
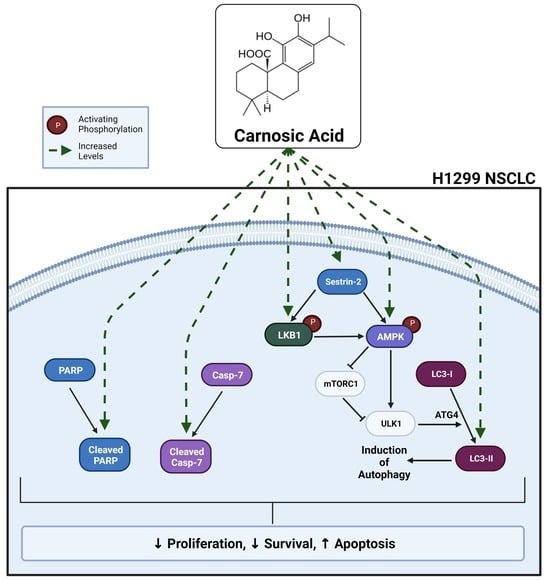
Carnosic Acid against Lung Cancer: Induction of Autophagy and Activation of Sestrin-2/LKB1/AMPK Signalling



Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
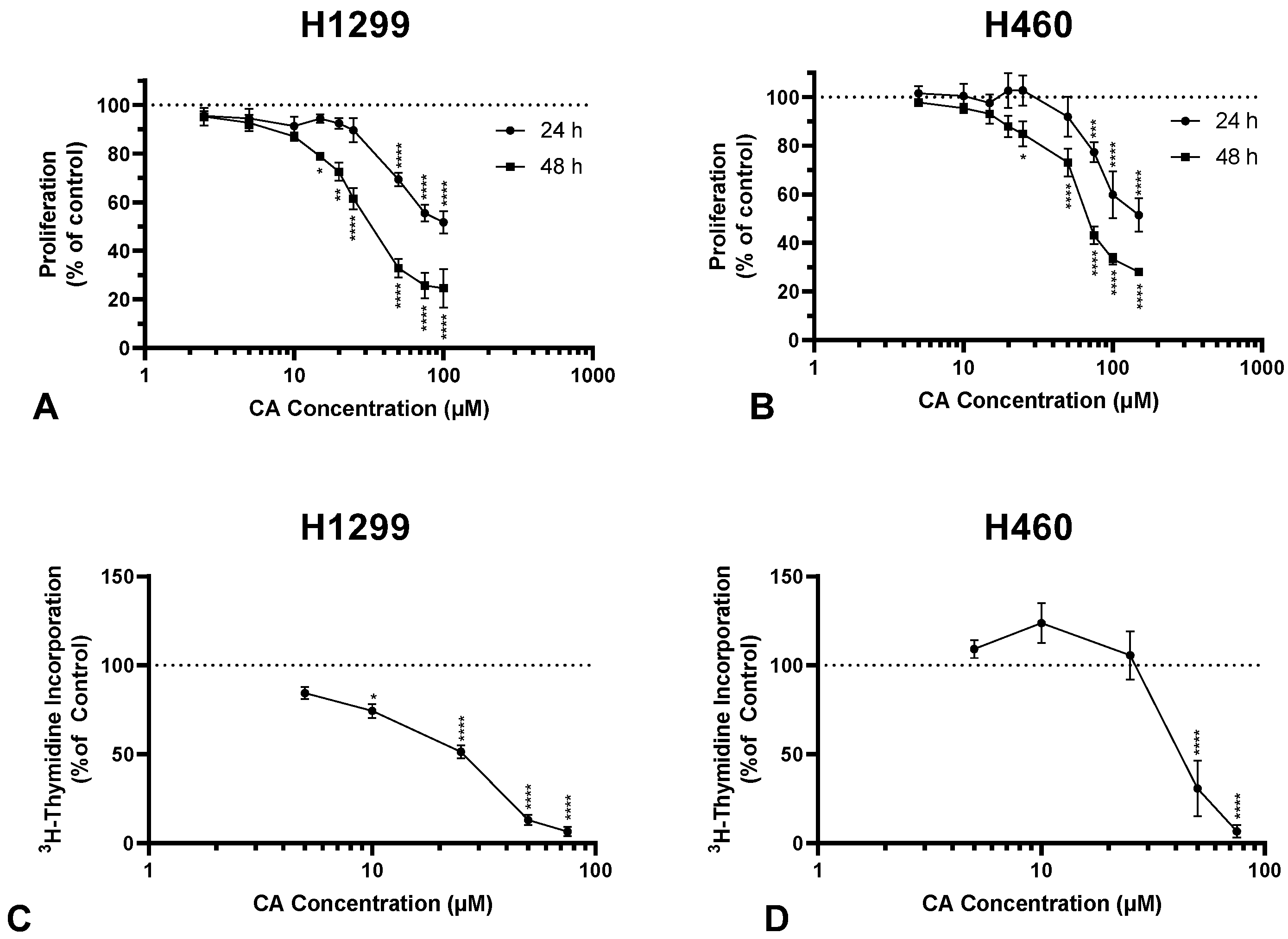
2.1. Carnosic Acid Inhibits Proliferation and Survival of NSCLC Cells
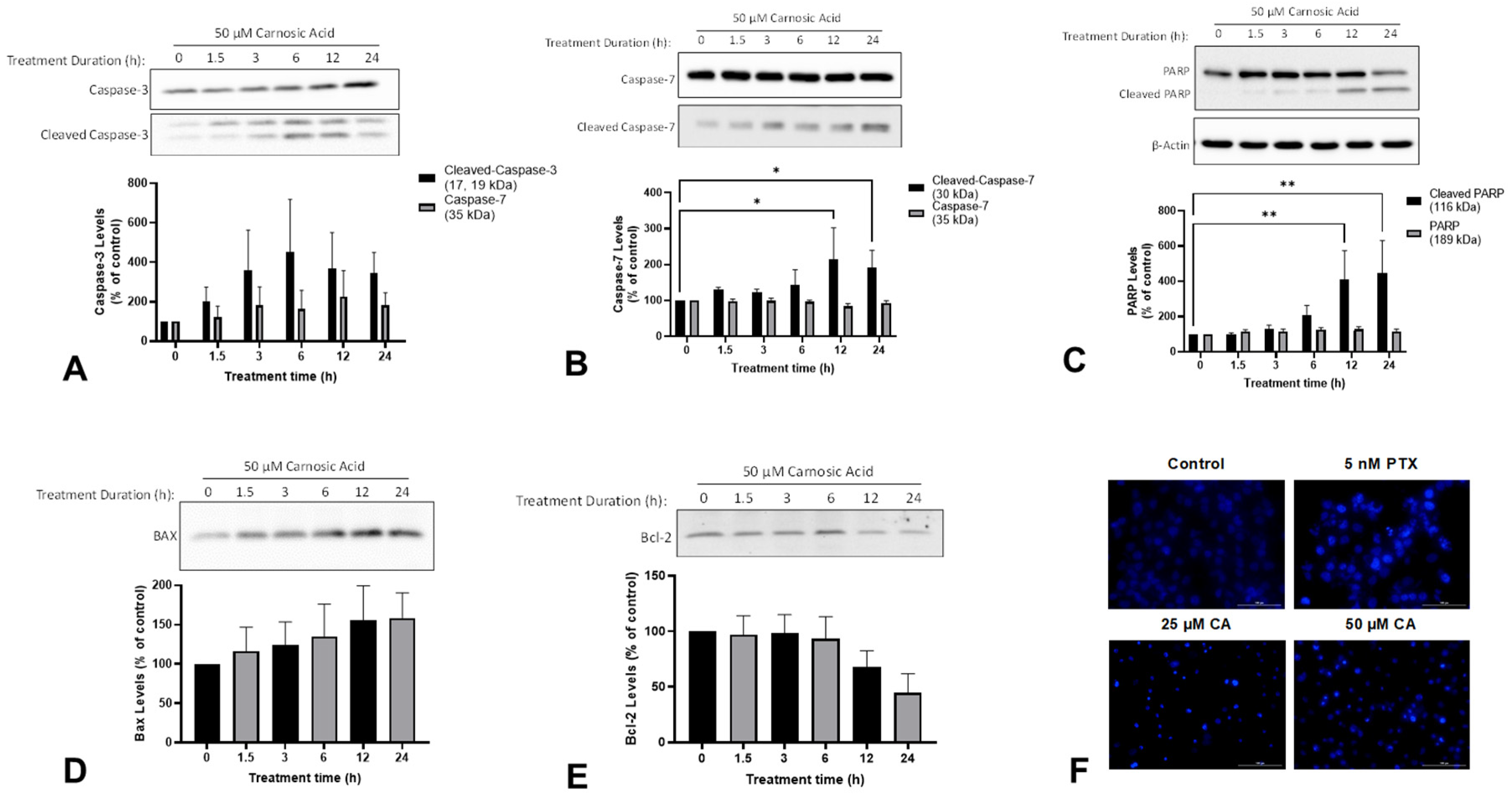
2.2. Carnosic Acid Induces Apoptosis of H1299 NSCLC Cells
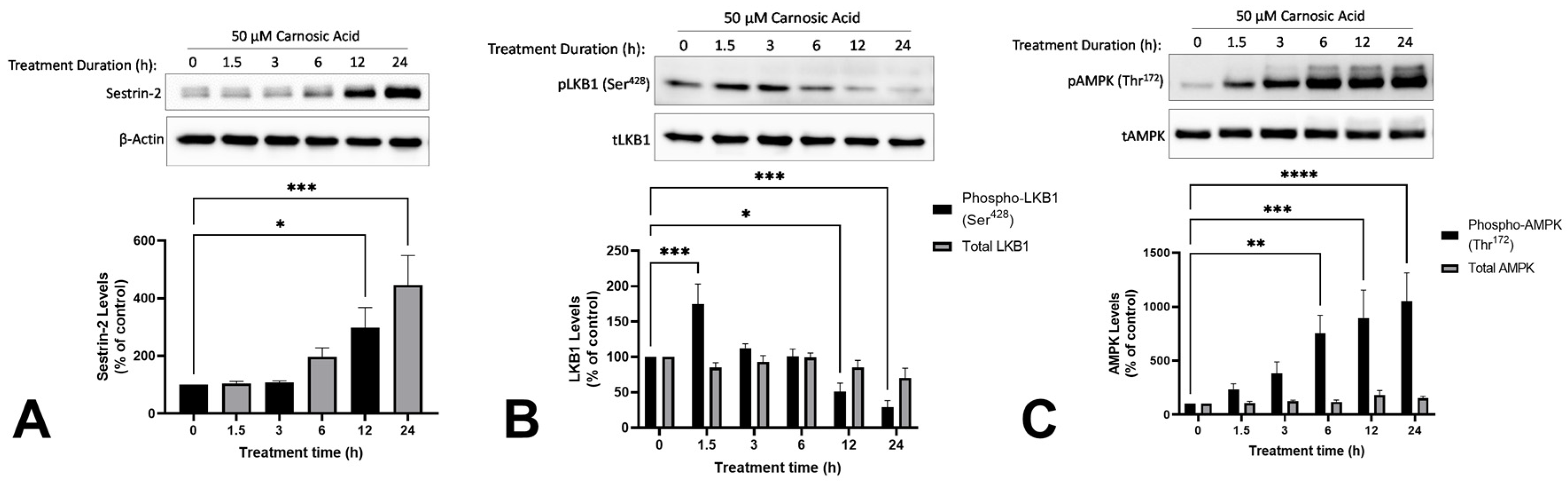
2.3. Carnosic Acid Activates Sestrin-2/LKB1/AMPK Signalling in H1299 NSCLC Cells
2.4. Carnosic Acid Activates Autophagy Signalling in H1299 NSCLC Cells
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Culture
4.3. Crystal Violet Proliferation Assay
4.4. [3H]-Thymidine Incorporation Assay
4.5. Clonogenic Survival Assay
4.6. SDS-PAGE and Western Blotting
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Parkin, D.M.; Piñeros, M.; Znaor, A.; Bray, F. Cancer Statistics for the Year 2020: An Overview. Int. J. Cancer 2021, 149, 778–789. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, A.G.; Tsao, M.S.; Beasley, M.B.; Borczuk, A.C.; Brambilla, E.; Cooper, W.A.; Dacic, S.; Jain, D.; Kerr, K.M.; Lantuejoul, S.; et al. The 2021 WHO Classification of Lung Tumors: Impact of Advances since 2015. J. Thorac. Oncol. 2022, 17, 362–387. [Google Scholar] [CrossRef] [PubMed]
- Ettinger, D.S.; Wood, D.E.; Aggarwal, C.; Aisner, D.L.; Akerley, W.; Bauman, J.R.; Bharat, A.; Bruno, D.S.; Chang, J.Y.; Chirieac, L.R.; et al. NCCN Guidelines Insights: Non-Small Cell Lung Cancer, Version 1.2020. J. Natl. Compr. Cancer Netw. JNCCN 2019, 17, 1464–1472. [Google Scholar] [CrossRef]
- National Cancer Institute. SEER*Explorer: An Interactive Website for SEER Cancer Statistics. Available online: https://seer.cancer.gov/statistics-network/explorer/ (accessed on 26 September 2023).
