
Most Monogenic Disorders Are Caused by Mutations Altering Protein Folding Free Energy



Abstract
1. Introduction
2. Results
2.1. Database of Monogenic Disorders (MOGEDO)
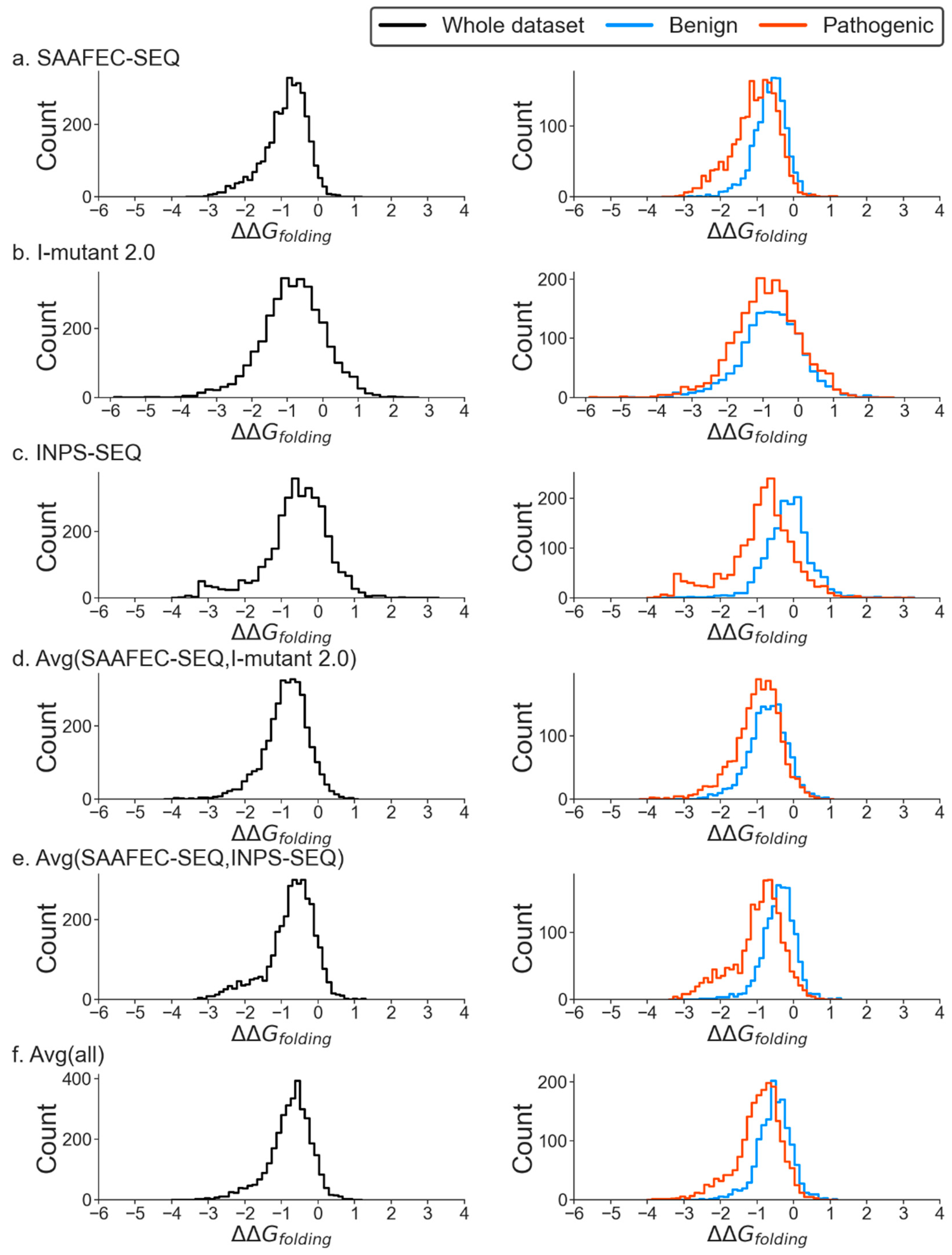
2.2. Change in Folding Free Energy and Pathogenicity
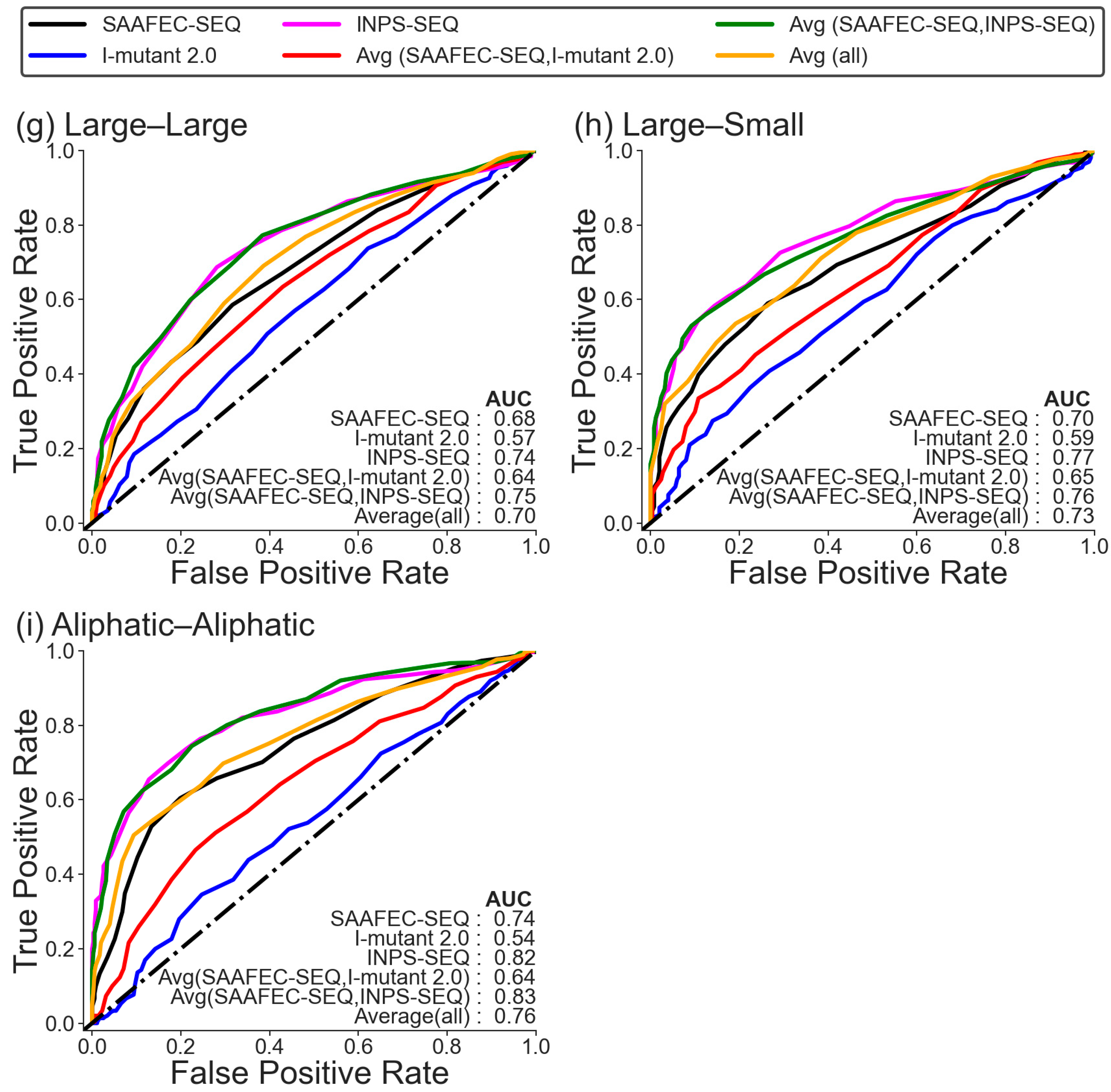
2.3. Folding Free Energy Change as a Measure of Pathogenicity Based on the Chemical Nature of Amino Acid Mutations
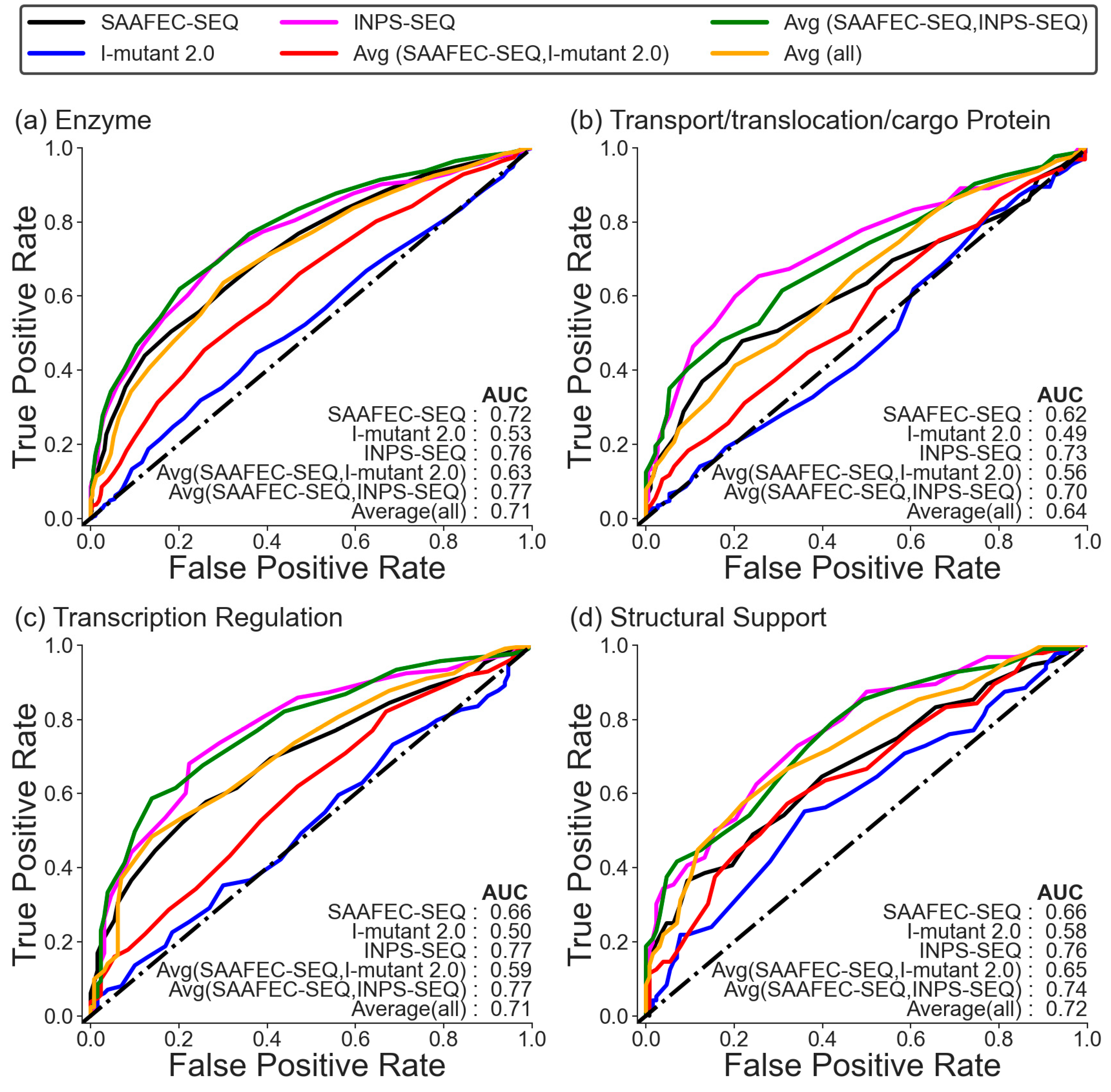
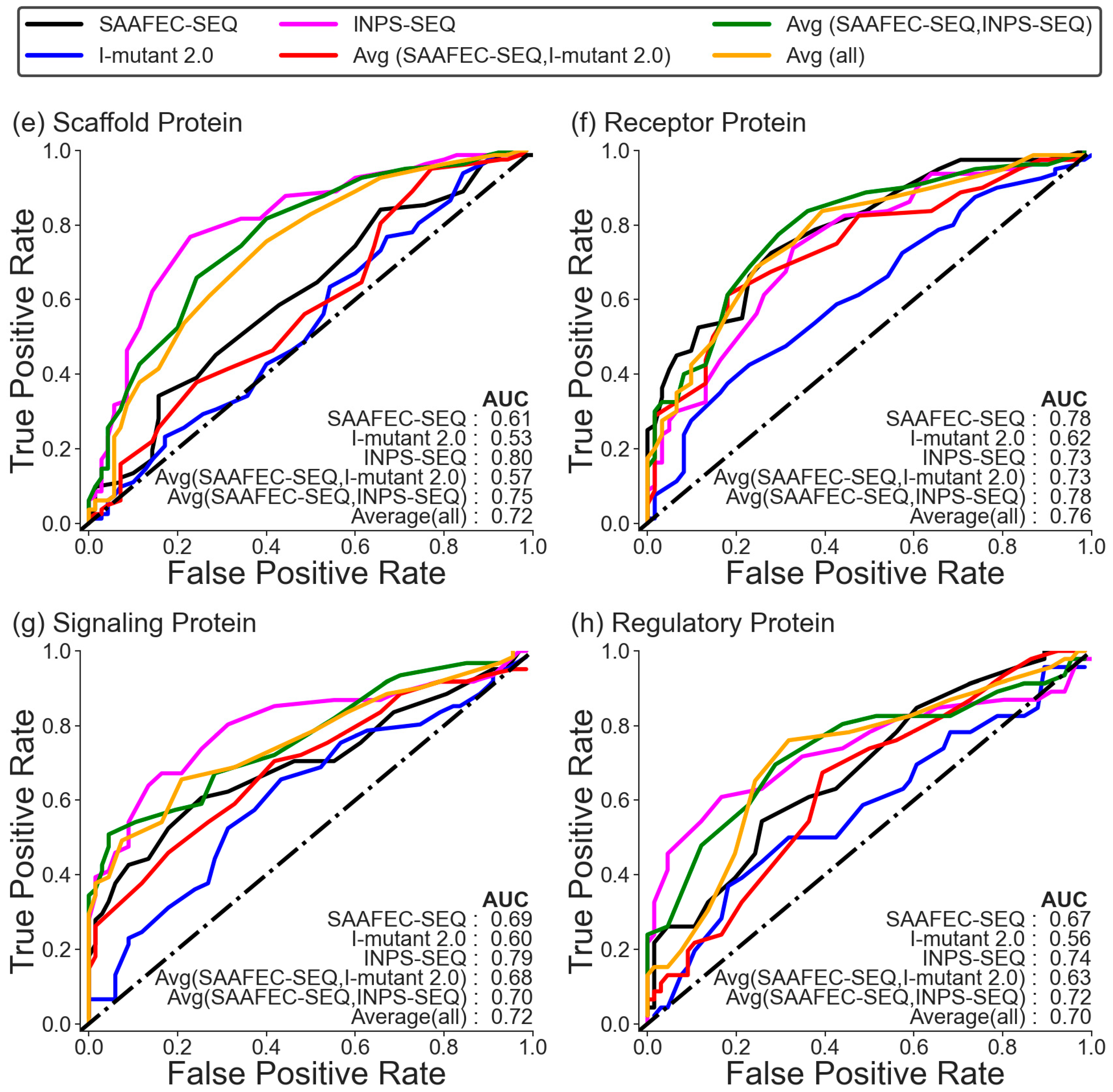
2.4. Folding Free Energy Change as a Measure of Pathogenicity Based on Functional Category
2.5. Relative Surface Area (RSA): A Measure of Pathogenicity
2.6. Logistic Regression Model Training and Testing
2.7. Performance Comparison of Change in Folding Free Energy Method, RSA Method, and Logistic Regression Model with Other Leading Pathogenicity Predictors
2.8. Profiling Pathogenic Mutations through Folding Free Energy Change Estimated Using SAAFEC-SEQ
3. Discussion
4. Materials and Methods
4.1. Database of Monogenic Disorders (MOGEDO)
4.2. Folding Free Energy Calculation
4.3. Solvent Accessible Surface Area Calculation
4.4. Regression Model Development
4.5. Methods for Predicting the Pathogenicity of Amino Acid Mutations
4.6. Receiver Operating Characteristics (ROC)
4.7. Sampling and Assessment of Predictions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gilissen, C.; Hoischen, A.; Brunner, H.G.; Veltman, J.A. Disease gene identification strategies for exome sequencing. Eur. J. Hum. Genet. 2012, 20, 490–497. [Google Scholar] [CrossRef]
- Tennessen, J.A.; Bigham, A.W.; O’Connor, T.D.; Fu, W.; Kenny, E.E.; Gravel, S.; McGee, S.; Do, R.; Liu, X.; Jun, G.; et al. Evolution and Functional Impact of Rare Coding Variation from Deep Sequencing of Human Exomes. Science 2012, 337, 64–69. [Google Scholar] [CrossRef]
- Ng, P.C.; Henikoff, S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003, 31, 3812–3814. [Google Scholar] [CrossRef]
- Vaser, R.; Adusumalli, S.; Leng, S.N.; Sikic, M.; Ng, P.C. SIFT missense predictions for genomes. Nat. Protoc. 2016, 11, 1–9. [Google Scholar] [CrossRef]
- Adzhubei, I.; Jordan, D.M.; Sunyaev, S.R. Predicting functional effect of human missense mutations using PolyPhen-2. Curr. Protoc. Hum. Genet. 2013. [Google Scholar] [CrossRef] [PubMed]
- Chun, S.; Fay, J.C. Identification of deleterious mutations within three human genomes. Genome Res. 2009, 19, 1553–1561. [Google Scholar] [CrossRef]
- Schwarz, J.M.; Rödelsperger, C.; Schuelke, M.; Seelow, D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat. Methods 2010, 7, 575–576. [Google Scholar] [CrossRef] [PubMed]
- Reva, B.; Antipin, Y.; Sander, C. Predicting the functional impact of protein mutations: Application to cancer genomics. Nucleic Acids Res. 2011, 39, e118. [Google Scholar] [CrossRef] [PubMed]
