
The XCL1-Mediated DNA Vaccine Targeting Type 1 Conventional Dendritic Cells Combined with Gemcitabine and Anti-PD1 Antibody Induces Potent Antitumor Immunity in a Mouse Lung Cancer Model

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
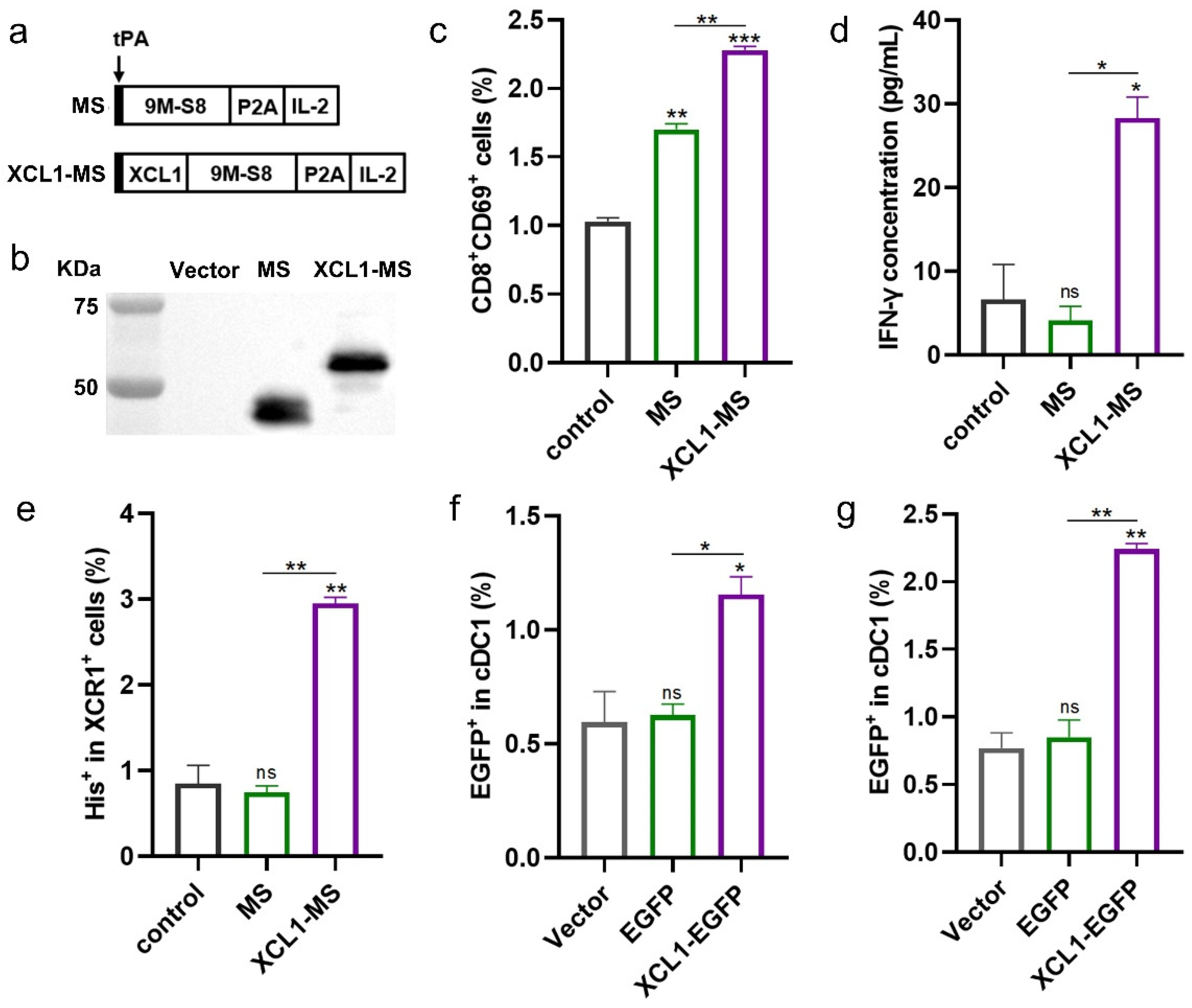
2.1. The Construction and Validation of DNA Vaccines
2.2. The Immunogenicity of DNA Vaccines
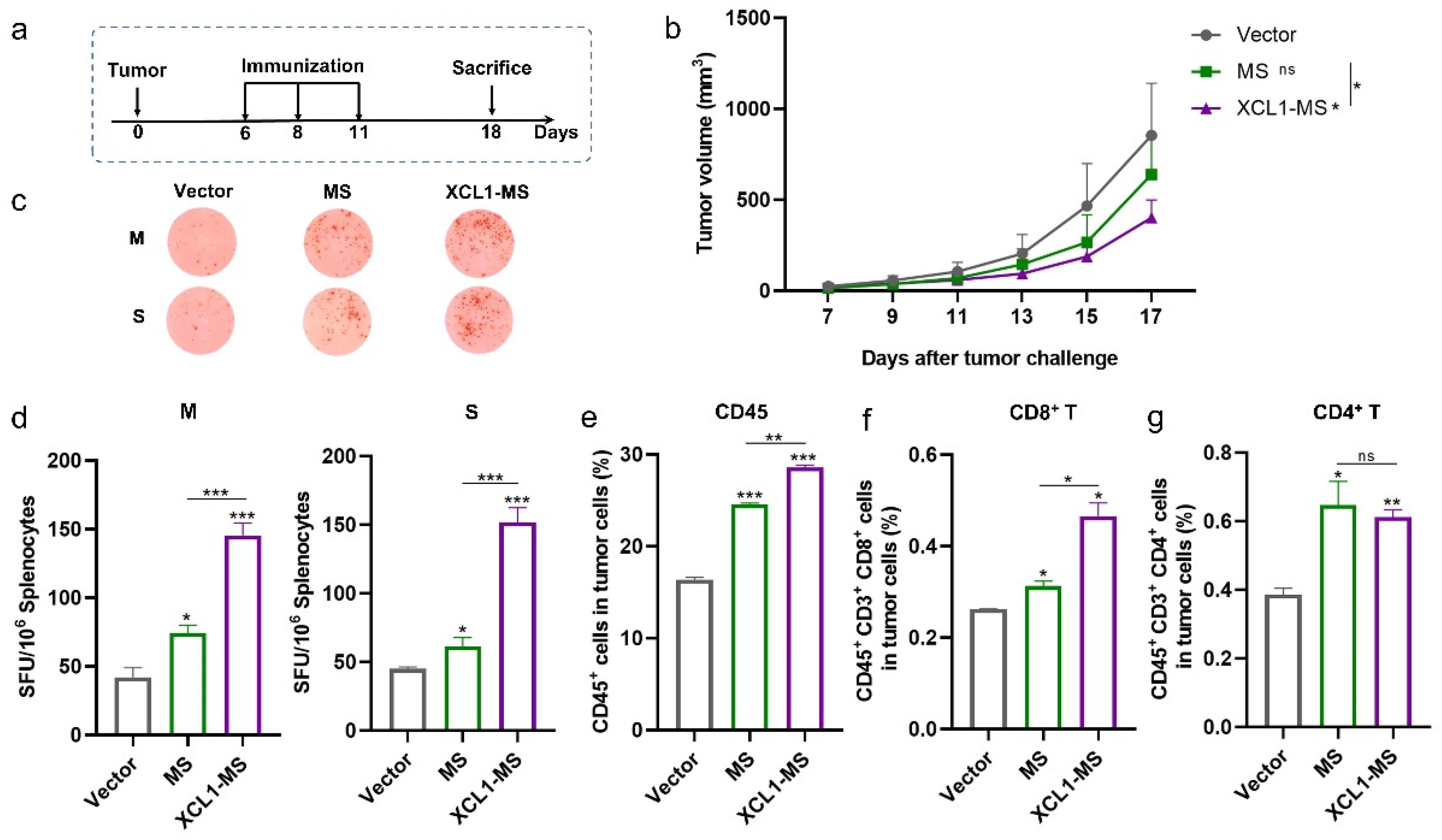
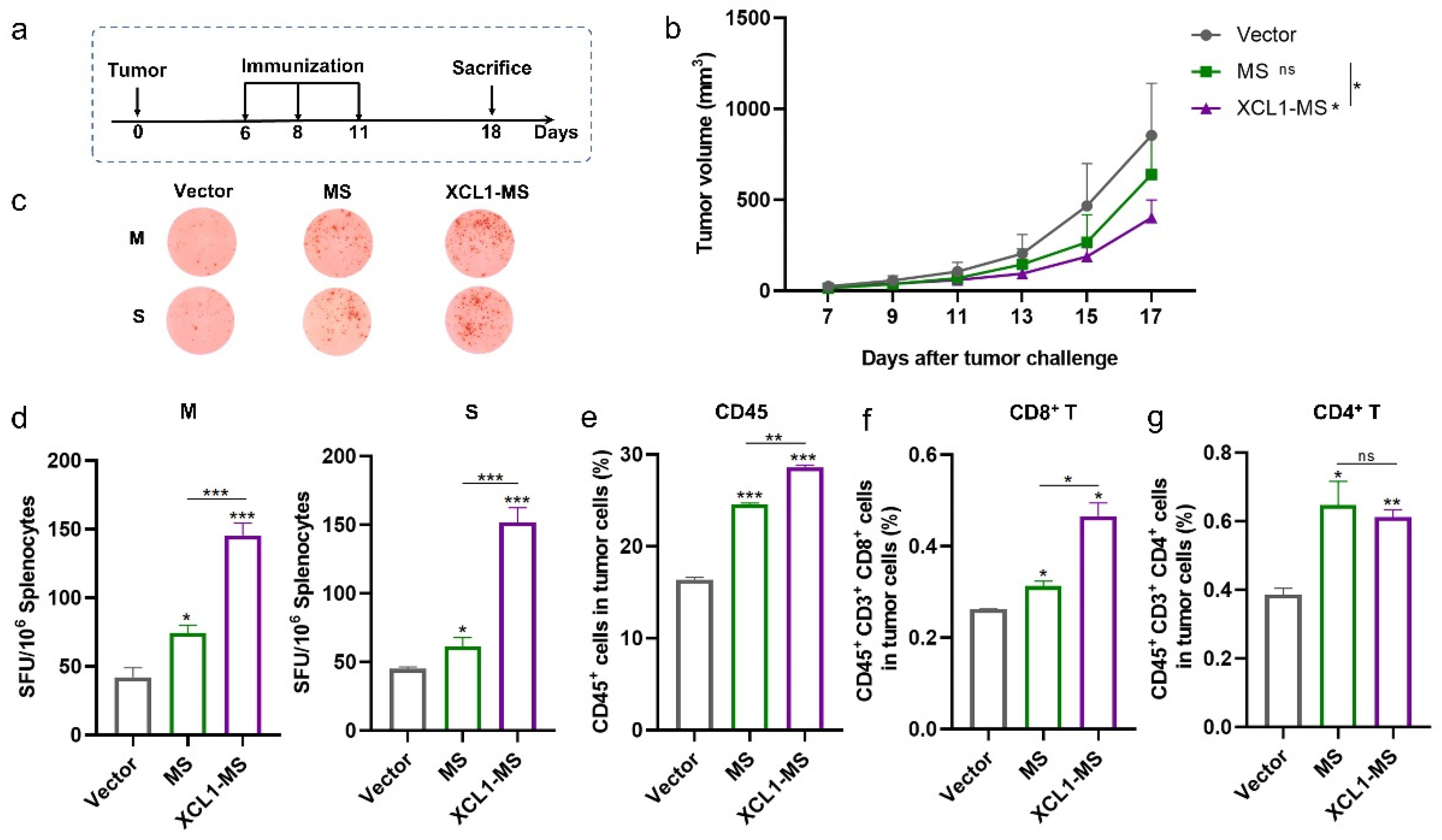
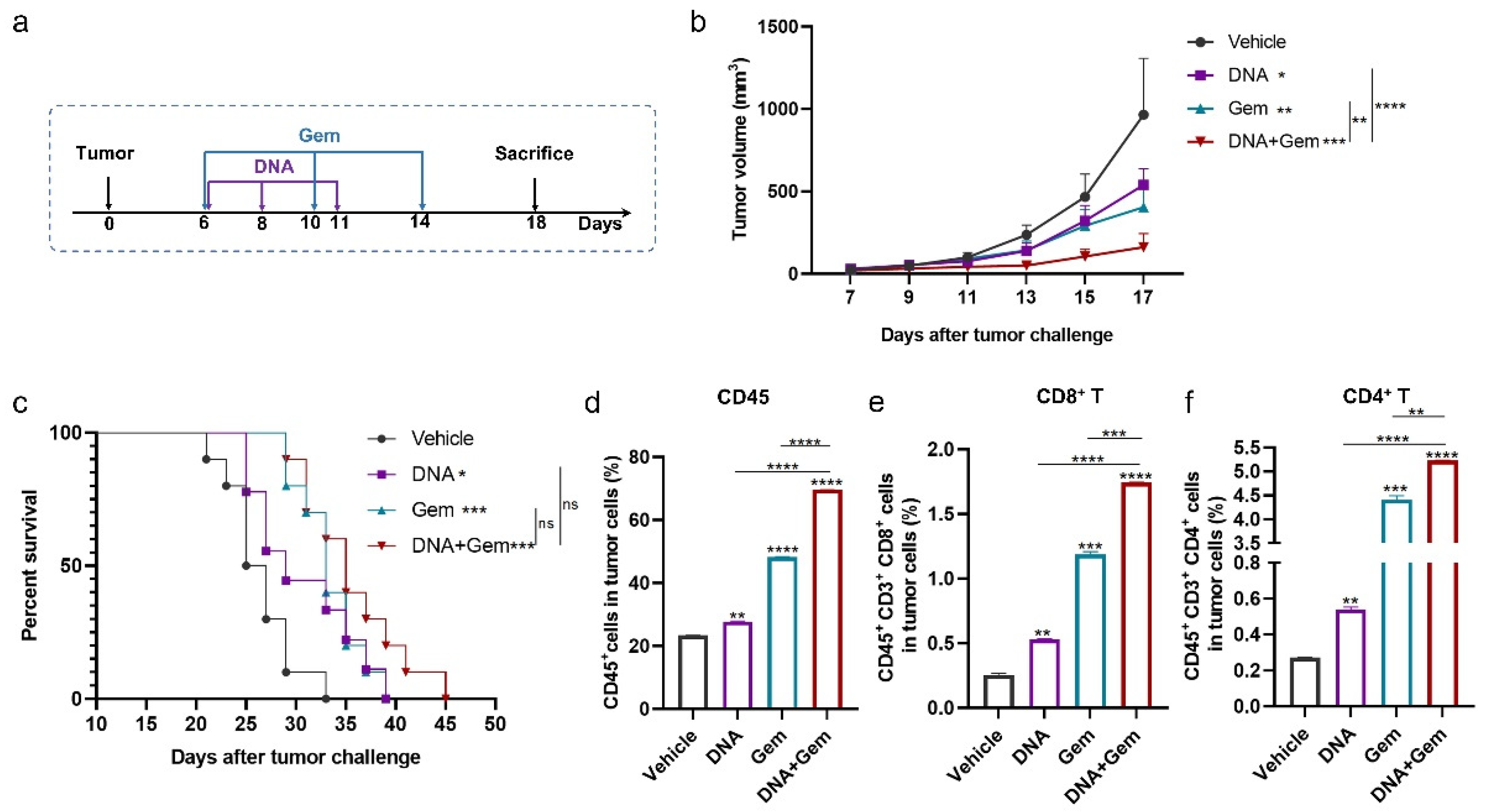
2.3. The Antitumor Effect of DNA Vaccines in a Mouse Lung Cancer Model
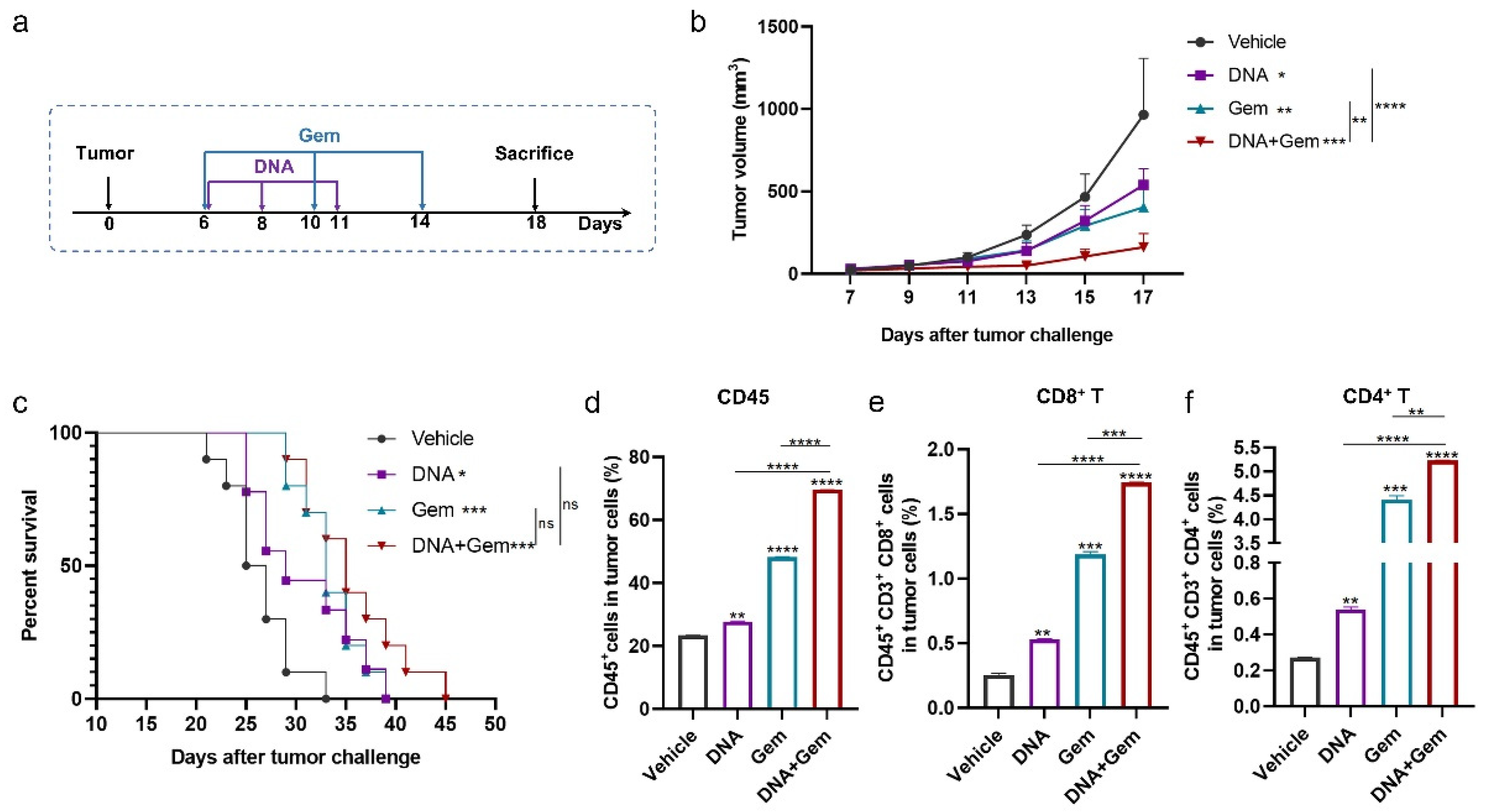
2.4. The Antitumor Effect of the DNA Vaccine (XCL1-MS) Combined with Gem
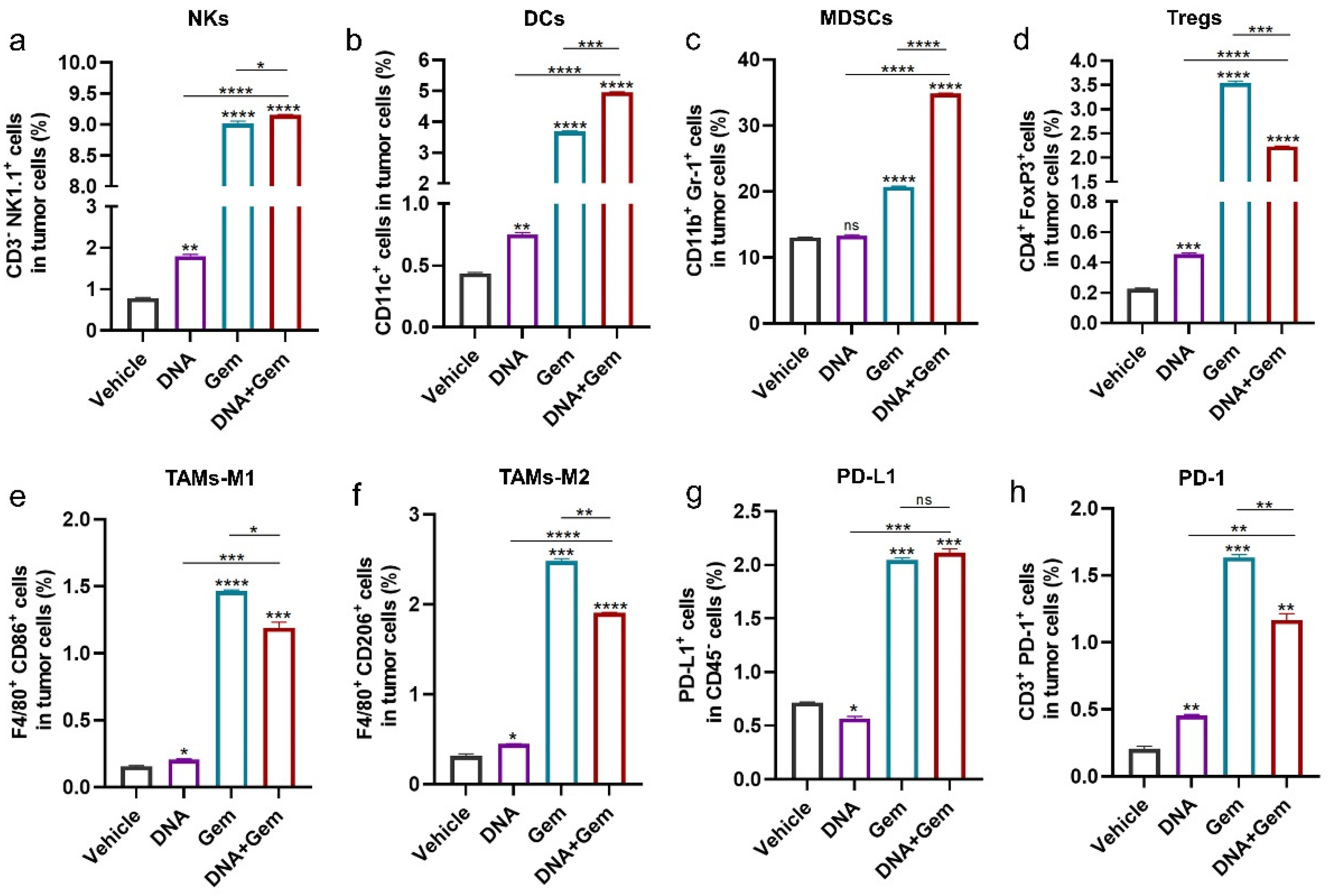
2.5. The Effect of Dual Therapy on the Tumor Immune Microenvironment
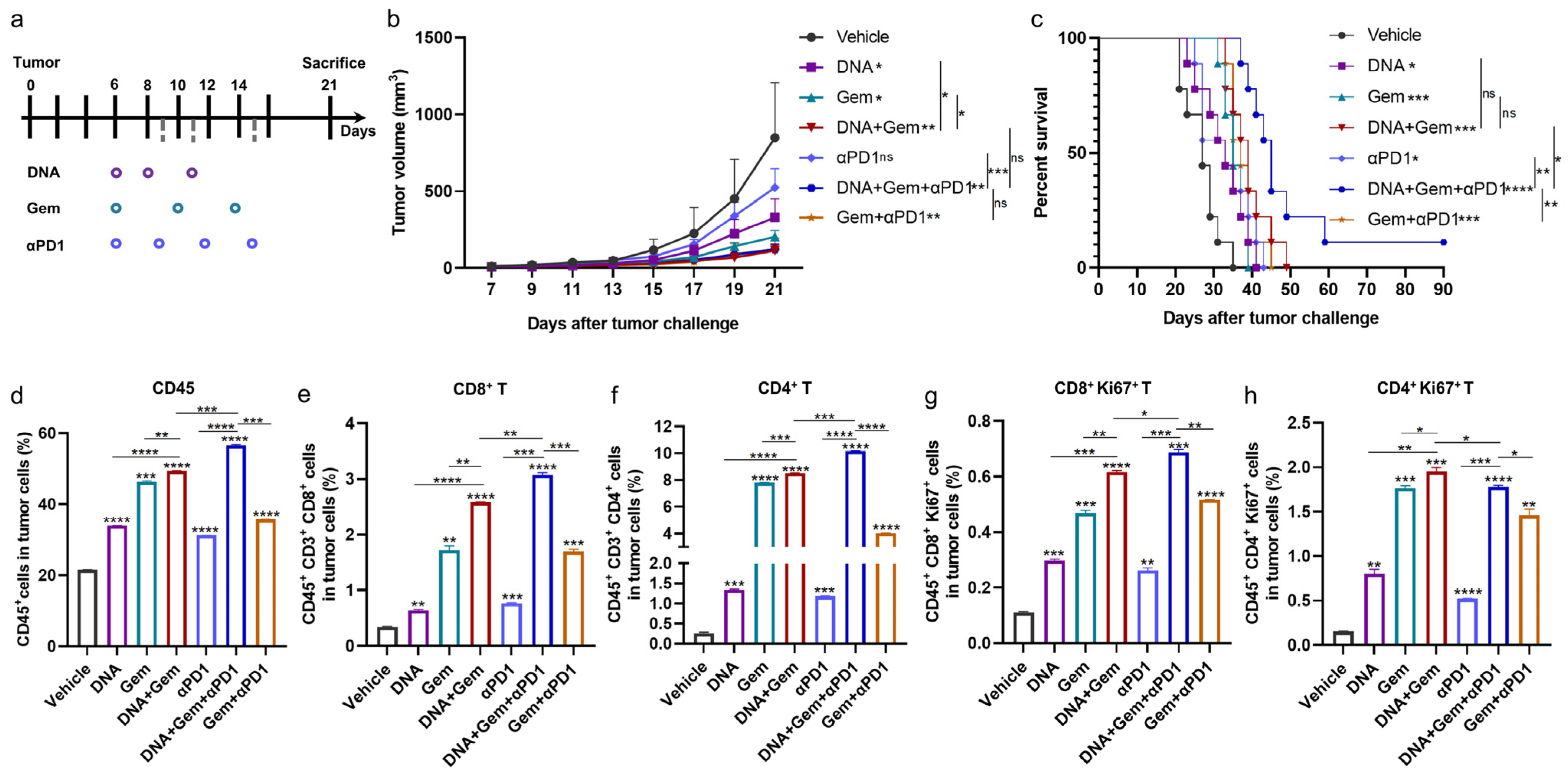
2.6. Anti-PD1 Antibody Further Enhances the Efficacy of Dual Therapy
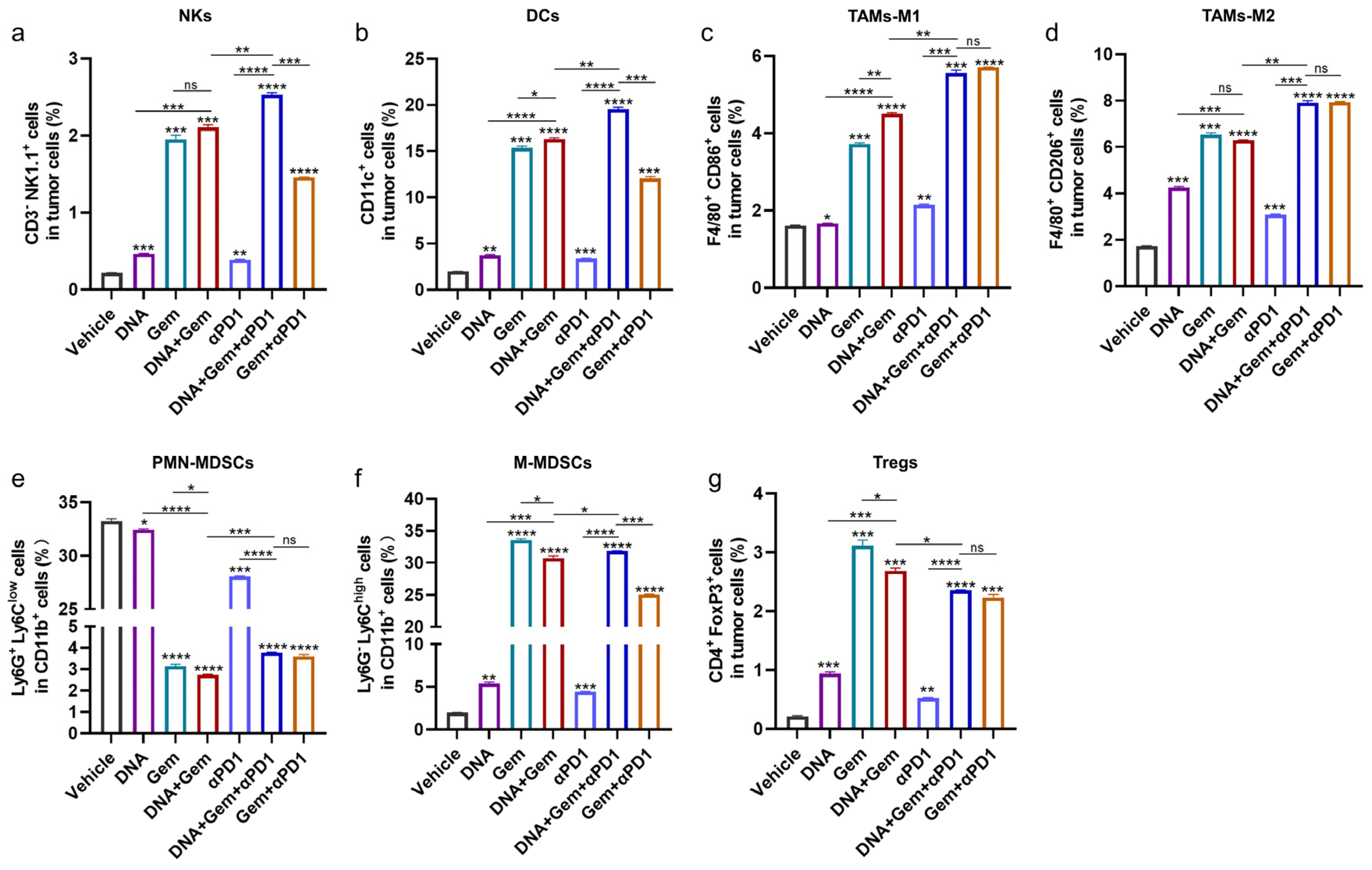
2.7. The Effect of Triple Therapy on the Tumor Microenvironment (TME)
3. Materials and Methods
3.1. Mice and Cell Lines
3.2. Vaccine Construction
3.3. Protein Purification
3.4. In Vitro Functional Verification
3.5. In Vivo Immune Strategies
3.6. ELISpot and Cytotoxicity Assays
3.7. Cell Staining and Flow Cytometry
3.8. Quantitative Real-Time PCR (qRT-PCR)
3.9. Statistical Analysis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Yang, L.; Ning, Q.; Tang, S.-S. Recent Advances and Next Breakthrough in Immunotherapy for Cancer Treatment. J. Immunol. Res. 2022, 2022, 8052212. [Google Scholar] [CrossRef] [PubMed]
- Schaft, N.; Dörrie, J.; Schuler, G.; Schuler-Thurner, B.; Sallam, H.; Klein, S.; Eisenberg, G.; Frankenburg, S.; Lotem, M.; Khatib, A. The future of affordable cancer immunotherapy. Front. Immunol. 2023, 14, 1248867. [Google Scholar] [CrossRef]
- Iwahori, K. Cytotoxic CD8+ Lymphocytes in the Tumor Microenvironment. In Tumor Microenvironment: Hematopoietic Cells—Part A; Birbrair, A., Ed.; Springer International Publishing: Cham, Switzerland, 2020; pp. 53–62. [Google Scholar] [CrossRef]
