
Modeling and Analysis of HIV-1 Pol Polyprotein as a Case Study for Predicting Large Polyprotein Structures



Abstract
1. Introduction
2. Results
2.1. Overview of the Workflow to Predict the Structure of the HIV-1 Pol Polyprotein
2.2. Comparing the Structure of Mature HIV-1 Proteins with Corresponding Segments in HIV-1 Pol Polyprotein
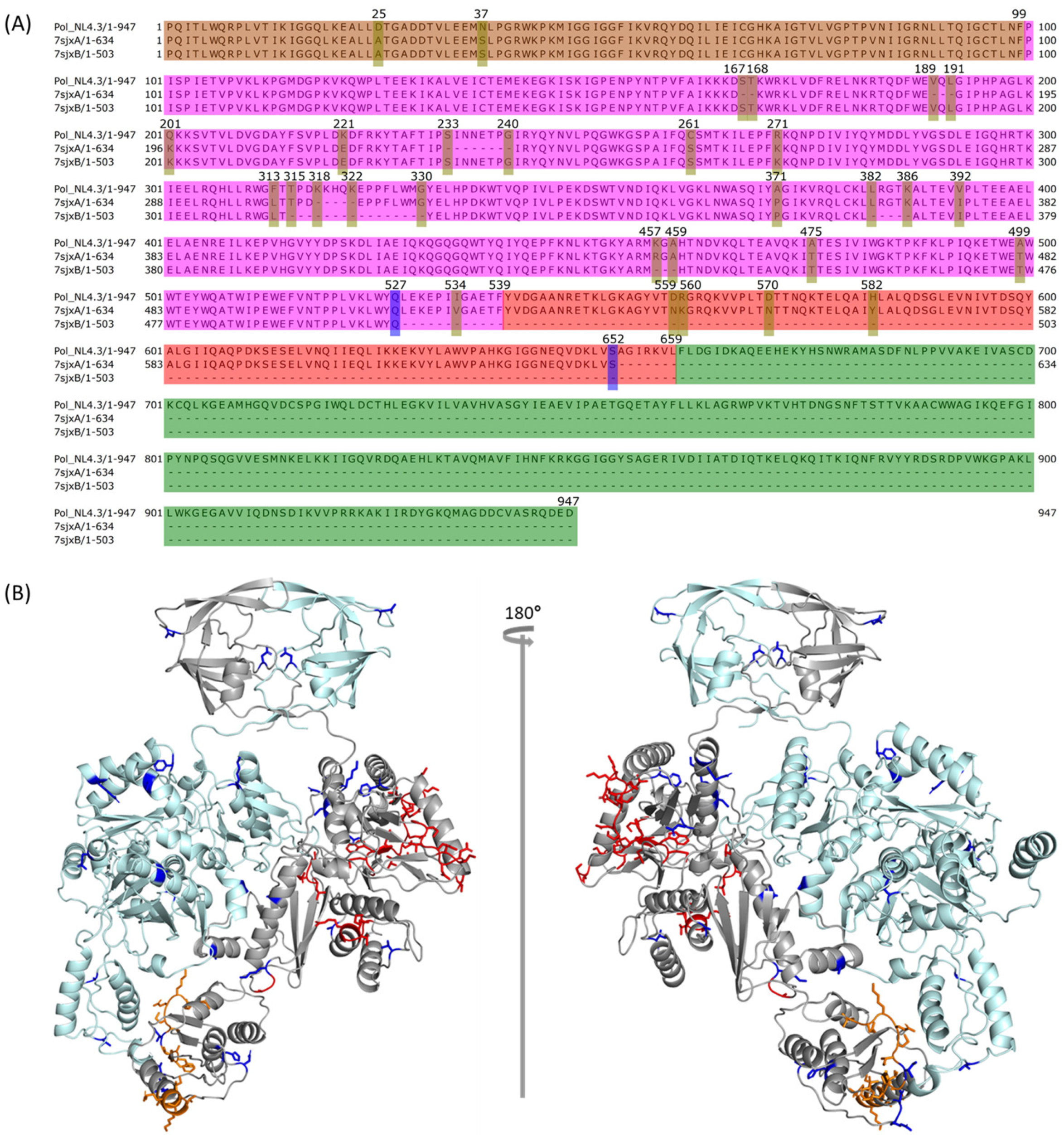
2.3. Comparison between NL4.3 HIV-1 Pol Sequence and That Extracted from the Crystalized Pol Structure
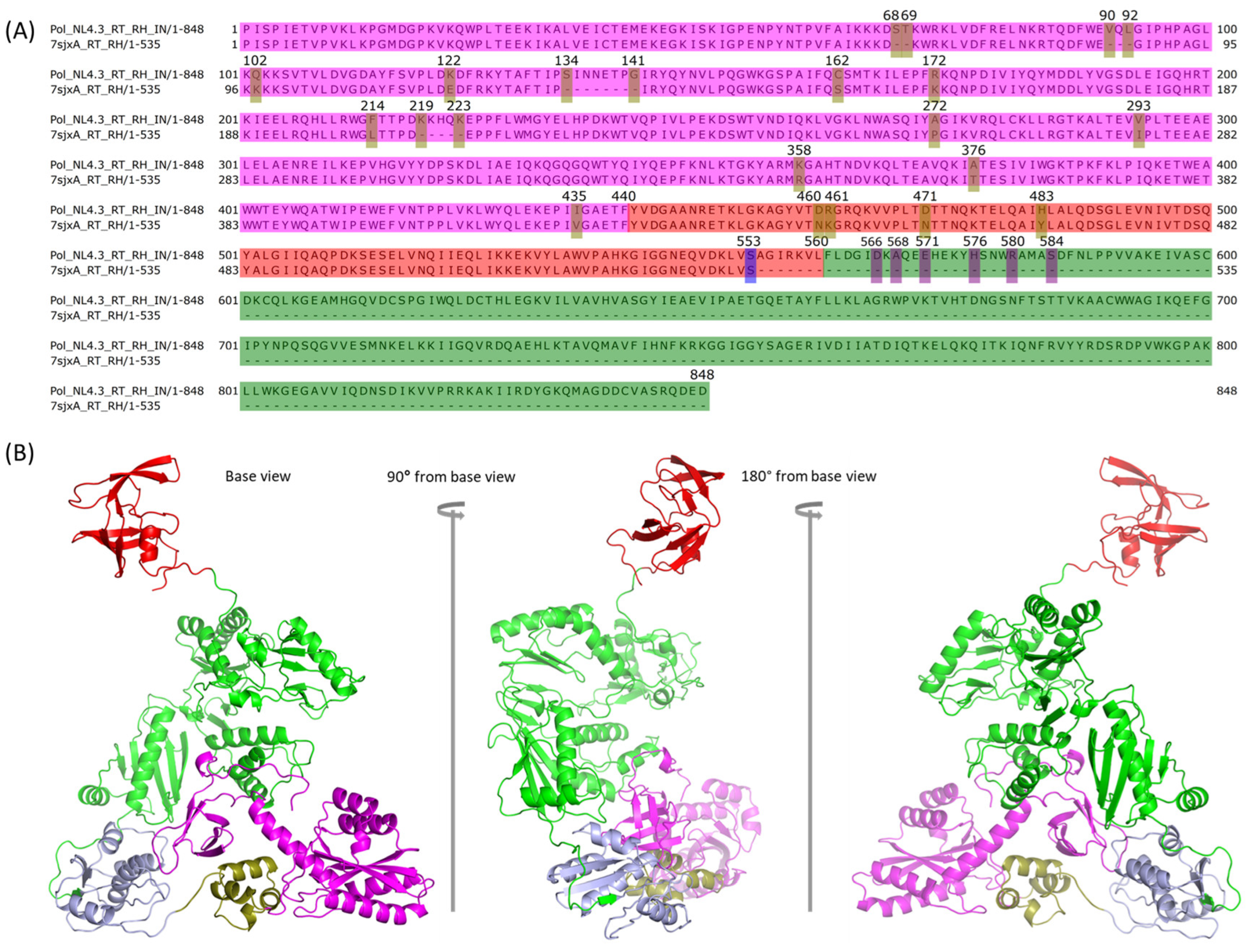
2.4. Modeling PR + RT + RH for Chain A and PR + RT for Chain B of Pol
2.5. Assembly of PR + RT + RH with IN in Chain A of Pol
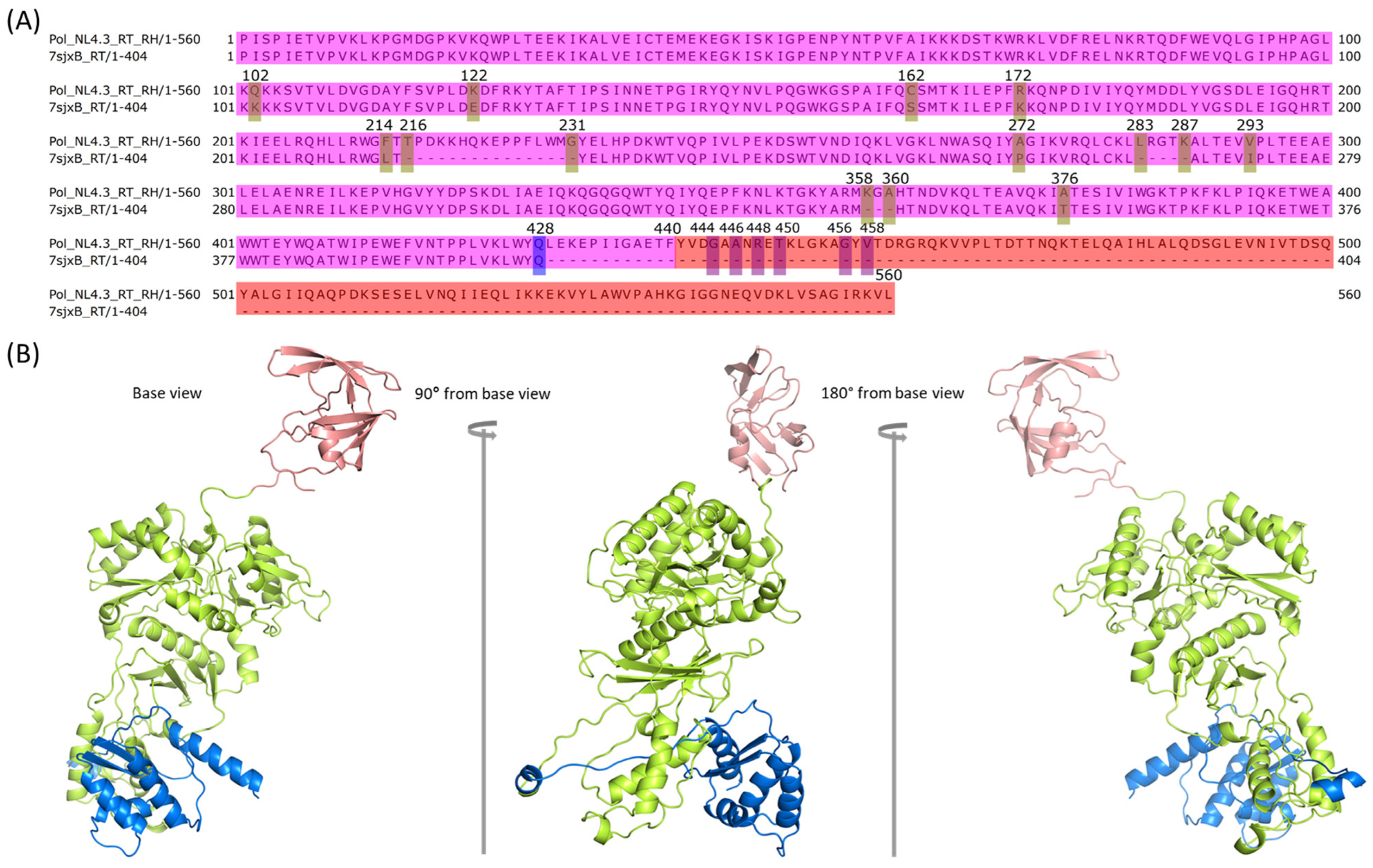
2.6. Assembly of RT with RH in Chain B of Pol
2.7. Assembly of RH with IN in Chain B of Pol
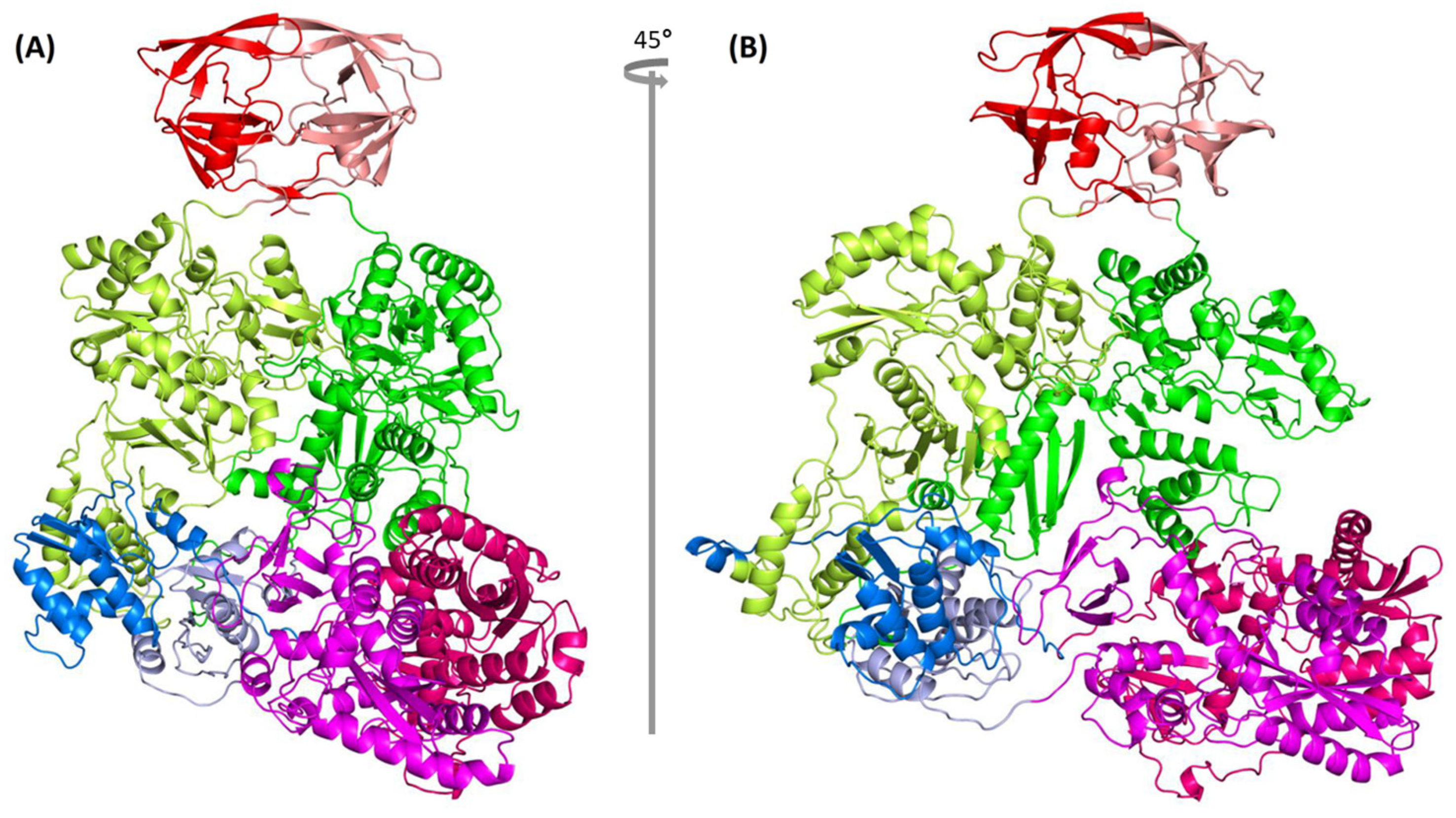
2.8. Complete Pol Dimer Construction and Optimization
2.9. Quality Measure of Modeled Pol Dimer
3. Discussion
4. Materials and Methods
4.1. Protein Sequences, Structures, and Sequence Alignment
4.2. Protein Structure Modeling, Optimization, and Domain Assembly Methods
4.3. Protein Model Quality Estimation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Burley, S.K.; Bhikadiya, C.; Bi, C.; Bittrich, S.; Chen, L.; Crichlow, G.V.; Christie, C.H.; Dalenberg, K.; Di Costanzo, L.; Duarte, J.M.; et al. RCSB protein data bank: Powerful new tools for exploring 3D structures of biological macromolecules for basic and applied research and education in fundamental biology, biomedicine, biotechnology, bioengineering and energy sciences. Nucleic Acids Res. 2020, 49, D437–D451. [Google Scholar] [CrossRef]
- Marsh, R.E.; Donohue, J. Crystal structure studies of amino acids and peptides. Adv. Protein Chem. 1967, 22, 235–256. [Google Scholar] [CrossRef]
- Blundell, T.L.; Dodson, G.G.; Dodson, E.; Hodgkin, D.C.; Vijayan, M. X-ray analysis and the structure of insulin. Recent. Prog. Horm. Res. 1971, 27, 1–40. [Google Scholar] [CrossRef]
- Bagby, S.; Tong, K.I.; Ikura, M. Optimization of protein solubility and stability for protein nuclear magnetic resonance. Methods Enzymol. 2001, 339, 20–41. [Google Scholar] [CrossRef]
- Pédelacq, J.-D.; Piltch, E.; Liong, E.C.; Berendzen, J.; Kim, C.-Y.; Rho, B.-S.; Park, M.S.; Terwilliger, T.C.; Waldo, G.S. Engineering soluble proteins for structural genomics. Nat. Biotechnol. 2002, 20, 927–932. [Google Scholar] [CrossRef]