- Brambilla, E.; Gazdar, A. Pathogenesis of Lung Cancer Signaling Pathways: Roadmap for Therapies. Eur. Respir. J. Off. J. Eur. Soc. Clin. Respir. Physiol. 2009, 33, 1485–1497. [Google Scholar] [CrossRef]
- Westover, D.; Zugazagoitia, J.; Cho, B.C.; Lovly, C.M.; Paz-Ares, L. Mechanisms of Acquired Resistance to First- and Second-Generation EGFR Tyrosine Kinase Inhibitors. Ann. Oncol. 2018, 29, i10–i19. [Google Scholar] [CrossRef]
- Stephenson, J. Potent Anti-Cancer Drug from Witches’ Umbrella. Lancet Oncol. 2000, 1, 8. [Google Scholar] [CrossRef]
- Renneberg, R. Biotech History: Yew Trees, Paclitaxel Synthesis and Fungi. Biotechnol. J. 2007, 2, 1207–1209. [Google Scholar] [CrossRef]
- Moreno, S.; Scheyer, T.; Romano, C.S.; Vojnov, A.A. Antioxidant and Antimicrobial Activities of Rosemary Extracts Linked to Their Polyphenol Composition. Free Radic. Res. 2006, 40, 223–231. [Google Scholar] [CrossRef]
- Nieto, G.; Ros, G.; Castillo, J. Antioxidant and Antimicrobial Properties of Rosemary (Rosmarinus officinalis, L.): A Review. Medicines 2018, 5, 98. [Google Scholar] [CrossRef] [PubMed]
- Naimi, M.; Vlavcheski, F.; Shamshoum, H.; Tsiani, E. Rosemary Extract as a Potential Anti-Hyperglycemic Agent: Current Evidence and Future Perspectives. Nutrients 2017, 9, 968. [Google Scholar] [CrossRef]
- Vlavcheski, F.; Tsiani, E. Attenuation of Free Fatty Acid-Induced Muscle Insulin Resistance by Rosemary Extract. Nutrients 2018, 10, 1623. [Google Scholar] [CrossRef]
- Moore, J.; Yousef, M.; Tsiani, E. Anticancer Effects of Rosemary (Rosmarinus officinalis L.) Extract and Rosemary Extract Polyphenols. Nutrients 2016, 8, 731. [Google Scholar] [CrossRef]
- O’Neill, E.J.; Den Hartogh, D.J.; Azizi, K.; Tsiani, E. Anticancer Properties of Carnosol: A Summary of In Vitro and In Vivo Evidence. Antioxidants 2020, 9, 961. [Google Scholar] [CrossRef]
- Moore, J.; Megaly, M.; MacNeil, A.J.; Klentrou, P.; Tsiani, E. Rosemary Extract Reduces Akt/MTOR/P70S6K Activation and Inhibits Proliferation and Survival of A549 Human Lung Cancer Cells. Biomed. Pharmacother. 2016, 83, 725–732. [Google Scholar] [CrossRef]