- Ott, P.A.; Wu, C.J. Cancer Vaccines: Steering T Cells Down the Right Path to Eradicate Tumors. Cancer Discov. 2019, 9, 476–481. [Google Scholar] [CrossRef] [PubMed]
- Saxena, M.; van der Burg, S.H.; Melief, C.J.M.; Bhardwaj, N. Therapeutic cancer vaccines. Nat. Rev. Cancer 2021, 21, 360–378. [Google Scholar] [CrossRef]
- Lopes, A.; Vandermeulen, G.; Préat, V. Cancer DNA vaccines: Current preclinical and clinical developments and future perspectives. J. Exp. Clin. Cancer Res. 2019, 38, 146. [Google Scholar] [CrossRef]
- Hu, Z.; Ott, P.A.; Wu, C.J. Towards personalized, tumour-specific, therapeutic vaccines for cancer. Nat. Rev. Immunol. 2018, 18, 168–182. [Google Scholar] [CrossRef]
- Santos, P.M.; Butterfield, L.H. Dendritic Cell-Based Cancer Vaccines. J. Immunol. 2018, 200, 443–449. [Google Scholar] [CrossRef]
- Fu, C.; Ma, T.; Zhou, L.; Mi, Q.-S.; Jiang, A. Dendritic Cell-Based Vaccines Against Cancer: Challenges, Advances and Future Opportunities. Immunol. Investig. 2022, 51, 2133–2158. [Google Scholar] [CrossRef]
- Matsuo, K.; Yoshie, O.; Kitahata, K.; Kamei, M.; Hara, Y.; Nakayama, T. Recent Progress in Dendritic Cell-Based Cancer Immunotherapy. Cancers 2021, 13, 2495. [Google Scholar] [CrossRef]
- Eisenbarth, S.C. Dendritic cell subsets in T cell programming: Location dictates function. Nat. Rev. Immunol. 2019, 19, 89–103. [Google Scholar] [CrossRef]
- Böttcher, J.P.; Reis e Sousa, C. The Role of Type 1 Conventional Dendritic Cells in Cancer Immunity. Trends Cancer 2018, 4, 784–792. [Google Scholar] [CrossRef]
- den Haan, J.M.M.; Lehar, S.M.; Bevan, M.J. Cd8+ but Not Cd8− Dendritic Cells Cross-Prime Cytotoxic T Cells in Vivo. J. Exp. Med. 2000, 192, 1685–1696. [Google Scholar] [CrossRef]
