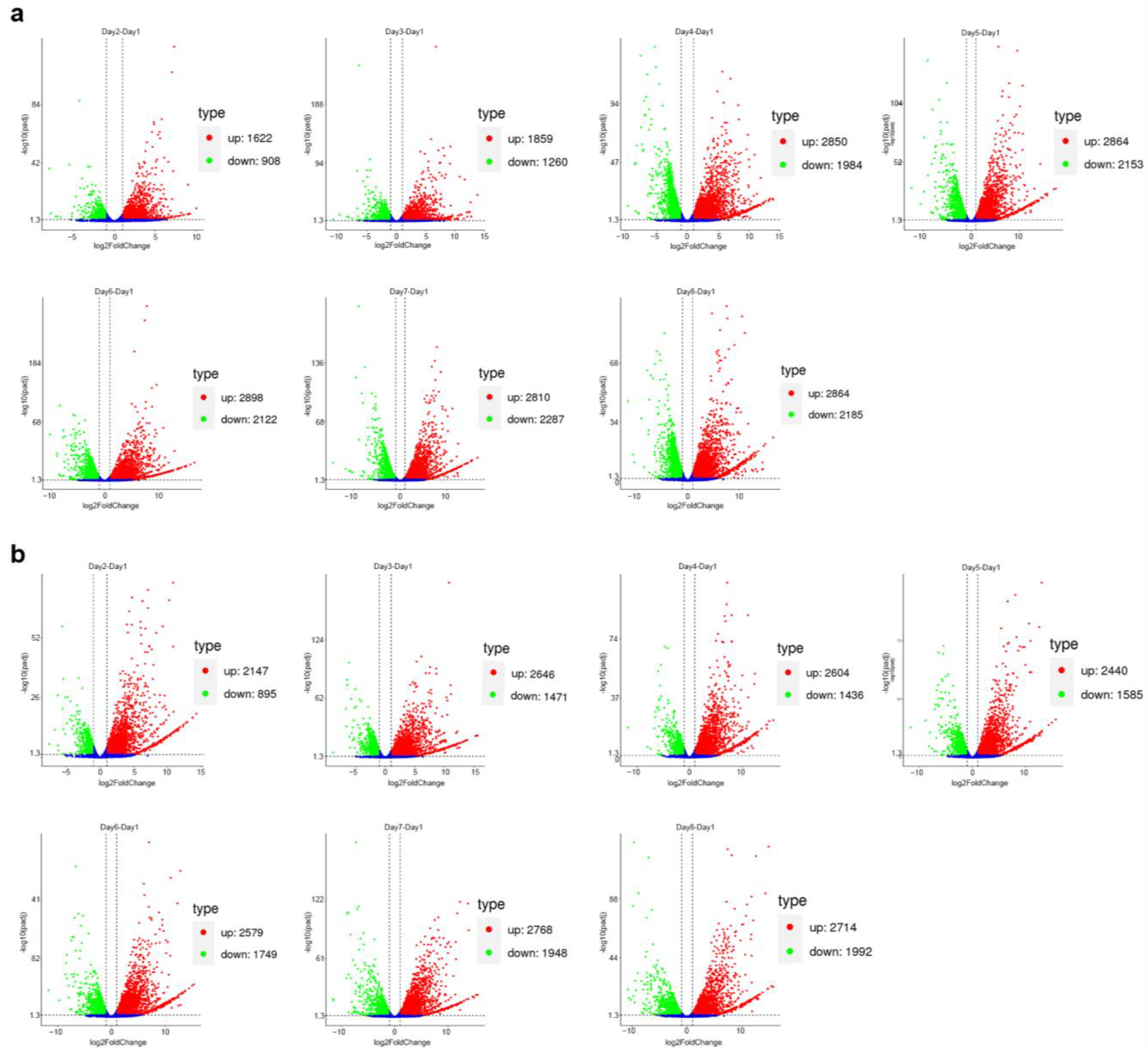
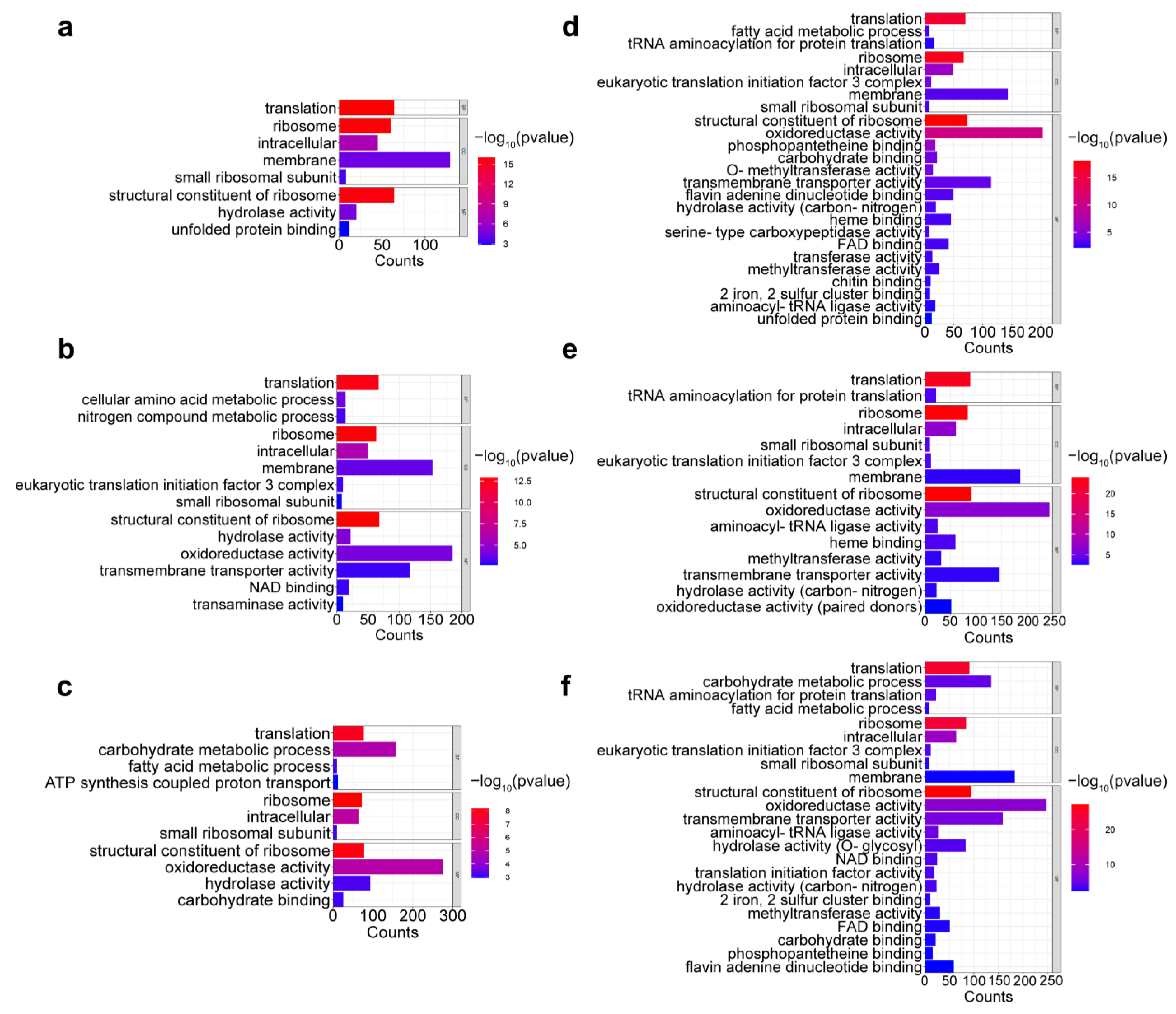
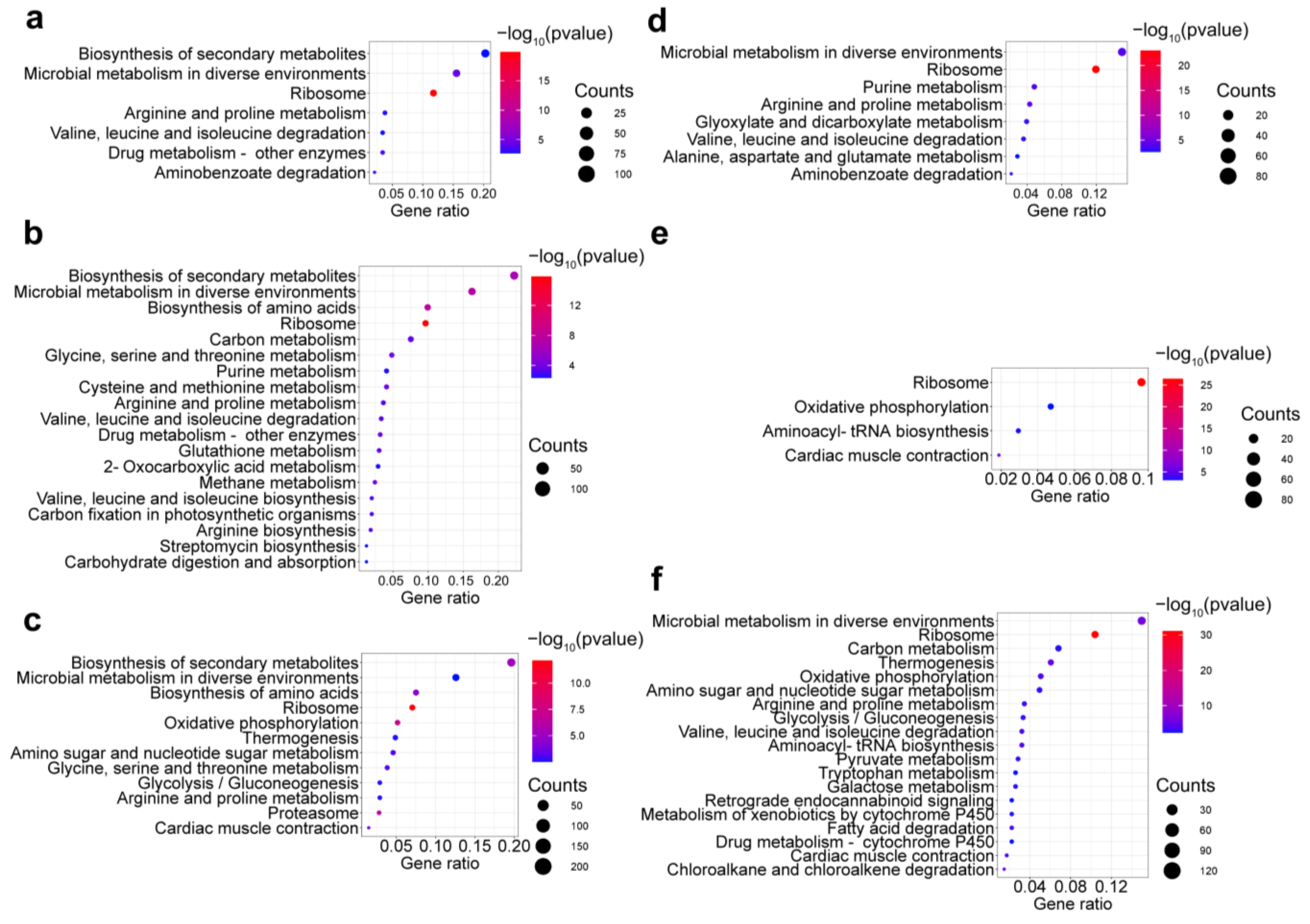
2.3. Transcriptomic Data Evaluation
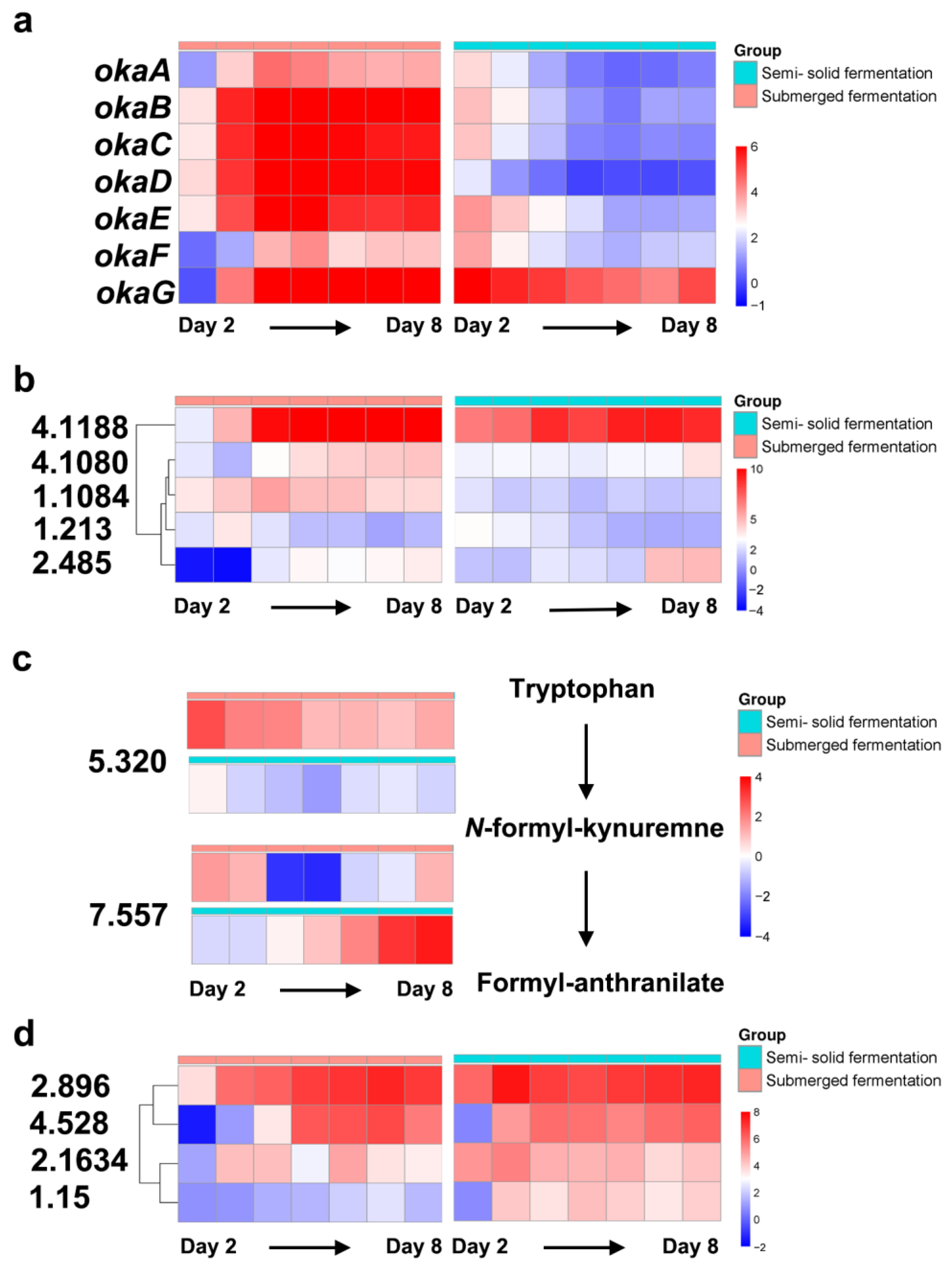
2.4. Differential Expression of Okaramine Biosynthesis Gene Cluster
2.5. Identification of the Differentially Expressed Regulator Genes
2.6. Identification of the DEG Involved in Tryptophan Metabolism
2.7. Identification of the DEG Involved in ATP-Binding Casette (ABC) Transporters
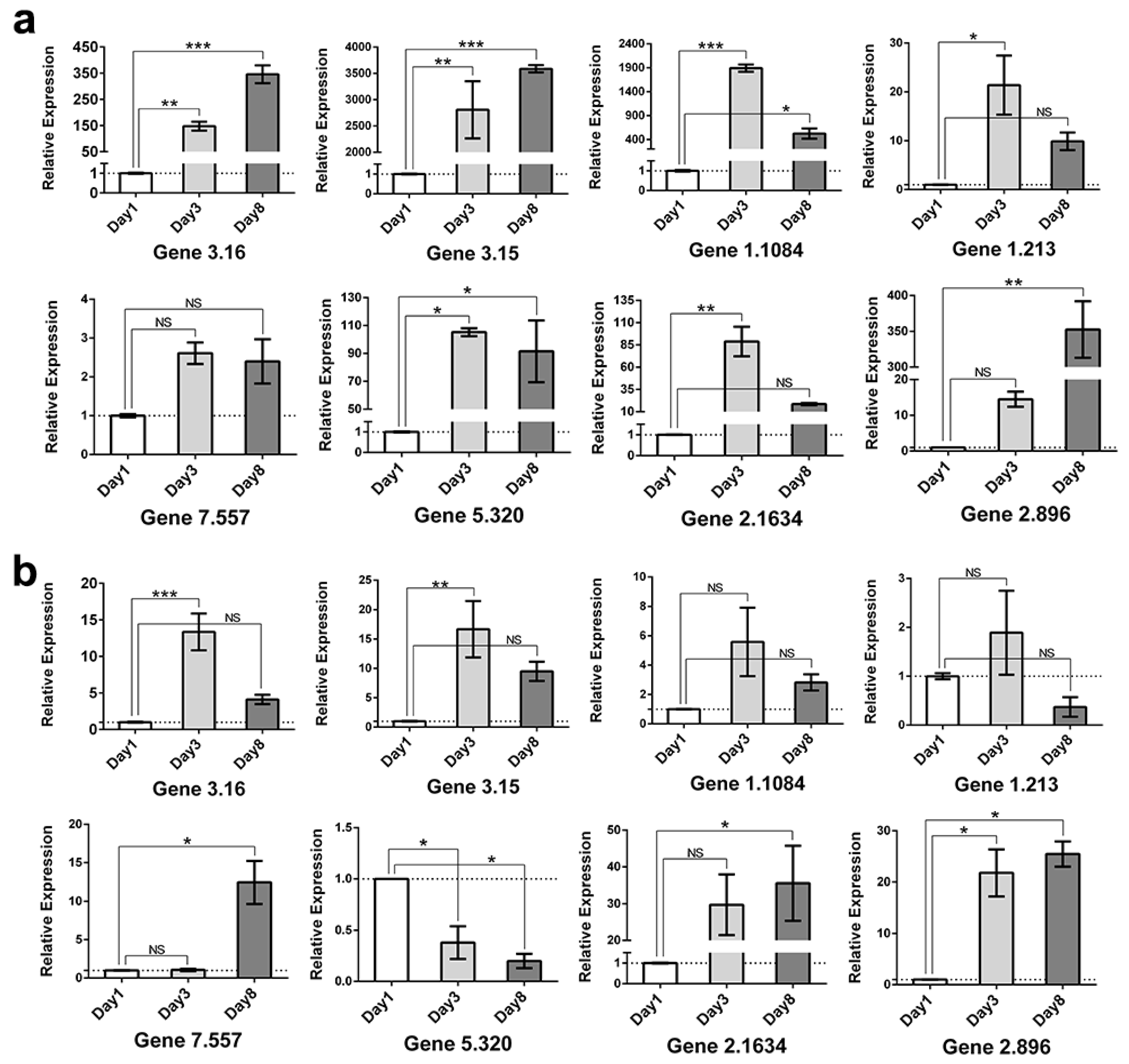
2.8. Validation of RNA-Seq Results by RT-qPCR
3. Discussion
4. Materials and Methods
4.1. Strains and Culture Conditions
4.2. Determination of Okaramine B Production and Mycelial Wet Weight
4.3. Genome DNA Isolation and Sequencing
4.4. Genome Assembly and Gene Annotation
4.5. RNA Isolation and Sequencing
4.6. Analysis of RNA-Seq Data
4.7. Real-Time Quantitative PCR Validation
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Nathiely Ramírez-Guzmán, K.; Torres-León, C.; Martínez-Terrazas, E.; De la Cruz-Quiroz, R.; Flores-Gallegos, A.C.; Rodríguez-Herrera, R.; Aguilar, C.N. Chapter 7—Biocontrol as an Efficient Tool for Food Control and Biosecurity. In Food Safety and Preservation; Grumezescu, A.M., Holban, A.M., Eds.; Academic Press: Cambridge, MA, USA, 2018; pp. 167–193. [Google Scholar]
- Mendez-Gonzalez, F.; Castillo-Minjarez, J.M.; Loera, O.; Favela-Torres, E. Current developments in the resistance, quality, and production of entomopathogenic fungi. World J. Microbiol. Biotechnol. 2022, 38, 115. [Google Scholar] [CrossRef]
- Sparks, T.C.; Storer, N.; Porter, A.; Slater, R.; Nauen, R. Insecticide resistance management and industry: The origins and evolution of the Insecticide Resistance Action Committee (IRAC) and the mode of action classification scheme. Pest. Manag. Sci. 2021, 77, 2609–2619. [Google Scholar] [CrossRef] [PubMed]
- Keller, N.P. Fungal secondary metabolism: Regulation, function and drug discovery. Nat. Rev. Microbiol. 2019, 17, 167–180. [Google Scholar] [CrossRef] [PubMed]
- Nicoletti, R.; Andolfi, A.; Becchimanzi, A.; Salvatore, M.M. Anti-Insect Properties of Penicillium Secondary Metabolites. Microorganisms 2023, 11, 1302. [Google Scholar] [CrossRef] [PubMed]