- Edwards, A.M.; Arrowsmith, C.H.; Christendat, D.; Dharamsi, A.; Friesen, J.D.; Greenblatt, J.F.; Vedadi, M. Protein production: Feeding the crystallographers and NMR spectroscopists. Nat. Struct. Biol. 2000, 7, 970–972. [Google Scholar] [CrossRef]
- Golovanov, A.P.; Hautbergue, G.M.; Wilson, S.A.; Lian, L.-Y. A Simple method for improving protein solubility and long-term stability. J. Am. Chem. Soc. 2004, 126, 8933–8939. [Google Scholar] [CrossRef]
- Kühlbrandt, W.; Williams, K.A. Analysis of macromolecular structure and dynamics by electron cryo-microscopy. Curr. Opin. Chem. Biol. 1999, 3, 537–543. [Google Scholar] [CrossRef]
- Auer, M. Three-dimensional electron cryo-microscopy as a powerful structural tool in molecular medicine. J. Mol. Med. 2000, 78, 191–202. [Google Scholar] [CrossRef]
- Tivol, W.F.; Briegel, A.; Jensen, G.J. An improved cryogen for plunge freezing. Microsc. Microanal. 2008, 14, 375–379. [Google Scholar] [CrossRef]
- Callaway, E. The revolution will not be crystallized: A new method sweeps through structural biology. Nature 2015, 525, 172–174. [Google Scholar] [CrossRef]
- Callaway, E. Revolutionary cryo-EM is taking over structural biology. Nature 2020, 578, 201. [Google Scholar] [CrossRef]
- Harrison, J.J.E.K.; Passos, D.O.; Bruhn, J.F.; Bauman, J.D.; Tuberty, L.; DeStefano, J.J.; Ruiz, F.X.; Lyumkis, D.; Arnold, E. Cryo-EM structure of the HIV-1 Pol polyprotein provides insights into virion maturation. Sci. Adv. 2022, 8, eabn9874. [Google Scholar] [CrossRef]
- Pettit, S.C.; Lindquist, J.N.; Kaplan, A.H.; Swanstrom, R. Processing sites in the human immunodeficiency virus type 1 (HIV-1) Gag-Pro-Pol precursor are cleaved by the viral protease at different rates. Retrovirology 2005, 2, 66. [Google Scholar] [CrossRef]
- Pettit, S.C.; Clemente, J.C.; Jeung, J.A.; Dunn, B.M.; Kaplan, A.H. Ordered processing of the human immunodeficiency virus type 1 GagPol precursor is influenced by the context of the embedded viral protease. J. Virol. 2005, 79, 10601–10607. [Google Scholar] [CrossRef]
- Pettit, S.C.; Everitt, L.E.; Choudhury, S.; Dunn, B.M.; Kaplan, A.H. Initial cleavage of the human immunodeficiency virus type 1 GagPol precursor by its activated protease occurs by an intramolecular mechanism. J. Virol. 2004, 78, 8477–8485. [Google Scholar] [CrossRef]