- O’Neill, E.J.; Moore, J.; Song, J.; Tsiani, E. Inhibition of Non-Small Cell Lung Cancer Proliferation and Survival by Rosemary Extract Is Associated with Activation of ERK and AMPK. Life 2021, 12, 52. [Google Scholar] [CrossRef] [PubMed]
- Jaglanian, A.; Tsiani, E. Rosemary Extract Inhibits Proliferation, Survival, Akt, and MTOR Signaling in Triple-Negative Breast Cancer Cells. Int. J. Mol. Sci. 2020, 21, 810. [Google Scholar] [CrossRef] [PubMed]
- Jaglanian, A.; Termini, D.; Tsiani, E. Rosemary (Rosmarinus officinalis L.) Extract Inhibits Prostate Cancer Cell Proliferation and Survival by Targeting Akt and MTOR. Biomed. Pharmacother. 2020, 131, 110717. [Google Scholar] [CrossRef] [PubMed]
- William, W.N.; Kim, J.-S.; Liu, D.D.; Solis, L.; Behrens, C.; Lee, J.J.; Lippman, S.M.; Kim, E.S.; Hong, W.K.; Wistuba, I.I.; et al. The Impact of Phosphorylated AMP-Activated Protein Kinase Expression on Lung Cancer Survival. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2012, 23, 78–85. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Ding, H.; Tang, X.; Liang, M.; Li, S.; Zhang, J.; Cao, J. Quercetin Induces Pro-Apoptotic Autophagy via SIRT1/AMPK Signaling Pathway in Human Lung Cancer Cell Lines A549 and H1299 in Vitro. Thorac. Cancer 2021, 12, 1415–1422. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Fan, Y.; Zhang, Y.; Liu, Y.; Yu, Y.; Ma, M. Resveratrol Induces Autophagy and Apoptosis in Non-Small-Cell Lung Cancer Cells by Activating the NGFR-AMPK-MTOR Pathway. Nutrients 2022, 14, 2413. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.-H.; Hsieh, C.-H.; Tsai, S.-Y.; Wang, C.-Y.; Wang, C.-C. Anticancer Effects of Epigallocatechin-3-Gallate Nanoemulsion on Lung Cancer Cells through the Activation of AMP-Activated Protein Kinase Signaling Pathway. Sci. Rep. 2020, 10, 5163. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Budanov, A.V.; Talukdar, S.; Park, E.J.; Park, H.L.; Park, H.-W.; Bandyopadhyay, G.; Li, N.; Aghajan, M.; Jang, I.; et al. Maintenance of Metabolic Homeostasis by Sestrin2 and Sestrin3. Cell Metab. 2012, 16, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Budanov, A.V.; Karin, M. The P53-Regulated Sestrin Gene Products Inhibit MTOR Signaling. Cell 2008, 134, 451–460. [Google Scholar] [CrossRef] [PubMed]
- Shackelford, D.B.; Shaw, R.J. The LKB1–AMPK Pathway: Metabolism and Growth Control in Tumour Suppression. Nat. Rev. Cancer 2009, 9, 563–575. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, S.; Iwakawa, R.; Takahashi, K.; Kohno, T.; Nakanishi, Y.; Matsuno, Y.; Suzuki, K.; Nakamoto, M.; Shimizu, E.; Minna, J.D.; et al. Prevalence and Specificity of LKB1 Genetic Alterations in Lung Cancers. Oncogene 2007, 26, 5911–5918. [Google Scholar] [CrossRef] [PubMed]
- Ji, H.; Ramsey, M.R.; Hayes, D.N.; Fan, C.; McNamara, K.; Kozlowski, P.; Torrice, C.; Wu, M.C.; Shimamura, T.; Perera, S.A.; et al. LKB1 Modulates Lung Cancer Differentiation and Metastasis. Nature 2007, 448, 807–810. [Google Scholar] [CrossRef]
- Morrison, A.; Chen, L.; Wang, J.; Zhang, M.; Yang, H.; Ma, Y.; Budanov, A.; Lee, J.H.; Karin, M.; Li, J. Sestrin2 Promotes LKB1-Mediated AMPK Activation in the Ischemic Heart. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2015, 29, 408–417. [Google Scholar] [CrossRef]
- Kullmann, L.; Krahn, M.P. Controlling the Master—Upstream Regulation of the Tumor Suppressor LKB1. Oncogene 2018, 37, 3045–3057. [Google Scholar] [CrossRef]
- Levy, J.M.M.; Towers, C.G.; Thorburn, A. Targeting Autophagy in Cancer. Nat. Rev. Cancer 2017, 17, 528–542. [Google Scholar] [CrossRef]
- Tamaddoni, A.; Mohammadi, E.; Sedaghat, F.; Qujeq, D.; As’Habi, A. The Anticancer Effects of Curcumin via Targeting the Mammalian Target of Rapamycin Complex 1 (MTORC1) Signaling Pathway. Pharmacol. Res. 2020, 156, 104798. [Google Scholar] [CrossRef]