- Shihab, H.A.; Gough, J.; Cooper, D.N.; Stenson, P.D.; Barker, G.L.A.; Edwards, K.J.; Day, I.N.M.; Gaunt, T.R. Predicting the Functional, Molecular, and Phenotypic Consequences of Amino Acid Substitutions using Hidden Markov Models. Hum. Mutat. 2013, 34, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.; Chan, A.P. PROVEAN web server: A tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics 2015, 31, 2745–2747. [Google Scholar] [CrossRef] [PubMed]
- Carter, H.; Douville, C.; Stenson, P.D.; Cooper, D.N.; Karchin, R. Identifying Mendelian disease genes with the Variant Effect Scoring Tool. BMC Genom. 2013, 14, S3. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Jhong, J.-H.; Lee, J.; Koo, J.-Y. Meta-analytic support vector machine for integrating multiple omics data. BioData Min. 2017, 10, 2. [Google Scholar] [CrossRef]
- Jagadeesh, K.A.; Wenger, A.M.; Berger, M.J.; Guturu, H.; Stenson, P.D.; Cooper, D.N.; Bernstein, J.A.; Bejerano, G. M-CAP eliminates a majority of variants of uncertain significance in clinical exomes at high sensitivity. Nat. Genet. 2016, 48, 1581–1586. [Google Scholar] [CrossRef] [PubMed]
- Ioannidis, N.M.; Rothstein, J.H.; Pejaver, V.; Middha, S.; McDonnell, S.K.; Baheti, S.; Musolf, A.; Li, Q.; Holzinger, E.; Karyadi, D.; et al. REVEL: An Ensemble Method for Predicting the Pathogenicity of Rare Missense Variants. Am. J. Hum. Genet. 2016, 99, 877–885. [Google Scholar] [CrossRef] [PubMed]
- Pejaver, V.; Urresti, J.; Lugo-Martinez, J.; Pagel, K.A.; Lin, G.N.; Nam, H.-J.; Mort, M.; Cooper, D.N.; Sebat, J.; Iakoucheva, L.M.; et al. Inferring the molecular and phenotypic impact of amino acid variants with MutPred2. Nat. Commun. 2020, 11, 5918. [Google Scholar] [CrossRef]
- Raimondi, D.; Tanyalcin, I.; Ferté, J.; Gazzo, A.; Orlando, G.; Lenaerts, T.; Rooman, M.; Vranken, W. DEOGEN2: Prediction and interactive visualization of single amino acid variant deleteriousness in human proteins. Nucleic Acids Res. 2017, 45, W201–W206. [Google Scholar] [CrossRef] [PubMed]
- Mathe, E.; Olivier, M.; Kato, S.; Ishioka, C.; Hainaut, P.; Tavtigian, S.V. Computational approaches for predicting the biological effect of p53 missense mutations: A comparison of three sequence analysis based methods. Nucleic Acids Res. 2006, 34, 1317–1325. [Google Scholar] [CrossRef]
- Tavtigian, S.V.; Deffenbaugh, A.M.; Yin, L.; Judkins, T.; Scholl, T.; Samollow, P.B.; de Silva, D.; Zharkikh, A.; Thomas, A. Comprehensive statistical study of 452 BRCA1 missense substitutions with classification of eight recurrent substitutions as neutral. J. Med. Genet. 2006, 43, 295–305. [Google Scholar] [CrossRef]
- Davydov, E.V.; Goode, D.L.; Sirota, M.; Cooper, G.M.; Sidow, A.; Batzoglou, S. Identifying a high fraction of the human genome to be under selective constraint using GERP++. PLoS Comput. Biol. 2010, 6, e1001025. [Google Scholar] [CrossRef]