- Könnyű, B.; Sadiq, S.K.; Turányi, T.; Hírmondó, R.; Müller, B.; Kräusslich, H.-G.; Coveney, P.V.; Müller, V. Gag-Pol processing during HIV-1 virion maturation: A systems biology approach. PLoS Comput. Biol. 2013, 9, e1003103. [Google Scholar] [CrossRef]
- Urano, E.; Timilsina, U.; Kaplan, J.A.; Ablan, S.; Ghimire, D.; Pham, P.; Kuruppu, N.; Mandt, R.; Durell, S.R.; Nitz, T.J.; et al. Resistance to second-generation HIV-1 maturation inhibitors. J. Virol. 2019, 93. [Google Scholar] [CrossRef]
- Imamichi, T. Action of Anti-HIV drugs and resistance: Reverse transcriptase inhibitors and protease inhibitors. Curr. Pharm. Des. 2004, 10, 4039–4053. [Google Scholar] [CrossRef]
- Yang, J.; Hao, M.; Khan, M.A.; Rehman, M.T.; Highbarger, H.C.; Chen, Q.; Goswami, S.; Sherman, B.T.; Rehm, C.A.; Dewar, R.L.; et al. A Combination of M50I and V151I polymorphic mutations in HIV-1 subtype B integrase results in defects in autoprocessing. Viruses 2021, 13, 2331. [Google Scholar] [CrossRef]
- Imamichi, T.; Bernbaum, J.G.; Laverdure, S.; Yang, J.; Chen, Q.; Highbarger, H.; Hao, M.; Sui, H.; Dewar, R.; Chang, W.; et al. Natural occurring polymorphisms in HIV-1 integrase and RNase H regulate viral release and autoprocessing. J. Virol. 2021, 95. [Google Scholar] [CrossRef]
- Imamichi, T.; Chen, Q.; Hao, M.; Chang, W.; Yang, J. The C-Terminal domain of RNase H and the C-terminus amino acid residue regulate virus release and autoprocessing of a defective HIV-1 possessing M50I and V151I changes in integrase. Viruses 2022, 14, 2687. [Google Scholar] [CrossRef]
- Chen, Y.; Lu, H.; Zhang, N.; Zhu, Z.; Wang, S.; Li, M. PremPS: Predicting the impact of missense mutations on protein stability. PLoS Comput. Biol. 2020, 16, e1008543. [Google Scholar] [CrossRef]
- Rodrigues, C.H.; Pires, D.E.; Ascher, D.B. DynaMut2: Assessing changes in stability and flexibility upon single and multiple point missense mutations. Protein Sci. 2020, 30, 60–69. [Google Scholar] [CrossRef]
- Zhou, Y.; Pan, Q.; Pires, D.E.V.; Rodrigues, C.H.M.; Ascher, D.B. DDMut: Predicting effects of mutations on protein stability using deep learning. Nucleic Acids Res. 2023, 51, W122–W128. [Google Scholar] [CrossRef]
- Hoyte, A.C.; Jamin, A.V.; Koneru, P.C.; Kobe, M.J.; Larue, R.C.; Fuchs, J.R.; Engelman, A.N.; Kvaratskhelia, M. Resistance to pyridine-based inhibitor KF116 reveals an unexpected role of integrase in HIV-1 Gag-Pol polyprotein proteolytic processing. J. Biol. Chem. 2017, 292, 19814–19825. [Google Scholar] [CrossRef]