- Pooley, J.L.; Heath, W.R.; Shortman, K. Cutting Edge: Intravenous Soluble Antigen Is Presented to CD4 T Cells by CD8− Dendritic Cells, but Cross-Presented to CD8 T Cells by CD8+ Dendritic Cells1. J. Immunol. 2001, 166, 5327–5330. [Google Scholar] [CrossRef]
- Audsley, K.M.; McDonnell, A.M.; Waithman, J. Cross-Presenting XCR1+ Dendritic Cells as Targets for Cancer Immunotherapy. Cells 2020, 9, 565. [Google Scholar] [CrossRef]
- Meyer, M.A.; Baer, J.M.; Knolhoff, B.L.; Nywening, T.M.; Panni, R.Z.; Su, X.; Weilbaecher, K.N.; Hawkins, W.G.; Ma, C.; Fields, R.C.; et al. Breast and pancreatic cancer interrupt IRF8-dependent dendritic cell development to overcome immune surveillance. Nat. Commun. 2018, 9, 1250. [Google Scholar] [CrossRef]
- Salmon, H.; Idoyaga, J.; Rahman, A.; Leboeuf, M.; Remark, R.; Jordan, S.; Casanova-Acebes, M.; Khudoynazarova, M.; Agudo, J.; Tung, N.; et al. Expansion and Activation of CD103(+) Dendritic Cell Progenitors at the Tumor Site Enhances Tumor Responses to Therapeutic PD-L1 and BRAF Inhibition. Immunity 2016, 44, 924–938. [Google Scholar] [CrossRef]
- Spranger, S.; Dai, D.; Horton, B.; Gajewski, T.F. Tumor-Residing Batf3 Dendritic Cells Are Required for Effector T Cell Trafficking and Adoptive T Cell Therapy. Cancer Cell 2017, 31, 711–723.e714. [Google Scholar] [CrossRef]
- Bachem, A.; Hartung, E.; Güttler, S.; Mora, A.; Zhou, X.; Hegemann, A.; Plantinga, M.; Mazzini, E.; Stoitzner, P.; Gurka, S.; et al. Expression of XCR1 Characterizes the Batf3-Dependent Lineage of Dendritic Cells Capable of Antigen Cross-Presentation. Front. Immunol. 2012, 3, 214. [Google Scholar] [CrossRef]
- Dorner, B.G.; Dorner, M.B.; Zhou, X.; Opitz, C.; Mora, A.; Güttler, S.; Hutloff, A.; Mages, H.W.; Ranke, K.; Schaefer, M.; et al. Selective expression of the chemokine receptor XCR1 on cross-presenting dendritic cells determines cooperation with CD8+ T cells. Immunity 2009, 31, 823–833. [Google Scholar] [CrossRef]