- Lu, W.; Ke, H.; Qianshan, D.; Zhen, W.; Guoan, X.; Honggang, Y. Apatinib Has Anti-Tumor Effects and Induces Autophagy in Colon Cancer Cells. Iran. J. Basic Med. Sci. 2017, 20, 990–995. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Zhang, J.; Fan, Y.; Li, Y. Antiproliferative Activity of Carnosic Acid Is Mediated via Inhibition of Cell Migration and Invasion, and Suppression of Phosphatidylinositol 3-Kinases (PI3K)/AKT/Mammalian Target of Rapamycin (MTOR) Signaling Pathway. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2019, 25, 7864–7871. [Google Scholar] [CrossRef]
- Corveloni, A.C.; Semprebon, S.C.; Baranoski, A.; Biazi, B.I.; Zanetti, T.A.; Mantovani, M.S. Carnosic Acid Exhibits Antiproliferative and Proapoptotic Effects in Tumoral NCI-H460 and Nontumoral IMR-90 Lung Cells. J. Toxicol. Environ. Health A 2020, 83, 412–421. [Google Scholar] [CrossRef] [PubMed]
- Yesil-Celiktas, O.; Sevimli, C.; Bedir, E.; Vardar-Sukan, F. Inhibitory Effects of Rosemary Extracts, Carnosic Acid and Rosmarinic Acid on the Growth of Various Human Cancer Cell Lines. Plant Foods Hum. Nutr. 2010, 65, 158–163. [Google Scholar] [CrossRef] [PubMed]
- El-Huneidi, W.; Bajbouj, K.; Muhammad, J.S.; Vinod, A.; Shafarin, J.; Khoder, G.; Saleh, M.A.; Taneera, J.; Abu-Gharbieh, E. Carnosic Acid Induces Apoptosis and Inhibits Akt/MTOR Signaling in Human Gastric Cancer Cell Lines. Pharmaceuticals 2021, 14, 230. [Google Scholar] [CrossRef]
- Barni, M.V.; Carlini, M.J.; Cafferata, E.G.; Puricelli, L.; Moreno, S. Carnosic Acid Inhibits the Proliferation and Migration Capacity of Human Colorectal Cancer Cells. Oncol. Rep. 2012, 27, 1041–1048. [Google Scholar] [CrossRef]
- Su, K.; Wang, C.; Zhang, Y.; Cai, Y.; Zhang, Y.; Zhao, Q. The Inhibitory Effects of Carnosic Acid on Cervical Cancer Cells Growth by Promoting Apoptosis via ROS-Regulated Signaling Pathway. Biomed. Pharmacother. 2016, 82, 180–191. [Google Scholar] [CrossRef]