- Adachi, A.; Gendelman, H.E.; Koenig, S.; Folks, T.; Willey, R.; Rabson, A.; Martin, M.A. Production of acquired immunodeficiency syndrome-associated retrovirus in human and nonhuman cells transfected with an infectious molecular clone. J. Virol. 1986, 59, 284–291. [Google Scholar] [CrossRef]
- Boffetta, P.; Hainaut, P. Encyclopedia of Cancer, 2nd ed.; Elsevier Science/Academic Press: Cambridge, MA, USA, 2002; pp. 167–172. [Google Scholar]
- Kirtipal, N.; Bharadwaj, S.; Kang, S.G. From SARS to SARS-CoV-2, insights on structure, pathogenicity and immunity aspects of pandemic human coronaviruses. Infect. Genet. Evol. 2020, 85, 104502. [Google Scholar] [CrossRef]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; De Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; DiMaio, F.; Wang, R.Y.-R.; Kim, D.; Miles, C.; Brunette, T.; Thompson, J.; Baker, D. High-Resolution Comparative Modeling with RosettaCM. Structure 2013, 21, 1735–1742. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.E.; Chivian, D.; Baker, D. Protein structure prediction and analysis using the Robetta server. Nucleic Acids Res. 2004, 32, W526–W531. [Google Scholar] [CrossRef] [PubMed]
- Heo, L.; Lee, H.; Seok, C. GalaxyRefineComplex: Refinement of protein-protein complex model structures driven by interface repacking. Sci. Rep. 2016, 6, 32153. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Park, T.; Lee, S.Y.; Yang, J.; Seok, C. GalaxyDomDock: An Ab initio domain–domain docking web server for multi-domain protein structure prediction. J. Mol. Biol. 2022, 434, 167508. [Google Scholar] [CrossRef] [PubMed]
- Krieger, E.; Joo, K.; Lee, J.; Lee, J.; Raman, S.; Thompson, J.; Tyka, M.; Baker, D.; Karplus, K. Improving physical realism, stereochemistry, and side-chain accuracy in homology modeling: Four approaches that performed well in CASP8. Proteins 2009, 77, 114–122. [Google Scholar] [CrossRef]
- Waterhouse, A.M.; Procter, J.B.; Martin, D.M.A.; Clamp, M.; Barton, G.J. Jalview Version 2—A multiple sequence alignment editor and analysis workbench. Bioinformatics 2009, 25, 1189–1191. [Google Scholar] [CrossRef]
- Notredame, C.; Higgins, D.G.; Heringa, J. T-coffee: A novel method for fast and accurate multiple sequence alignment. J. Mol. Biol. 2000, 302, 205–217. [Google Scholar] [CrossRef]