- Choi, Y.; Sims, G.E.; Murphy, S.; Miller, J.R.; Chan, A.P. Predicting the Functional Effect of Amino Acid Substitutions and Indels. PLoS ONE 2012, 7, e46688. [Google Scholar] [CrossRef]
- Peng, Y.; Alexov, E. Investigating the linkage between disease-causing amino acid variants and their effect on protein stability and binding. Proteins 2016, 84, 232–239. [Google Scholar] [CrossRef]
- Yue, P.; Li, Z.; Moult, J. Loss of protein structure stability as a major causative factor in monogenic disease. J. Mol. Biol. 2005, 353, 459–473. [Google Scholar] [CrossRef] [PubMed]
- Gerasimavicius, L.; Liu, X.; Marsh, J.A. Identification of pathogenic missense mutations using protein stability predictors. Sci. Rep. 2020, 10, 15387. [Google Scholar] [CrossRef] [PubMed]
- Aledo, P.; Aledo, J.C. Proteome-Wide Structural Computations Provide Insights into Empirical Amino Acid Substitution Matrices. Int. J. Mol. Sci. 2023, 24, 796. [Google Scholar] [CrossRef] [PubMed]
- Jemimah, S.; Gromiha, M.M. Insights into changes in binding affinity caused by disease mutations in protein-protein complexes. Comput. Biol. Med. 2020, 123, 103829. [Google Scholar] [CrossRef] [PubMed]
- Sahni, N.; Yi, S.; Taipale, M.; Fuxman Bass, J.I.; Coulombe-Huntington, J.; Yang, F.; Peng, J.; Weile, J.; Karras, G.I.; Wang, Y.; et al. Widespread macromolecular interaction perturbations in human genetic disorders. Cell 2015, 161, 647–660. [Google Scholar] [CrossRef] [PubMed]
- Zaucha, J.; Heinzinger, M.; Kulandaisamy, A.; Kataka, E.; Salvádor, Ó.L.; Popov, P.; Rost, B.; Gromiha, M.M.; Zhorov, B.S.; Frishman, D. Mutations in transmembrane proteins: Diseases, evolutionary insights, prediction and comparison with globular proteins. Brief. Bioinform. 2021, 22, bbaa132. [Google Scholar] [CrossRef] [PubMed]
- Jackson, M.; Marks, L.; May, G.H.W.; Wilson, J.B. The genetic basis of disease. Essays Biochem. 2018, 62, 643–723. [Google Scholar] [CrossRef]
- Takano, K.; Liu, D.; Tarpey, P.; Gallant, E.; Lam, A.; Witham, S.; Alexov, E.; Chaubey, A.; Stevenson, R.E.; Schwartz, C.E.; et al. An X-linked channelopathy with cardiomegaly due to a CLIC2 mutation enhancing ryanodine receptor channel activity. Hum. Mol. Genet. 2012, 21, 4497–4507. [Google Scholar] [CrossRef]
- Witham, S.; Takano, K.; Schwartz, C.; Alexov, E. A missense mutation in CLIC2 associated with intellectual disability is predicted by in silico modeling to affect protein stability and dynamics. Proteins Struct. Funct. Bioinform. 2011, 79, 2444–2454. [Google Scholar] [CrossRef]
- Yang, Y.; Kucukkal, T.G.; Li, J.; Alexov, E.; Cao, W. Binding Analysis of Methyl-CpG Binding Domain of MeCP2 and Rett Syndrome Mutations. ACS Chem. Biol. 2016, 11, 2706–2715. [Google Scholar] [CrossRef] [PubMed]
- Pandey, P.; Ghimire, S.; Wu, B.; Alexov, E. On the linkage of thermodynamics and pathogenicity. Curr. Opin. Struct. Biol. 2023, 80, 102572. [Google Scholar] [CrossRef] [PubMed]