- Pistritto, G.; Trisciuoglio, D.; Ceci, C.; Garufi, A.; D’Orazi, G. Apoptosis as Anticancer Mechanism: Function and Dysfunction of Its Modulators and Targeted Therapeutic Strategies. Aging 2016, 8, 603–619. [Google Scholar] [CrossRef]
- Danial, N.N.; Korsmeyer, S.J. Cell Death: Critical Control Points. Cell 2004, 116, 205–219. [Google Scholar] [CrossRef]
- Degterev, A.; Boyce, M.; Yuan, J. A Decade of Caspases. Oncogene 2003, 22, 8543–8567. [Google Scholar] [CrossRef]
- Lee, S.-O.; Andey, T.; Jin, U.-H.; Kim, K.; Sachdeva, M.; Safe, S. The Nuclear Receptor TR3 Regulates MTORC1 Signaling in Lung Cancer Cells Expressing Wild-Type P53. Oncogene 2012, 31, 3265–3276. [Google Scholar] [CrossRef]
- Liu, X.; Yang, Y.; Shao, H.; Liu, S.; Niu, Y.; Fu, L. Globular Adiponectin Ameliorates Insulin Resistance in Skeletal Muscle by Enhancing the LKB1-Mediated AMPK Activation via SESN2. Sports Med. Health Sci. 2022, 5, 34–41. [Google Scholar] [CrossRef]
- Quan, N.; Sun, W.; Wang, L.; Chen, X.; Bogan, J.S.; Zhou, X.; Cates, C.; Liu, Q.; Zheng, Y.; Li, J. Sestrin2 Prevents Age-Related Intolerance to Ischemia and Reperfusion Injury by Modulating Substrate Metabolism. FASEB J. 2017, 31, 4153–4167. [Google Scholar] [CrossRef]
- Kim, G.T.; Lee, S.H.; Kim, J.I.; Kim, Y.M. Quercetin Regulates the Sestrin 2-AMPK-P38 MAPK Signaling Pathway and Induces Apoptosis by Increasing the Generation of Intracellular ROS in a P53-Independent Manner. Int. J. Mol. Med. 2014, 33, 863. [Google Scholar] [CrossRef]
- Nagalingam, A.; Arbiser, J.L.; Bonner, M.Y.; Saxena, N.K.; Sharma, D. Honokiol Activates AMP-Activated Protein Kinase in Breast Cancer Cells via an LKB1-Dependent Pathway and Inhibits Breast Carcinogenesis. Breast Cancer Res. BCR 2012, 14, R35. [Google Scholar] [CrossRef]