- Nakamura, A.; Tamura, N.; Yasutake, Y. Structure of the HIV-1 reverse transcriptase Q151M mutant: Insights into the inhibitor resistance of HIV-1 reverse transcriptase and the structure of the nucleotide-binding pocket of Hepatitis B virus polymerase. Acta Crystallogr. F Struct. Biol. Commun. 2015, 71, 1384–1390. [Google Scholar] [CrossRef]
- Sweeney, Z.K.; Harris, S.F.; Arora, N.; Javanbakht, H.; Li, Y.; Fretland, J.; Davidson, J.P.; Billedeau, J.R.; Gleason, S.K.; Hirschfeld, D.; et al. Design of annulated pyrazoles as inhibitors of HIV-1 reverse transcriptase. J. Med. Chem. 2008, 51, 7449–7458. [Google Scholar] [CrossRef]
- Jones, L.H.; Allan, G.; Barba, O.; Burt, C.; Corbau, R.; Dupont, T.; Knöchel, T.; Irving, S.; Middleton, D.S.; Mowbray, C.E.; et al. Novel indazole non-nucleoside reverse transcriptase inhibitors using molecular hybridization based on crystallographic overlays. J. Med. Chem. 2009, 52, 1219–1223. [Google Scholar] [CrossRef]
- Ren, J.; Diprose, J.; Warren, J.; Esnouf, R.M.; Bird, L.E.; Ikemizu, S.; Slater, M.; Milton, J.; Balzarini, J.; Stuart, D.I. Phenylethylthiazolylthiourea (PETT) non-nucleoside inhibitors of HIV-1 and HIV-2 reverse transcriptases: Structural and biochemical analyses. J. Biol. Chem. 2000, 275, 5633–5639. [Google Scholar] [CrossRef]
- Ren, J.; Esnouf, R.M.; Hopkins, A.L.; Stuart, D.I.; Stammers, D.K. Crystallographic analysis of the binding modes of thiazoloisoindolinone non-nucleoside inhibitors to HIV-1 reverse transcriptase and comparison with modeling studies. J. Med. Chem. 1999, 42, 3845–3851. [Google Scholar] [CrossRef]
- Hopkins, A.L.; Ren, J.; Tanaka, H.; Baba, M.; Okamato, M.; Stuart, D.I.; Stammers, D.K. Design of MKC-442 (emivirine) analogues with improved activity against drug-resistant HIV mutants. J. Med. Chem. 1999, 42, 4500–4505. [Google Scholar] [CrossRef]
- Cai, M.; Zheng, R.; Caffrey, M.; Craigie, R.; Clore, G.M.; Gronenborn, A.M. Solution structure of the N-terminal zinc binding domain of HIV-1 integrase. Nat. Struct. Biol. 1997, 4, 567–577. [Google Scholar] [CrossRef]
- Wang, J.; Ling, H.; Yang, W.; Craigie, R. Structure of a two-domain fragment of HIV-1 integrase: Implications for domain organization in the intact protein. EMBO J. 2001, 20, 7333–7343. [Google Scholar] [CrossRef]
- Bujacz, G.; Alexandratos, J.; Zhou-Liu, Q.; Clément-Mella, C.; Wlodawer, A. The catalytic domain of human immunodeficiency virus integrase: Ordered active site in the F185H mutant. FEBS Lett. 1996, 398, 175–178. [Google Scholar] [CrossRef]