- Savojardo, C.; Manfredi, M.; Martelli, P.L.; Casadio, R. Solvent Accessibility of Residues Undergoing Pathogenic Variations in Humans: From Protein Structures to Protein Sequences. Front. Mol. Biosci. 2021, 7, 626363. [Google Scholar] [CrossRef] [PubMed]
- Martelli, P.L.; Fariselli, P.; Savojardo, C.; Babbi, G.; Aggazio, F.; Casadio, R. Large scale analysis of protein stability in OMIM disease related human protein variants. BMC Genom. 2016, 17, 397. [Google Scholar] [CrossRef] [PubMed]
- Casadio, R.; Vassura, M.; Tiwari, S.; Fariselli, P.; Luigi Martelli, P. Correlating disease-related mutations to their effect on protein stability: A large-scale analysis of the human proteome. Hum. Mutat. 2011, 32, 1161–1170. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Zhou, H.X. Prediction of solvent accessibility and sites of deleterious mutations from protein sequence. Nucleic Acids Res. 2005, 33, 3193–3199. [Google Scholar] [CrossRef] [PubMed]
- Savojardo, C.; Babbi, G.; Martelli, P.L.; Casadio, R. Functional and Structural Features of Disease-Related Protein Variants. Int. J. Mol. Sci. 2019, 20, 1530. [Google Scholar] [CrossRef] [PubMed]
- Durairaj, G.; Demir, Ö.; Lim, B.; Baronio, R.; Tifrea, D.; Hall, L.V.; DeForest, J.C.; Lauinger, L.; Jebril Fallatah, M.M.; Yu, C.; et al. Discovery of compounds that reactivate p53 mutants in vitro and in vivo. Cell Chem. Biol. 2022, 29, 1381–1395.e13. [Google Scholar] [CrossRef]
- Fersht, A.R.; Jackson, S.E.; Serrano, L. Protein stability: Experimental data from protein engineering. Philos. Trans. R. Soc. Lond. Ser. A Phys. Eng. Sci. 1993, 345, 141–151. [Google Scholar] [CrossRef]
- Sherafatizangeneh, M.; Farshadfar, C.; Mojahed, N.; Noorbakhsh, A.; Ardalan, N. Blockage of the Monoamine Oxidase by a Natural Compound to Overcome Parkinson’s Disease via Computational Biology. J. Comput. Biophys. Chem. 2022, 21, 373–387. [Google Scholar] [CrossRef]
- Mishra, P.; Biswas, S.; Baur, S.; Basak, S.; Mukherjee, A.; Basu, A. Development of Pyrazole Harbouring Novel Leads Against β-Amyloid Protein Fibrillation by in silico Drug Design. J. Comput. Biophys. Chem. 2022, 21, 541–553. [Google Scholar] [CrossRef]
- Nedeljković, N.V.; Bojović, D.N.; Živanović, A.S.; Mijajlović, M.Ž.; Nikolić, M.V. Virtual Screening of Potential In Silico Hits for the Prevention of Neuroinflammation: Arylalkanoic Acid Derivatives of NSAIDs as Selective Dual Inhibitors of Microsomal Prostaglandin E Synthase-2 (mPGES-2) and 5-Lipoxygenase-Activating Protein (FLAP). J. Comput. Biophys. Chem. 2022, 21, 797–819. [Google Scholar] [CrossRef]
- Mali, S.N.; Pandey, A. Balanced QSAR and Molecular Modeling to Identify Structural Requirements of Imidazopyridine Analogues as Anti-infective Agents Against Trypanosomiases. J. Comput. Biophys. Chem. 2022, 21, 83–114. [Google Scholar] [CrossRef]