- Crozat, K.; Guiton, R.; Contreras, V.; Feuillet, V.; Dutertre, C.A.; Ventre, E.; Vu Manh, T.P.; Baranek, T.; Storset, A.K.; Marvel, J.; et al. The XC chemokine receptor 1 is a conserved selective marker of mammalian cells homologous to mouse CD8alpha+ dendritic cells. J. Exp. Med. 2010, 207, 1283–1292. [Google Scholar] [CrossRef]
- Bachem, A.; Güttler, S.; Hartung, E.; Ebstein, F.; Schaefer, M.; Tannert, A.; Salama, A.; Movassaghi, K.; Opitz, C.; Mages, H.W.; et al. Superior antigen cross-presentation and XCR1 expression define human CD11c+CD141+ cells as homologues of mouse CD8+ dendritic cells. J. Exp. Med. 2010, 207, 1273–1281. [Google Scholar] [CrossRef]
- Jongbloed, S.L.; Kassianos, A.J.; McDonald, K.J.; Clark, G.J.; Ju, X.; Angel, C.E.; Chen, C.J.; Dunbar, P.R.; Wadley, R.B.; Jeet, V.; et al. Human CD141+ (BDCA-3)+ dendritic cells (DCs) represent a unique myeloid DC subset that cross-presents necrotic cell antigens. J. Exp. Med. 2010, 207, 1247–1260. [Google Scholar] [CrossRef]
- Poulin, L.F.; Salio, M.; Griessinger, E.; Anjos-Afonso, F.; Craciun, L.; Chen, J.L.; Keller, A.M.; Joffre, O.; Zelenay, S.; Nye, E.; et al. Characterization of human DNGR-1+ BDCA3+ leukocytes as putative equivalents of mouse CD8alpha+ dendritic cells. J. Exp. Med. 2010, 207, 1261–1271. [Google Scholar] [CrossRef]
- Qi, H.; Sun, Z.; Yao, Y.; Chen, L.; Su, X. Immunogenicity of the Xcl1-SARS-CoV-2 Spike Fusion DNA Vaccine for COVID-19. Vaccines 2022, 10, 407. [Google Scholar] [CrossRef]
- Fossum, E.; Grødeland, G.; Terhorst, D.; Tveita, A.A.; Vikse, E.; Mjaaland, S.; Henri, S.; Malissen, B.; Bogen, B. Vaccine molecules targeting Xcr1 on cross-presenting DCs induce protective CD8+ T-cell responses against influenza virus. Eur. J. Immunol. 2015, 45, 624–635. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Wu, Z.; Zhao, H.; Wang, Y.; Ge, Y.; Wang, D.; Li, Z.; An, C.; Liu, Y.; Wang, F.; et al. XCL1/Glypican-3 Fusion Gene Immunization Generates Potent Antitumor Cellular Immunity and Enhances Anti-PD-1 Efficacy. Cancer Immunol. Res. 2020, 8, 81–93. [Google Scholar] [CrossRef] [PubMed]