- Fan, Y.; Chiu, J.-F.; Liu, J.; Deng, Y.; Xu, C.; Zhang, J.; Li, G. Resveratrol Induces Autophagy-Dependent Apoptosis in HL-60 Cells. BMC Cancer 2018, 18, 581. [Google Scholar] [CrossRef] [PubMed]
- Kaur, H.; Moreau, R. Curcumin Represses MTORC1 Signaling in Caco-2 Cells by a Two-Sided Mechanism Involving the Loss of IRS-1 and Activation of AMPK. Cell. Signal. 2021, 78, 109842. [Google Scholar] [CrossRef] [PubMed]
- Nemec, M.J.; Kim, H.; Marciante, A.B.; Barnes, R.C.; Hendrick, E.D.; Bisson, W.H.; Talcott, S.T.; Mertens-Talcott, S.U. Polyphenolics from Mango (Mangifera indica L.) Suppress Breast Cancer Ductal Carcinoma in Situ Proliferation through Activation of AMPK Pathway and Suppression of MTOR in Athymic Nude Mice. J. Nutr. Biochem. 2017, 41, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Fan, D.; Yu, S.; Yang, Y.; Qu, S. Pyoluteorin Induces Apoptosis and Autophagy in NSCLC Cells. Biol. Pharm. Bull. 2021, 44, 976–983. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Y.; Xin, M.; Xu, J.; Xiang, X.; Li, X.; Jiang, J.; Jia, X. Polyphyllin II Induced Apoptosis of NSCLC Cells by Inhibiting Autophagy through the MTOR Pathway. Pharm. Biol. 2022, 60, 1781–1789. [Google Scholar] [CrossRef]
- Hua, X.; Xu, J.; Deng, X.; Xu, J.; Li, J.; Zhu, D.Q.; Zhu, J.; Jin, H.; Tian, Z.; Huang, H. New Compound ChlA-F Induces Autophagy-Dependent Anti-Cancer Effect via Upregulating Sestrin-2 in Human Bladder Cancer. Cancer Lett. 2018, 436, 38–51. [Google Scholar] [CrossRef]
- Liang, Y.; Zhu, J.; Huang, H.; Xiang, D.; Li, Y.; Zhang, D.; Li, J.; Wang, Y.; Jin, H.; Jiang, G.; et al. SESN2/Sestrin 2 Induction-Mediated Autophagy and Inhibitory Effect of Isorhapontigenin (ISO) on Human Bladder Cancers. Autophagy 2016, 12, 1229–1239. [Google Scholar] [CrossRef]
- Jeong, S.; Kim, D.Y.; Kang, S.H.; Yun, H.K.; Kim, J.L.; Kim, B.R.; Park, S.H.; Na, Y.J.; Jo, M.J.; Jeong, Y.A. Docosahexaenoic Acid Enhances Oxaliplatin-Induced Autophagic Cell Death via the ER Stress/Sesn2 Pathway in Colorectal Cancer. Cancers 2019, 11, 982. [Google Scholar] [CrossRef]