- Saba, A.; Muhammad, S.; Khera, R.A.; Alhujaily, M.; Iqbal, J.; Chaudhry, A.R.; Alarfaji, S.S.; Belali, T.M. Identification of Halogen-Based Derivatives as Potent Inhibitors of Estrogen Receptor Alpha of Breast Cancer: An In-Silico Investigation. J. Comput. Biophys. Chem. 2022, 21, 181–205. [Google Scholar] [CrossRef]
- Ramuthai, M.; Jeyavijayan, S.; Premkumar, R.; Uma Priya, M.; Jayram, N.D. Structure, Spectroscopic Investigation, Molecular Docking and In vitro Cytotoxicity Studies on 4,7-dihydroxycoumarin: A Breast Cancer Drug. J. Comput. Biophys. Chem. 2022, 21, 219–236. [Google Scholar] [CrossRef]
- Gopal, S. Virtual Screening and Molecular Docking Analysis of Degradation Products of Curcumin as Inhibitors of EGFR. J. Comput. Biophys. Chem. 2022, 21, 629–646. [Google Scholar] [CrossRef]
- Agostini, L.; Morosetti, S. In silico Design of Glyco-D,L-Peptide Antiviral Molecules. J. Comput. Biophys. Chem. 2022, 21, 349–360. [Google Scholar] [CrossRef]
- Abdullah, N.N.; Imran, S.; Lam, K.W.; Ismail, N.H. Computational Screening of Styryl Lactone Compounds Isolated from Goniothalamus Species to Identify Potential Inhibitors for Dengue Virus. J. Comput. Biophys. Chem. 2022, 21, 821–843. [Google Scholar] [CrossRef]
- Jana, A.; Roy, T.; Layek, S.; Ghosal, S.; Banerjee, D.R. Computational Investigation on Natural Quinazoline Alkaloids as Potential Inhibitors of the Main Protease (Mpro) of SARS-CoV-2. J. Comput. Biophys. Chem. 2022, 21, 65–82. [Google Scholar] [CrossRef]
- Azerang, P.; Yazdani, M.; RayatSanati, K.; Tahghighi, A. Newly Identified COVID-19 Drug Candidates Based on Computational Strategies. J. Comput. Biophys. Chem. 2022, 21, 123–137. [Google Scholar] [CrossRef]
- Qaisar, M.; Muhammad, S.; Iqbal, J.; Khera, R.A.; Al-Sehemi, A.G.; Alarfaji, S.S.; Khalid, M.; Hussain, F. Identification of Marine Fungi-Based Antiviral Agents as Potential Inhibitors of SARS-CoV-2 by Molecular Docking, ADMET and Molecular Dynamic Study. J. Comput. Biophys. Chem. 2022, 21, 139–153. [Google Scholar] [CrossRef]
- Vahabzadeh, T.; Miran, M.; Razzaghi-Asl, N. Molecular Dynamics Simulation of Privileged Biflavonoids as SARS-CoV2 3CLpro Targeting Agents. J. Comput. Biophys. Chem. 2022, 21, 569–582. [Google Scholar] [CrossRef]
- Ounissi, M.; Rachedi, F.Z. Targeting the SARS-CoV-2 Main Protease: In Silico Study Contributed to Exploring Potential Natural Compounds as Candidate Inhibitors. J. Comput. Biophys. Chem. 2022, 21, 663–682. [Google Scholar] [CrossRef]
- Zangeneh, J.; Shirvani, P.; Etebari, M.; Saghaie, L. In Silico Screening for Novel Tyrosine Kinase Inhibitors with Oxindole Scaffold as Anti-Cancer Agents: Design, QSAR Analysis, Molecular Docking and ADMET Studies. J. Comput. Biophys. Chem. 2022, 21, 583–598. [Google Scholar] [CrossRef]