- Ashtekar, N.; Anand, G.; Thulasiram, H.V.; Rajeshkumar, K.C. Chapter 14—Genus Penicillium: Advances and application in the modern era. In New and Future Developments in Microbial Biotechnology and Bioengineering; Singh, J., Gehlot, P., Eds.; Elsevier: Amsterdam, The Netherlands, 2021; pp. 201–213. [Google Scholar] [CrossRef]
- Hayashi, H.; Sakaguchi, A. Okaramine G, a New Okaramine Congener from Penicillium simplicissimum ATCC 90288. Biosci. Biotechnol. Biochem. 1998, 62, 804–806. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, H.; Asabu, Y.; Murao, S.; Arai, M. New Okaramine Congeners, Okaramines D, E, and F, from Penicillium simplicissimum ATCC 90288. Biosci. Biotechnol. Biochem. 1995, 59, 246–250. [Google Scholar] [CrossRef]
- Hayashi, H.; Fujiwara, T.; Murao, S.; Arai, M. Okaramine C, a New Insecticidal Indole Alkaloid from Penicillium simplicissimum. Agric. Biol. Chem. 1991, 55, 3143–3145. [Google Scholar] [CrossRef]
- Hayashi, H.; Takiuchi, K.; Murao, S.; Arai, M. Structure and Insecticidal Activity of New Indole Alkaloids, Okaramines A and B, from Penicillium simplicissimum AK-40. Agric. Biol. Chem. 1989, 53, 461–469. [Google Scholar] [CrossRef]
- Shiono, Y.; Akiyama, K.; Hayashi, H. Okaramines N, O, P, Q and R, new okaramine congeners, from Penicillium simplicissimum ATCC 90288. Biosci. Biotechnol. Biochem. 2000, 64, 103–110. [Google Scholar] [CrossRef]
- Hayashi, H.; Furutsuka, K.; Shiono, Y. Okaramines H and I, new okaramine congeners, from aspergillus aculeatus. J. Nat. Prod. 1999, 62, 315–317. [Google Scholar] [CrossRef]
- Cai, S.; Sun, S.; Peng, J.; Kong, X.; Zhou, H.; Zhu, T.; Gu, Q.; Li, D. Okaramines S–U, three new indole diketopiperazine alkaloids from Aspergillus taichungensis ZHN-7-07. Tetrahedron 2015, 71, 3715–3719. [Google Scholar] [CrossRef]
- Liu, L.; Qian, X.; Yang, T.; Fang, D.; Qin, Z.; Ren, B.; Li, G. Cyclopiazonic Acid and Okaramine Analogues, Including Chlorinated Compounds, from Chrysosporium undulatum YT-1. J. Nat. Prod. 2022, 85, 2547–2556. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, K. Chemical and biological studies of natural and synthetic products for the highly selective control of pest insect species. Biosci. Biotechnol. Biochem. 