- Wang, N.; Pan, W.; Zhu, M.; Zhang, M.; Hao, X.; Liang, G.; Feng, Y. Fangchinoline Induces Autophagic Cell Death via P53/Sestrin2/AMPK Signalling in Human Hepatocellular Carcinoma Cells. Br. J. Pharmacol. 2011, 164, 731–742. [Google Scholar] [CrossRef]
- Yen, J.-H.; Huang, S.-T.; Huang, H.-S.; Fong, Y.-C.; Wu, Y.-Y.; Chiang, J.-H.; Su, Y.-C. HGK-Sestrin 2 Signaling-Mediated Autophagy Contributes to Antitumor Efficacy of Tanshinone IIA in Human Osteosarcoma Cells. Cell Death Dis. 2018, 9, 1003. [Google Scholar] [CrossRef]
- Barron, C.C.; Moore, J.; Tsakiridis, T.; Pickering, G.; Tsiani, E. Inhibition of Human Lung Cancer Cell Proliferation and Survival by Wine. Cancer Cell Int. 2014, 14, 6. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
O’Neill, E.J.; Sze, N.S.K.; MacPherson, R.E.K.; Tsiani, E. Carnosic Acid against Lung Cancer: Induction of Autophagy and Activation of Sestrin-2/LKB1/AMPK Signalling. Int. J. Mol. Sci. 2024, 25, 1950. https://doi.org/10.3390/ijms25041950
O’Neill EJ, Sze NSK, MacPherson REK, Tsiani E. Carnosic Acid against Lung Cancer: Induction of Autophagy and Activation of Sestrin-2/LKB1/AMPK Signalling. International Journal of Molecular Sciences. 2024; 25(4):1950. https://doi.org/10.3390/ijms25041950
Chicago/Turabian StyleO’Neill, Eric J., Newman Siu Kwan Sze, Rebecca E. K. MacPherson, and Evangelia Tsiani. 2024. "Carnosic Acid against Lung Cancer: Induction of Autophagy and Activation of Sestrin-2/LKB1/AMPK Signalling" International Journal of Molecular Sciences 25, no. 4: 1950. https://doi.org/10.3390/ijms25041950
APA StyleO’Neill, E. J., Sze, N. S. K., MacPherson, R. E. K., & Tsiani, E. (2024). Carnosic Acid against Lung Cancer: Induction of Autophagy and Activation of Sestrin-2/LKB1/AMPK Signalling. International Journal of Molecular Sciences, 25(4), 1950. https://doi.org/10.3390/ijms25041950