- Greenwald, J.; Le, V.; Butler, S.L.; Bushman, F.D.; Choe, S. The mobility of an HIV-1 integrase active site loop is correlated with catalytic activity. Biochemistry 1999, 38, 8892–8898. [Google Scholar] [CrossRef]
- Dyda, F.; Hickman, A.B.; Jenkins, T.M.; Engelman, A.; Craigie, R.; Davies, D.R. Crystal structure of the catalytic domain of HIV-1 integrase: Similarity to other polynucleotidyl transferases. Science 1994, 266, 1981–1986. [Google Scholar] [CrossRef]
- Gupta, K.; Turkki, V.; Sherrill-Mix, S.; Hwang, Y.; Eilers, G.; Taylor, L.; McDanal, C.; Wang, P.; Temelkoff, D.; Nolte, R.T.; et al. Structural basis for inhibitor-induced aggregation of HIV integrase. PLoS Biol. 2016, 14, e1002584. [Google Scholar] [CrossRef]
- Chen, J.C.-H.; Krucinski, J.; Miercke, L.J.W.; Finer-Moore, J.S.; Tang, A.H.; Leavitt, A.D.; Stroud, R.M. Crystal structure of the HIV-1 integrase catalytic core and C-terminal domains: A model for viral DNA binding. Proc. Natl. Acad. Sci. USA 2000, 97, 8233–8238. [Google Scholar] [CrossRef]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef]
- Lemkul, J. From proteins to perturbed hamiltonians: A suite of tutorials for the GROMACS-2018 molecular simulation package [article v1.0]. Living J. Comput. Mol. Sci. 2019, 1, 5068. [Google Scholar] [CrossRef]
- Williams, C.J.; Headd, J.J.; Moriarty, N.W.; Prisant, M.G.; Videau, L.L.; Deis, L.N.; Verma, V.; Keedy, D.A.; Hintze, B.J.; Chen, V.B.; et al. MolProbity: More and better reference data for improved all-atom structure validation. Protein Sci. 2018, 27, 293–315. [Google Scholar] [CrossRef]
- Krissinel, E.; Henrick, K. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 2007, 372, 774–797. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Bertoline, L.M.F.; Lima, A.N.; Krieger, J.E.; Teixeira, S.K. Before and after AlphaFold2: An overview of protein structure prediction. Front. Bioinform. 2023, 3, 1120370. [Google Scholar] [CrossRef]
- Bayar, E.; Ren, Y.; Chen, Y.; Hu, Y.; Zhang, S.; Yu, X.; Fan, J. Construction, investigation and application of TEV protease variants with improved oxidative stability. J. Microbiol. Biotechnol. 2021, 31, 1732–1740. [Google Scholar] [CrossRef]