2021, 86, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, K. Okaramines and other plant fungal products as new insecticide leads. Curr. Opin. Insect Sci. 2018, 30, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, H.; Takiuchi, K.; Murao, S.; Arai, M. Okaramine B, an Insecticidal Indole Alkaloid, Produced by Penicillium simplicissimum AK-40. Agric. Biol. Chem. 1988, 52, 2131–2133. [Google Scholar] [CrossRef]
- Furutani, S.; Nakatani, Y.; Miura, Y.; Ihara, M.; Kai, K.; Hayashi, H.; Matsuda, K. GluCl a target of indole alkaloid okaramines: A 25 year enigma solved. Sci. Rep. 2014, 4, 6190. [Google Scholar] [CrossRef] [PubMed]
- Furutani, S.; Ihara, M.; Lees, K.; Buckingham, S.D.; Partridge, F.A.; David, J.A.; Patel, R.; Warchal, S.; Mellor, I.R.; Matsuda, K.; et al. The fungal alkaloid Okaramine-B activates an L-glutamate-gated chloride channel from Ixodes scapularis, a tick vector of Lyme disease. Int. J. Parasitol. Drugs Drug Resist. 2018, 8, 350–360. [Google Scholar] [CrossRef]
- Murao, S.; Hayashi, H.; Takiuchi, K.; Arai, M. Okaramine A, a Novel Indole Alkaloid with Insecticidal Activity, from Penicillium simplicissimum AK-40. Agric. Biol. Chem. 1988, 52, 885–886. [Google Scholar] [CrossRef]
- Lai, C.Y.; Lo, I.W.; Hewage, R.T.; Chen, Y.C.; Chen, C.T.; Lee, C.F.; Lin, S.; Tang, M.C.; Lin, H.C. Biosynthesis of Complex Indole Alkaloids: Elucidation of the Concise Pathway of Okaramines. Angew. Chem. Int. Ed. 2017, 56, 9478–9482. [Google Scholar] [CrossRef]
- Houbraken, J.; Kocsube, S.; Visagie, C.M.; Yilmaz, N.; Wang, X.C.; Meijer, M.; Kraak, B.; Hubka, V.; Bensch, K.; Samson, R.A.; et al. Classification of Aspergillus, Penicillium, Talaromyces and related genera (Eurotiales): An overview of families, genera, subgenera, sections, series and species. Stud. Mycol. 2020, 95, 5–169. [Google Scholar] [CrossRef]
- Hewitt, P.R.; Cleator, E.; Ley, S.V. A concise total synthesis of (+)-okaramine C. Org. Biomol. Chem. 2004, 2, 2415–2417. [Google Scholar] [CrossRef] [PubMed]
- Iizuka, T.; Takiguchi, S.; Kumakura, Y.-s.; Tsukioka, N.; Higuchi, K.; Kawasaki, T. First total synthesis and stereochemical revision of okaramine M. Tetrahedron Lett. 2010, 51, 6003–6005. [Google Scholar] [CrossRef]