- Le Gall, C.; Cammarata, A.; de Haas, L.; Ramos-Tomillero, I.; Cuenca-Escalona, J.; Schouren, K.; Wijfjes, Z.; Becker, A.M.D.; Bödder, J.; Dölen, Y.; et al. Efficient targeting of NY-ESO-1 tumor antigen to human cDC1s by lymphotactin results in cross-presentation and antigen-specific T cell expansion. J. Immunother. Cancer 2022, 10, e004309. [Google Scholar] [CrossRef] [PubMed]
- Hartung, E.; Becker, M.; Bachem, A.; Reeg, N.; Jäkel, A.; Hutloff, A.; Weber, H.; Weise, C.; Giesecke, C.; Henn, V.; et al. Induction of Potent CD8 T Cell Cytotoxicity by Specific Targeting of Antigen to Cross-Presenting Dendritic Cells In Vivo via Murine or Human XCR1. J. Immunol. 2015, 194, 1069–1079. [Google Scholar] [CrossRef] [PubMed]
- Gao, T.; Cen, Q.; Lei, H. A review on development of MUC1-based cancer vaccine. Biomed. Pharmacother. Biomed. Pharmacother. 2020, 132, 110888. [Google Scholar] [CrossRef] [PubMed]
- Garg, H.; Suri, P.; Gupta, J.C.; Talwar, G.P.; Dubey, S. Survivin: A unique target for tumor therapy. Cancer Cell Int. 2016, 16, 49. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Liu, C.; Zhang, F.; Geng, F.; Xia, Q.; Lu, Z.; Xu, P.; Xie, Y.; Wu, H.; Yu, B.; et al. MUC1 and survivin combination tumor gene vaccine generates specific immune responses and anti-tumor effects in a murine melanoma model. Vaccine 2016, 34, 2648–2655. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q.; Wang, L.; Xu, P.; Geng, F.; Guo, J.; Dong, L.; Bao, X.; Zhou, Y.; Feng, M.; Wu, J.; et al. Heterologous prime-boost immunization co-targeting dual antigens inhibit tumor growth and relapse. Oncoimmunology 2020, 9, 1841392. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Lu, Z.; Xie, Y.; Guo, Q.; Geng, F.; Sun, B.; Wu, H.; Yu, B.; Wu, J.; Zhang, H.; et al. Soluble PD-1-based vaccine targeting MUC1 VNTR and survivin improves anti-tumor effect. Immunol. Lett. 2018, 200, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Kerr, M.D.; McBride, D.A.; Chumber, A.K.; Shah, N.J. Combining therapeutic vaccines with chemo- and immunotherapies in the treatment of cancer. Expert. Opin. Drug Discov. 