- Broom, A.; Jacobi, Z.; Trainor, K.; Meiering, E.M. Computational tools help improve protein stability but with a solubility tradeoff. J. Biol. Chem. 2017, 292, 14349–14361. [Google Scholar] [CrossRef]
- Trevino, S.R.; Scholtz, J.M.; Pace, C.N. Amino acid contribution to protein solubility: Asp, Glu, and Ser contribute more favorably than the other hydrophilic amino acids in RNase Sa. J. Mol. Biol. 2007, 366, 449–460. [Google Scholar] [CrossRef] [PubMed]
- McNutt, A.T.; Francoeur, P.; Aggarwal, R.; Masuda, T.; Meli, R.; Ragoza, M.; Sunseri, J.; Koes, D.R. GNINA 1.0: Molecular docking with deep learning. J. Cheminform. 2021, 13, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Ragoza, M.; Hochuli, J.; Idrobo, E.; Sunseri, J.; Koes, D.R. Protein–Ligand scoring with convolutional neural networks. J. Chem. Inf. Model. 2017, 57, 942–957. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Jaroszewski, L.; Li, Z.; Godzik, A. AIDA: Ab initio domain assembly server. Nucleic Acids Res. 2014, 42, W308–W313. [Google Scholar] [CrossRef]
- Zhou, X.; Peng, C.; Zheng, W.; Li, Y.; Zhang, G.; Zhang, Y. DEMO2: Assemble multi-domain protein structures by coupling analogous template alignments with deep-learning inter-domain restraint prediction. Nucleic Acids Res. 2022, 50, W235–W245. [Google Scholar] [CrossRef]
- Xu, D.; Zhang, Y. Improving the physical realism and structural accuracy of protein models by a two-step atomic-level energy minimization. Biophys. J. 2011, 101, 2525–2534. [Google Scholar] [CrossRef]
- Sui, H.; Hao, M.; Chang, W.; Imamichi, T. The Role of Ku70 as a cytosolic DNA sensor in innate immunity and beyond. Front. Cell. Infect. Microbiol. 2021, 11. [Google Scholar] [CrossRef]
- Henes, M.; Lockbaum, G.J.; Kosovrasti, K.; Leidner, F.; Nachum, G.S.; Nalivaika, E.A.; Lee, S.-K.; Spielvogel, E.; Zhou, S.; Swanstrom, R. Picomolar to micromolar: Elucidating the role of distalmutations in HIV-1 protease in conferring drug resistance. ACS Chem. Biol. 2019, 14, 2441–2452. [Google Scholar] [CrossRef]
- Ren, J.; Chamberlain, P.P.; Stamp, A.; Short, S.A.; Weaver, K.L.; Romines, K.R.; Hazen, R.; Freeman, A.; Ferris, R.G.; Andrews, C.W. Structural basis for the improved drug resistance profileof new generation benzophenone non-nucleoside HIV-1 reverse transcriptase inhibitors. J. Med.Chem. 2008, 51, 5000–5008. [Google Scholar] [CrossRef]