- Baran, P.S.; Guerrero, C.A.; Corey, E.J. Short, enantioselective total synthesis of okaramine N. J. Am. Chem. Soc. 2003, 125, 5628–5629. [Google Scholar] [CrossRef] [PubMed]
- Li, X.W.; Si, T.X.; Liu, Y.P.; Wang, M.Z.; Chan, A.S.C. Divergent syntheses of okaramines C, J, L, and S-U. Org. Biomol. Chem. 2020, 18, 3848–3852. [Google Scholar] [CrossRef] [PubMed]
- Kato, N.; Furutani, S.; Otaka, J.; Noguchi, A.; Kinugasa, K.; Kai, K.; Hayashi, H.; Ihara, M.; Takahashi, S.; Matsuda, K.; et al. Biosynthesis and Structure-Activity Relationship Studies of Okaramines That Target Insect Glutamate-Gated Chloride Channels. ACS Chem. Biol. 2018, 13, 561–566. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.; Bao, L.; Gao, M.; Chen, M.; Lei, Y.; Liu, G.; Qu, Y. Penicillium decumbens BrlA extensively regulates secondary metabolism and functionally associates with the expression of cellulase genes. Appl. Microbiol. Biotechnol. 2013, 97, 10453–10467. [Google Scholar] [CrossRef]
- Yazaki, K. ABC transporters involved in the transport of plant secondary metabolites. FEBS Lett. 2006, 580, 1183–1191. [Google Scholar] [CrossRef]
- Viglas, J.; Olejnikova, P. An update on ABC transporters of filamentous fungi—From physiological substrates to xenobiotics. Microbiol. Res. 2021, 246, 126684. [Google Scholar] [CrossRef]
- Holighaus, G.; Rohlfs, M. Fungal allelochemicals in insect pest management. Appl. Microbiol. Biotechnol. 2016, 100, 5681–5689. [Google Scholar] [CrossRef]
- Li, G.-H.; Zhang, K.-Q. Natural nematicidal metabolites and advances in their biocontrol capacity on plant parasitic nematodes. Nat. Product. Rep. 2023, 40, 646–675. [Google Scholar] [CrossRef]
- González, M.C.; Lull, C.; Moya, P.; Ayala, I.; Primo, J.; Primo Yúfera, E. Insecticidal activity of penitrems, including penitrem G, a new member of the family isolated from Penicillium crustosum. J. Agric. Food Chem. 2003, 51, 2156–2160. [Google Scholar] [CrossRef]
- Horikoshi, R.; Goto, K.; Mitomi, M.; Oyama, K.; Hirose, T.; Sunazuka, T.; Omura, S. Afidopyropen, a novel insecticide originating from microbial secondary extracts. Sci. Rep. 2022, 12, 2827. [Google Scholar] [CrossRef]