- Fragoza, R.; Das, J.; Wierbowski, S.D.; Liang, J.; Tran, T.N.; Liang, S.; Beltran, J.F.; Rivera-Erick, C.A.; Ye, K.; Wang, T.-Y.; et al. Extensive disruption of protein interactions by genetic variants across the allele frequency spectrum in human populations. Nat. Commun. 2019, 10, 4141. [Google Scholar] [CrossRef] [PubMed]
- Amberger, J.S.; Bocchini, C.A.; Schiettecatte, F.; Scott, A.F.; Hamosh, A. OMIM.org: Online Mendelian Inheritance in Man (OMIM®), an online catalog of human genes and genetic disorders. Nucleic Acids Res. 2015, 43, D789–D798. [Google Scholar] [CrossRef] [PubMed]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.R.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; et al. ClinVar: Improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018, 46, D1062–D1067. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, F.; Allen, J.E.; Allen, J.; Alvarez-Jarreta, J.; Ridwan Amode, M.; Armean, I.M.; Austine-Orimoloye, O.; Azov, A.G.; Barnes, I.; Bennett, R.; et al. Ensembl 2022. Nucleic Acids Res. 2022, 50, D988–D995. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Panday, S.K.; Alexov, E. SAAFEC-SEQ: A Sequence-Based Method for Predicting the Effect of Single Point Mutations on Protein Thermodynamic Stability. Int. J. Mol. Sci. 2021, 22, 606. [Google Scholar] [CrossRef]
- Capriotti, E.; Fariselli, P.; Casadio, R. I-Mutant2.0: Predicting stability changes upon mutation from the protein sequence or structure. Nucleic Acids Res. 2005, 33, W306–W310. [Google Scholar] [CrossRef]
- Savojardo, C.; Fariselli, P.; Martelli, P.L.; Casadio, R. INPS-MD: A web server to predict stability of protein variants from sequence and structure. Bioinformatics 2016, 32, 2542–2544. [Google Scholar] [CrossRef]
- Klausen, M.S.; Jespersen, M.C.; Nielsen, H.; Jensen, K.K.; Jurtz, V.I.; Sønderby, C.K.; Sommer, M.O.A.; Nielsen, M.; Petersen, B.; Marcatili, P. NetSurfP-2.0: Improved prediction of protein structural features by integrated deep learning. Proteins 2019, 87, 520–527. [Google Scholar] [CrossRef]
- Capriotti, E.; Calabrese, R.; Casadio, R. Predicting the insurgence of human genetic diseases associated to single point protein mutations with support vector machines and evolutionary information. Bioinformatics 2006, 22, 2729–2734. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Methods | Dataset 1 | |||||
| Cut-off > 2 kcal/mol | Cut-off > 1 kcal/mol | Cut-off > 1.1 kcal/mol | ||||
| No. of stabilizing mutations | No. of destabilizing mutations | No. of stabilizing mutations | No. of destabilizing mutations | No. of stabilizing mutations | No. of destabilizing mutations | |
| SAAFEC-SEQ | 0 | 199 | 1 | 1222 | 1 | 1021 |
| I-mutant 2.0 | 3 | 309 | 44 | 1374 | 34 | 1223 |
| INPS-SEQ | 5 | 252 | 52 | 826 | 40 | 674 |
| Avg (SAAFEC-SEQ, I-mutant 2.0) | 0 | 141 | 0 | 1211 | 0 | 1000 |
| Avg (SAAFEC-SEQ, INPS-SEQ) | 0 | 214 | 2 | 883 | 2 | 734 |
| Avg (all) | 0 | 153 | 1 | 943 | 1 | 766 |
| Dataset 2 | ||||||