2021, 16, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; He, X.; Fang, D.; Wang, X.; Guan, J.; Shi, Z.W.; Chen, X. Gemcitabine-facilitated modulation of the tumor microenvironment and PD-1/PD-L1 blockade generate a synergistic antitumor effect in a murine hepatocellular carcinoma model. Clin. Res. Hepatol. Gastroenterol. 2022, 46, 101853. [Google Scholar] [CrossRef] [PubMed]
- Tomihara, K.; Fuse, H.; Heshiki, W.; Takei, R.; Zhang, B.; Arai, N.; Nakamori, K.; Noguchi, M. Gemcitabine chemotherapy induces phenotypic alterations of tumor cells that facilitate antitumor T cell responses in a mouse model of oral cancer. Oral Oncol. 2014, 50, 457–467. [Google Scholar] [CrossRef] [PubMed]
- Du, B.; Wen, X.; Wang, Y.; Lin, M.; Lai, J. Gemcitabine and checkpoint blockade exhibit synergistic anti-tumor effects in a model of murine lung carcinoma. Int. Immunopharmacol. 2020, 86, 106694. [Google Scholar] [CrossRef]
- Geng, F.; Dong, L.; Bao, X.; Guo, Q.; Guo, J.; Zhou, Y.; Yu, B.; Wu, H.; Wu, J.; Zhang, H.; et al. CAFs/tumor cells co-targeting DNA vaccine in combination with low-dose gemcitabine for the treatment of Panc02 murine pancreatic cancer. Mol. Ther. Oncolytics 2022, 26, 304–313. [Google Scholar] [CrossRef]
- Macri, C.; Jenika, D.; Ouslinis, C.; Mintern, J.D. Targeting dendritic cells to advance cross-presentation and vaccination outcomes. Semin. Immunol. 2023, 68, 101762. [Google Scholar] [CrossRef]
- Baljon, J.J.; Wilson, J.T. Bioinspired vaccines to enhance MHC class-I antigen cross-presentation. Curr. Opin. Immunol. 2022, 77, 102215. [Google Scholar] [CrossRef]