- Sharma, A.; Slaughter, A.; Jena, N.; Feng, L.; Kessl, J.J.; Fadel, H.J.; Malani, N.; Male, F.; Wu, L.; Poeschla, E. A new class of multimerization selective inhibitors of HIV-1 integrase. PLoS Pathog. 2014, 10, e1004171. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Version/Accession Date | URL | Representative Function | References |
|---|---|---|---|---|
| GalaxyDomDock | 04-2023 | https://galaxy.seoklab.org/cgi-bin/submit.cgi?type=DOMDOCK_INTRO | Domain assembly | [34] |
| SWISS-MODEL | 01-2023 | https://swissmodel.expasy.org/ | Structural modeling/optimization | [30] |
| Robetta | 01-2023 | https://robetta.bakerlab.org/ | Structural modeling | [31,32] |
| GalaxyRefineComplex | 02-2023 | https://galaxy.seoklab.org/cgi-bin/submit.cgi?type=COMPLEX | Structural optimization | [33] |
| YASARA Energy Minimization Server | 08-2023 | http://www.yasara.org/minimizationserver.htm | Structural optimization | [35] |
| YASARA View | v.23.5.19 | http://www.yasara.org/viewdl.htm | YASARA file operation | [35] |
| Open-Source PyMOL | v.2.5.0 | https://github.com/schrodinger/pymol-open-source | Structural modeling/visualization | - |
| Jalview | v.2.11.3.1 | https://www.jalview.org/ | Sequence alignment | [36,37] |
| PISA Radar Interface Parameters | Our Modeled Pol Dimer | 7SJX |
|---|---|---|
| NSB | 90% | 58% |
| NHB | 81% | 94% |
| HYP | 77% | 84% |
| TBE | 90% | 97% |
| SOE | 96% | 96% |
| INA | 96% | 96% |
| NDB | 22% | 22% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hao, M.; Imamichi, T.; Chang, W. Modeling and Analysis of HIV-1 Pol Polyprotein as a Case Study for Predicting Large Polyprotein Structures. Int. J. Mol. Sci. 2024, 25, 1809. https://doi.org/10.3390/ijms25031809
Hao M, Imamichi T, Chang W. Modeling and Analysis of HIV-1 Pol Polyprotein as a Case Study for Predicting Large Polyprotein Structures. International Journal of Molecular Sciences. 2024; 25(3):1809. https://doi.org/10.3390/ijms25031809
Chicago/Turabian StyleHao, Ming, Tomozumi Imamichi, and Weizhong Chang. 2024. "Modeling and Analysis of HIV-1 Pol Polyprotein as a Case Study for Predicting Large Polyprotein Structures" International Journal of Molecular Sciences 25, no. 3: 1809. https://doi.org/10.3390/ijms25031809
APA StyleHao, M., Imamichi, T., & Chang, W. (2024). Modeling and Analysis of HIV-1 Pol Polyprotein as a Case Study for Predicting Large Polyprotein Structures. International Journal of Molecular Sciences, 25(3), 1809. https://doi.org/10.3390/ijms25031809