| Cut-off > 2 kcal/mol | Cut-off > 1 kcal/mol | Cut-off > 1.1 kcal/mol | ||||
| No. of stabilizing mutations | No. of destabilizing mutations | No. of stabilizing mutations | No. of destabilizing mutations | No. of stabilizing mutations | No. of destabilizing mutations | |
| SAAFEC-SEQ | 0 | 251 | 1 | 1564 | 1 | 1312 |
| I-mutant 2.0 | 4 | 417 | 66 | 1756 | 48 | 1564 |
| INPS-SEQ | 6 | 327 | 67 | 1101 | 50 | 885 |
| Avg (SAAFEC-SEQ, I-mutant 2.0) | 0 | 190 | 1 | 1554 | 0 | 1285 |
| Avg (SAAFEC-SEQ, INPS-SEQ) | 0 | 280 | 2 | 1160 | 2 | 968 |
| Avg (all) | 0 | 207 | 1 | 1227 | 1 | 1002 |
| Se. No | Regression Model | Dataset 1 | |||
| Training set | Test set | ||||
| AUC | MCC | AUC | MCC | ||
| 1. | PSAAFEC-SEQ,RSA | 0.82 | 0.52 | 0.81 | 0.52 |
| 2. | PI-mutant 2.0,RSA | 0.80 | 0.52 | 0.76 | 0.52 |
| 3. | PINPS-SEQ,RSA | 0.84 | 0.54 | 0.85 | 0.54 |
| 4. | PAvg (SAAFEC-SEQ, I-mutant 2.0),RSA | 0.80 | 0.49 | 0.81 | 0.49 |
| 5. | PAvg (SAAFEC-SEQ, INPS-SEQ),RSA | 0.84 | 0.55 | 0.85 | 0.55 |
| 6. | PAvg (All),RSA | 0.82 | 0.51 | 0.81 | 0.51 |
| Dataset 2 | |||||
| Training set | Test set | ||||
| AUC | MCC | AUC | MCC | ||
| 1. | PSAAFEC-SEQ,RSA | 0.81 | 0.51 | 0.81 | 0.51 |
| 2. | PI-mutant 2.0,RSA | 0.80 | 0.51 | 0.78 | 0.51 |
| 3. | PINPS-SEQ,RSA | 0.84 | 0.53 | 0.83 | 0.53 |
| 4. | PAvg (SAAFEC-SEQ, I-mutant 2.0),RSA | 0.79 | 0.47 | 0.82 | 0.47 |
| 5. | PAvg (SAAFEC-SEQ, INPS-SEQ),RSA | 0.84 | 0.54 | 0.83 | 0.54 |
| 6. | PAvg (All),RSA | 0.82 | 0.51 | 0.80 | 0.51 |
| Se. No. | Methods | Dataset 1 | ||||
| Cut-off | Avg TPR | Avg FPR | Avg FNR | Avg Accuracy | ||
| 1. | SAAFEC-SEQ | −1.1 | 0.44 ± 0.02 | 0.14 ± 0.01 | 0.56 ± 0.02 | 0.65 ± 0.01 |
| 2. | I-mutant 2.0 | −1.7 | 0.18 ± 0.01 | 0.12 ± 0.01 | 0.82 ± 0.01 | 0.53 ± 0.01 |
| 3. | INPS-SEQ | −0.5 | 0.68 ± 0.01 | 0.25 ± 0.01 | 0.32 ± 0.01 | 0.72 ± 0.01 |
| 4. | Avg (SAAFEC-SEQ, I-mutant 2.0) | −1.0 | 0.45 ± 0.02 | 0.26 ± 0.01 | 0.55 ± 0.02 | 0.59 ± 0.01 |
| 5. | Avg (SAAFEC-SEQ, INPS-SEQ) | −0.6 | 0.69 ± 0.01 | 0.28 ± 0.01 | 0.31 ± 0.01 | 0.71 ± 0.01 |
| 6. | Avg (all) | −0.7 | 0.64 ± 0.01 | 0.32 ± 0.01 | 0.36 ± 0.01 | 0.66 ± 0.01 |
| 7. | RSA | 0.35 | 0.77 ± 0.01 | 0.27 ± 0.01 | 0.23 ± 0.01 | 0.75 ± 0.01 |
| 8. | PSAAFEC-SEQ,RSA | -- | 0.74 ± 0.01 | 0.24 ± 0.01 | 0.26 ± 0.01 | 0.75 ± 0.01 |
| 9. | PI-mutant 2.0,RSA | -- | 0.75 ± 0.01 | 0.25 ± 0.01 | 0.25 ± 0.01 | 0.75 ± 0.01 |
| 10. | PINPS-SEQ,RSA | -- | 0.76 ± 0.01 | 0.23 ± 0.01 | 0.24 ± 0.01 | 0.77 ± 0.01 |
| 11. | PAvg (SAAFEC-SEQ, I-mutant 2.0),RSA | -- | 0.75 ± 0.01 | 0.26 ± 0.01 | 0.25 ± 0.01 | 0.74 ± 0.01 |
| 12. | PAvg (SAAFEC-SEQ, INPS-SEQ),RSA | -- | 0.75 ± 0.01 | 0.21 ± 0.01 | 0.25 ± 0.01 | 0.77 ± 0.01 |
| 13. | PAvg (All),RSA | -- | 0.76 ± 0.01 | 0.25 ± 0.01 | 0.24 ± 0.01 | 0.75 ± 0.01 |
| 14. | PhD-SNP | -- | 0.68 ± 0.01 | 0.22 ± 0.01 | 0.32 ± 0.01 | 0.73 ± 0.01 |
| 15. | PolyPhen (HumDiv) | -- | 0.93 ± 0.01 | 0.27 ± 0.01 | 0.07 ± 0.01 | 0.83 ± 0.01 |