- Lei, H.M.; Wang, J.T.; Hu, Q.Y.; Li, C.Q.; Mo, M.H.; Zhang, K.Q.; Li, G.H.; Zhao, P.J. 2-Furoic acid associated with the infection of nematodes by Dactylellina haptotyla and its biocontrol potential on plant root-knot nematodes. Microbiol. Spectr. 2023, 11, e0189623. [Google Scholar] [CrossRef]
- Fernandes, M.; Keller, N.P.; Adams, T.H. Sequence-specific binding by Aspergillus nidulans AflR, a C6 zinc cluster protein regulating mycotoxin biosynthesis. Mol. Microbiol. 1998, 28, 1355–1365. [Google Scholar] [CrossRef]
- Kumar, D.; Barad, S.; Chen, Y.; Luo, X.; Tannous, J.; Dubey, A.; Glam Matana, N.; Tian, S.; Li, B.; Keller, N.; et al. LaeA regulation of secondary metabolism modulates virulence in Penicillium expansum and is mediated by sucrose. Mol. Plant Pathol. 2017, 18, 1150–1163. [Google Scholar] [CrossRef]
- Oakley, C.E.; Ahuja, M.; Sun, W.W.; Entwistle, R.; Akashi, T.; Yaegashi, J.; Guo, C.J.; Cerqueira, G.C.; Russo Wortman, J.; Wang, C.C.; et al. Discovery of McrA, a master regulator of Aspergillus secondary metabolism. Mol. Microbiol. 2017, 103, 347–365. [Google Scholar] [CrossRef] [PubMed]
- Arakawa, G.Y.; Kudo, H.; Yanase, A.; Eguchi, Y.; Kodama, H.; Ogawa, M.; Koyama, Y.; Shindo, H.; Hosaka, M.; Tokuoka, M. A unique Zn(II)2-Cys6-type protein, KpeA, is involved in secondary metabolism and conidiation in Aspergillus oryzae. Fungal Genet. Biol. 2019, 127, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Hu, P.; Li, H.; Wang, Y.; Long, L.K.; Li, K.; Zhang, X.; Pan, Y.; Liu, G. A Myb transcription factor represses conidiation and cephalosporin C production in Acremonium chrysogenum. Fungal Genet. Biol. 2018, 118, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Baba, S.; Abe, Y.; Ono, C.; Hosobuchi, M. Targeted disruption of the genes, mlcR and ariB, which encode GAL4-type proteins in Penicillium citrinum. Biochim. Biophys. Acta (BBA) Gene Struct. Expr. 2006, 1759, 410–416. [Google Scholar] [CrossRef] [PubMed]
- Cheng, M.; Zhao, S.; Liu, H.; Liu, Y.; Lin, C.; Song, J.; Thawai, C.; Charoensettasilp, S.; Yang, Q. Functional analysis of a chaetoglobosin A biosynthetic regulator in Chaetomium globosum. Fungal Biol. 2021, 125, 201–210. [Google Scholar] [CrossRef] [PubMed]