- Macri, C.; Dumont, C.; Johnston, A.P.; Mintern, J.D. Targeting dendritic cells: A promising strategy to improve vaccine effectiveness. Clin. Transl. Immunol. 2016, 5, e66. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Jin, Y.; Li, W.; Zhang, B.; He, Y.; Liu, H.; Xia, N.; Wei, H.; Yan, J. DNA vaccines targeting the encoded antigens to dendritic cells induce potent antitumor immunity in mice. BMC Immunol. 2013, 14, 39. [Google Scholar] [CrossRef] [PubMed]
- Sancho, D.; Mourão-Sá, D.; Joffre, O.P.; Schulz, O.; Rogers, N.C.; Pennington, D.J.; Carlyle, J.R.; Reis e Sousa, C. Tumor therapy in mice via antigen targeting to a novel, DC-restricted C-type lectin. J. Clin. Investig. 2008, 118, 2098–2110. [Google Scholar] [CrossRef] [PubMed]
- Roberts, E.W.; Broz, M.L.; Binnewies, M.; Headley, M.B.; Nelson, A.E.; Wolf, D.M.; Kaisho, T.; Bogunovic, D.; Bhardwaj, N.; Krummel, M.F. Critical Role for CD103(+)/CD141(+) Dendritic Cells Bearing CCR7 for Tumor Antigen Trafficking and Priming of T Cell Immunity in Melanoma. Cancer Cell 2016, 30, 324–336. [Google Scholar] [CrossRef] [PubMed]
- Böttcher, J.P.; Bonavita, E.; Chakravarty, P.; Blees, H.; Cabeza-Cabrerizo, M.; Sammicheli, S.; Rogers, N.C.; Sahai, E.; Zelenay, S.; Reis e Sousa, C. NK Cells Stimulate Recruitment of cDC1 into the Tumor Microenvironment Promoting Cancer Immune Control. Cell 2018, 172, 1022–1037.e14. [Google Scholar] [CrossRef]
- Cancel, J.C.; Crozat, K.; Dalod, M.; Mattiuz, R. Are Conventional Type 1 Dendritic Cells Critical for Protective Antitumor Immunity and How? Front. Immunol. 2019, 10, 9. [Google Scholar] [CrossRef]
- Bergamaschi, C.; Pandit, H.; Nagy, B.A.; Stellas, D.; Jensen, S.M.; Bear, J.; Cam, M.; Valentin, A.; Fox, B.A.; Felber, B.K.; et al. Heterodimeric IL-15 delays tumor growth and promotes intratumoral CTL and dendritic cell accumulation by a cytokine network involving XCL1, IFN-γ, CXCL9 and CXCL10. J. Immunother. Cancer 2020, 8, e000599. [Google Scholar] [CrossRef]