| 16. | PolyPhen (HumVar) | -- | 0.89 ± 0.01 | 0.17 ± 0.01 | 0.11 ± 0.01 | 0.86 ± 0.01 |
| 17. | Align GVGD | -- | 0.65 ± 0.02 | 0.36 ± 0.01 | 0.35 ± 0.02 | 0.65 ± 0.01 |
| 18. | SIFT 4G | 0.05 | 0.98 ± 0.00 | 0.49 ± 0.01 | 0.02 ± 0.0 | 0.75 ± 0.01 |
| Dataset 2 | ||||||
| Cut-off | Avg TPR | Avg FPR | Avg FNR | Avg Accuracy | ||
| 1. | SAAFEC-SEQ | −1.0 | 0.42 ± 0.01 | 0.14 ± 0.01 | 0.58 ± 0.01 | 0.64 ± 0.01 |
| 2. | I-mutant 2.0 | −1.3 | 0.31 ± 0.01 | 0.24 ± 0.01 | 0.69 ± 0.01 | 0.54 ± 0.01 |
| 3. | INPS-SEQ | −0.4 | 0.74 ± 0.01 | 0.31 ± 0.01 | 0.26 ± 0.01 | 0.71 ± 0.01 |
| 4. | Avg (SAAFEC-SEQ, I-mutant 2.0) | −1.0 | 0.44 ± 0.01 | 0.26 ± 0.01 | 0.56 ± 0.01 | 0.59 ± 0.01 |
| 5. | Avg (SAAFEC-SEQ, INPS-SEQ) | −0.6 | 0.70 ± 0.01 | 0.28 ± 0.01 | 0.30 ± 0.01 | 0.71 ± 0.01 |
| 6. | Avg (all) | −0.7 | 0.64 ± 0.01 | 0.32 ± 0.01 | 0.36 ± 0.01 | 0.66 ± 0.01 |
| 7. | RSA | 0.35 | 0.77 ± 0.01 | 0.27 ± 0.01 | 0.23 ± 0.01 | 0.75 ± 0.01 |
| 8. | PSAAFEC-SEQ,RSA | -- | 0.75 ± 0.01 | 0.24 ± 0.01 | 0.25 ± 0.01 | 0.75 ± 0.01 |
| 9. | PI-mutant 2.0,RSA | -- | 0.76 ± 0.01 | 0.26 ± 0.01 | 0.24 ± 0.01 | 0.75 ± 0.01 |
| 10. | PINPS-SEQ,RSA | -- | 0.76 ± 0.01 | 0.22 ± 0.01 | 0.24 ± 0.01 | 0.77 ± 0.01 |
| 11. | PAvg (SAAFEC-SEQ, I-mutant 2.0),RSA | -- | 0.75 ± 0.01 | 0.26 ± 0.01 | 0.25 ± 0.01 | 0.75 ± 0.01 |
| 12. | PAvg (SAAFEC-SEQ, INPS-SEQ),RSA | -- | 0.76 ± 0.01 | 0.22 ± 0.01 | 0.24 ± 0.01 | 0.77 ± 0.01 |
| 13. | PAvg (All),RSA | -- | 0.76 ± 0.01 | 0.25 ± 0.01 | 0.24 ± 0.01 | 0.76 ± 0.01 |
| 14. | PhD-SNP | -- | 0.69 ± 0.01 | 0.21 ± 0.01 | 0.31 ± 0.01 | 0.74 ± 0.01 |
| 15. | PolyPhen (HumDiv) | -- | 0.93 ± 0.01 | 0.29 ± 0.01 | 0.07 ± 0.01 | 0.82 ± 0.01 |
| 16. | PolyPhen (HumVar) | -- | 0.89 ± 0.01 | 0.18 ± 0.01 | 0.11 ± 0.01 | 0.86 ± 0.01 |
| 17. | Align GVGD | -- | 0.66 ± 0.01 | 0.36 ± 0.01 | 0.34 ± 0.01 | 0.65 ± 0.01 |
| 18. | SIFT 4G | 0.05 | 0.98 ± 0.00 | 0.50 ± 0.01 | 0.02 ± 0.00 | 0.74 ± 0.01 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pandey, P.; Alexov, E. Most Monogenic Disorders Are Caused by Mutations Altering Protein Folding Free Energy. Int. J. Mol. Sci. 2024, 25, 1963. https://doi.org/10.3390/ijms25041963
Pandey P, Alexov E. Most Monogenic Disorders Are Caused by Mutations Altering Protein Folding Free Energy. International Journal of Molecular Sciences. 2024; 25(4):1963. https://doi.org/10.3390/ijms25041963
Chicago/Turabian StylePandey, Preeti, and Emil Alexov. 2024. "Most Monogenic Disorders Are Caused by Mutations Altering Protein Folding Free Energy" International Journal of Molecular Sciences 25, no. 4: 1963. https://doi.org/10.3390/ijms25041963
APA StylePandey, P., & Alexov, E. (2024). Most Monogenic Disorders Are Caused by Mutations Altering Protein Folding Free Energy. International Journal of Molecular Sciences, 25(4), 1963. https://doi.org/10.3390/ijms25041963