- Bröker, J.N.; Müller, B.; van Deenen, N.; Prüfer, D.; Schulze Gronover, C. Upregulating the mevalonate pathway and repressing sterol synthesis in Saccharomyces cerevisiae enhances the production of triterpenes. Appl. Microbiol. Biotechnol. 2018, 102, 6923–6934. [Google Scholar] [CrossRef]
- Rogers, S.O.; Bendich, A.J. Extraction of total cellular DNA from plants, algae and fungi. In Plant Molecular Biology Manual; Gelvin, S.B., Schilperoort, R.A., Eds.; Springer: Dordrecht, The Netherlands, 1994; pp. 183–190. [Google Scholar]
- Visagie, C.M.; Houbraken, J.; Frisvad, J.C.; Hong, S.B.; Klaassen, C.H.; Perrone, G.; Seifert, K.A.; Varga, J.; Yaguchi, T.; Samson, R.A. Identification and nomenclature of the genus Penicillium. Stud. Mycol. 2014, 78, 343–371. [Google Scholar] [CrossRef]
- Wick, R.R.; Judd, L.M.; Holt, K.E. Performance of neural network basecalling tools for Oxford Nanopore sequencing. Genome Biol. 2019, 20, 129. [Google Scholar] [CrossRef]
- Hu, J.; Fan, J.; Sun, Z.; Liu, S. NextPolish: A fast and efficient genome polishing tool for long-read assembly. Bioinformatics 2020, 36, 2253–2255. [Google Scholar] [CrossRef]
- Seppey, M.; Manni, M.; Zdobnov, E.M. BUSCO: Assessing Genome Assembly and Annotation Completeness. Methods Mol. Biol. 2019, 1962, 227–245. [Google Scholar] [CrossRef]
- Keilwagen, J.; Hartung, F.; Grau, J. GeMoMa: Homology-Based Gene Prediction Utilizing Intron Position Conservation and RNA-seq Data. Methods Mol. Biol. 2019, 1962, 161–177. [Google Scholar] [CrossRef]
- Hoff, K.J.; Lange, S.; Lomsadze, A.; Borodovsky, M.; Stanke, M. BRAKER1: Unsupervised RNA-Seq-Based Genome Annotation with GeneMark-ET and AUGUSTUS. Bioinformatics 2016, 32, 767–769. [Google Scholar] [CrossRef] [PubMed]
- Haas, B.J.; Salzberg, S.L.; Zhu, W.; Pertea, M.; Allen, J.E.; Orvis, J.; White, O.; Buell, C.R.; Wortman, J.R. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 2008, 9, R7. [Google Scholar] [CrossRef] [PubMed]
- Nawrocki, E.P.; Eddy, S.R. Infernal 1.1: 100-Fold faster RNA homology searches. Bioinformatics 2013, 29, 2933–2935. [Google Scholar] [CrossRef] [PubMed]