- Pitt, J.M.; Marabelle, A.; Eggermont, A.; Soria, J.C.; Kroemer, G.; Zitvogel, L. Targeting the tumor microenvironment: Removing obstruction to anticancer immune responses and immunotherapy. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2016, 27, 1482–1492. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Tan, X.; Hu, X.; Zhou, M.; Yan, J.; Ding, C. Tumor Microenvironment following Gemcitabine Treatment Favors Differentiation of Immunosuppressive Ly6C(high) Myeloid Cells. J. Immunol. 2020, 204, 212–223. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, K.; Wuri, Q.; Cai, Z.; Qu, X.; Zhang, S.; Wu, H.; Wu, J.; Wang, C.; Yu, X.; Kong, W.; et al. The XCL1-Mediated DNA Vaccine Targeting Type 1 Conventional Dendritic Cells Combined with Gemcitabine and Anti-PD1 Antibody Induces Potent Antitumor Immunity in a Mouse Lung Cancer Model. Int. J. Mol. Sci. 2024, 25, 1880. https://doi.org/10.3390/ijms25031880
Zhang K, Wuri Q, Cai Z, Qu X, Zhang S, Wu H, Wu J, Wang C, Yu X, Kong W, et al. The XCL1-Mediated DNA Vaccine Targeting Type 1 Conventional Dendritic Cells Combined with Gemcitabine and Anti-PD1 Antibody Induces Potent Antitumor Immunity in a Mouse Lung Cancer Model. International Journal of Molecular Sciences. 2024; 25(3):1880. https://doi.org/10.3390/ijms25031880
Chicago/Turabian StyleZhang, Ke, Qimuge Wuri, Zongyu Cai, Xueli Qu, Shiqi Zhang, Hui Wu, Jiaxin Wu, Chu Wang, Xianghui Yu, Wei Kong, and et al. 2024. "The XCL1-Mediated DNA Vaccine Targeting Type 1 Conventional Dendritic Cells Combined with Gemcitabine and Anti-PD1 Antibody Induces Potent Antitumor Immunity in a Mouse Lung Cancer Model" International Journal of Molecular Sciences 25, no. 3: 1880. https://doi.org/10.3390/ijms25031880
APA StyleZhang, K., Wuri, Q., Cai, Z., Qu, X., Zhang, S., Wu, H., Wu, J., Wang, C., Yu, X., Kong, W., & Zhang, H. (2024). The XCL1-Mediated DNA Vaccine Targeting Type 1 Conventional Dendritic Cells Combined with Gemcitabine and Anti-PD1 Antibody Induces Potent Antitumor Immunity in a Mouse Lung Cancer Model. International Journal of Molecular Sciences, 25(3), 1880. https://doi.org/10.3390/ijms25031880