- Nawrocki, E.P.; Burge, S.W.; Bateman, A.; Daub, J.; Eberhardt, R.Y.; Eddy, S.R.; Floden, E.W.; Gardner, P.P.; Jones, T.A.; Tate, J.; et al. Rfam 12.0: Updates to the RNA families database. Nucleic Acids Res. 2015, 43, D130–D137. [Google Scholar] [CrossRef] [PubMed]
- Chan, P.P.; Lowe, T.M. tRNAscan-SE: Searching for tRNA Genes in Genomic Sequences. In Gene Prediction; Methods in Molecular Biology Book Series (MIMB); Humana: New York, NY, USA, 2019; Volume 1962, pp. 1–14. [Google Scholar] [CrossRef]
- Lagesen, K.; Hallin, P.; Rodland, E.A.; Staerfeldt, H.H.; Rognes, T.; Ussery, D.W. RNAmmer: Consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 2007, 35, 3100–3108. [Google Scholar] [CrossRef]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [PubMed]
- Urban, M.; Cuzick, A.; Seager, J.; Wood, V.; Rutherford, K.; Venkatesh, S.Y.; Sahu, J.; Iyer, S.V.; Khamari, L.; De Silva, N.; et al. PHI-base in 2022: A multi-species phenotype database for Pathogen–Host Interactions. Nucleic Acids Res. 2022, 50, D837–D847. [Google Scholar] [CrossRef] [PubMed]
- Lombard, V.; Golaconda Ramulu, H.; Drula, E.; Coutinho, P.M.; Henrissat, B. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 2014, 42, D490–D495. [Google Scholar] [CrossRef]
- Park, J.; Lee, S.; Choi, J.; Ahn, K.; Park, B.; Park, J.; Kang, S.; Lee, Y.H. Fungal cytochrome P450 database. BMC Genom. 2008, 9, 402. [Google Scholar] [CrossRef]
- Almagro Armenteros, J.J.; Tsirigos, K.D.; Sonderby, C.K.; Petersen, T.N.; Winther, O.; Brunak, S.; von Heijne, G.; Nielsen, H. SignalP 5.0 improves signal peptide predictions using deep neural networks. Nat. Biotechnol. 2019, 37, 420–423. [Google Scholar] [CrossRef] [PubMed]
- Krogh, A.; Larsson, B.; von Heijne, G.; Sonnhammer, E.L. Predicting transmembrane protein topology with a hidden Markov model: Application to complete genomes. J. Mol. Biol. 2001, 305, 567–580. [Google Scholar] [CrossRef]
- Wang, J.; Chitsaz, F.; Derbyshire, M.K.; Gonzales, N.R.; Gwadz, M.; Lu, S.; Marchler, G.H.; Song, J.S.; Thanki, N.; Yamashita, R.A.; et al. The conserved domain database in 2023. Nucleic Acids Res. 2023, 51, D384–D388. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control tool for High Throughput Sequence Data. Babraham Bioinformatics. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 2 February 2024).
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Kovaka, S.; Zimin, A.V.; Pertea, G.M.; Razaghi, R.; Salzberg, S.L.; Pertea, M. Transcriptome assembly from long-read RNA-seq alignments with StringTie2. Genome Biol. 2019, 20, 278. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef]
- Koressaar, T.; Lepamets, M.; Kaplinski, L.; Raime, K.; Andreson, R.; Remm, M. Primer3_masker: Integrating masking of template sequence with primer design software. Bioinformatics 2018, 34, 1937–1938. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genome Features | Value |
|---|---|
| Genome size (bp) | 37,366,436 |
| Number of chromosomes | 8 |
| N50 scaffold length (bp) | 4,967,589 |
| N90 scaffold length (bp) | 2,981,452 |
| Maximum scaffold length (bp) | 7,888,045 |
| Minimum scaffold length (bp) | 2,295,385 |
| G + C content (%) | 49.07 |
| Complete BUSCOs of genome (%) | 99.47 |
| Number of protein-coding genes | 10,131 |
| Avg gene length (bp) | 1663.62 |
| Avg CDS length (bp) | 1479.65 |
| Average number of exons per gene | 3.24 |
| Average exon length (bp) | 456.45 |
| Average intron length (bp) | 82.07 |
| rRNAs | 58 |
| tRNAs | 164 |
| Complete BUSCOs of genes (%) | 99.08 |
| Number of annotated genes by NR | 9983 |
| Number of annotated genes by SWISS-PROT | 7676 |
| Number of annotated genes by GO | 6025 |
| Number of annotated genes by KEGG | 3514 |
| Number of annotated genes by KOG | 3347 |
| Number of annotated genes by PHI-base | 2771 |
| Number of annotated genes by CAZy | 1435 |
| Number of annotated genes by P450 | 177 |
| Number of secreted proteins | 715 |
| Gene ID | Primer Name | Primer Sequence 5′→3′ |
|---|---|---|
| 4.104 | actin-F | ACAGTCCAAGCGTGGTATCC |
| actin-R | CACACGGAGCTCGTTGTAGA | |
| 3.16 | okaA-F | CTTGATCCGAGCGAGGTTAG |
| okaA-R | CGGCTATCGAAGCACTTAGG | |
| 3.15 | okaB-F | CAAACCAACAAGGCCAAAGT |
| okaB-R | TATACCCTCGACCCTCGTTG | |
| 1.1084 | reg2-F | ACCAGATCAGCTCACCCAAA |
| reg2-R | CGATCAAAGCCTGGAACGAG | |
| 1.213 | reg19-F | TCAGCTCACCAAGTTCCAGT |
| reg19-R | CTTCTCGATCTCCTCTGCGT | |
| 7.557 | try7-F | TGATCTCTGGTGCCCTCAAG |
| try7-R | GCTGGCTTAGAGTTACCCCA | |
| 5.320 | try9-F | CTTCTGTGGAAACTCGCTGG |
| try9-R | ACAAACACCAATGGAAGCCC | |
| 2.1634 | trans4-F | GCACTGTTGAGCGAAAGGAA |
| trans4-R | TGTTTCAGGTTGGTGGCATG | |
| 2.896 | trans5-F | TCCTTGCTCGTTCCCTCATT |
| trans5-R | AGTACATCCACGACTGCCAA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Chen, L.; Fang, W.; Zeng, Z.; Wu, Z.; Liu, F.; Liu, X.; Gong, Y.; Zhu, L.; Wang, K. Genomic and Comparative Transcriptomic Analyses Reveal Key Genes Associated with the Biosynthesis Regulation of Okaramine B in Penicillium daleae NBP-49626. Int. J. Mol. Sci. 2024, 25, 1965. https://doi.org/10.3390/ijms25041965
Wang Y, Chen L, Fang W, Zeng Z, Wu Z, Liu F, Liu X, Gong Y, Zhu L, Wang K. Genomic and Comparative Transcriptomic Analyses Reveal Key Genes Associated with the Biosynthesis Regulation of Okaramine B in Penicillium daleae NBP-49626. International Journal of Molecular Sciences. 2024; 25(4):1965. https://doi.org/10.3390/ijms25041965
Chicago/Turabian StyleWang, Yueying, Ling Chen, Wei Fang, Zhen Zeng, Zhaoyuan Wu, Fang Liu, Xiaoyan Liu, Yan Gong, Lei Zhu, and Kaimei Wang. 2024. "Genomic and Comparative Transcriptomic Analyses Reveal Key Genes Associated with the Biosynthesis Regulation of Okaramine B in Penicillium daleae NBP-49626" International Journal of Molecular Sciences 25, no. 4: 1965. https://doi.org/10.3390/ijms25041965
APA StyleWang, Y., Chen, L., Fang, W., Zeng, Z., Wu, Z., Liu, F., Liu, X., Gong, Y., Zhu, L., & Wang, K. (2024). Genomic and Comparative Transcriptomic Analyses Reveal Key Genes Associated with the Biosynthesis Regulation of Okaramine B in Penicillium daleae NBP-49626. International Journal of Molecular Sciences, 25(4), 1965. https://doi.org/10.3390/ijms25041965