
Making Use of Averaging Methods in MODELLER for Protein Structure Prediction



Abstract
1. Introduction
2. Results
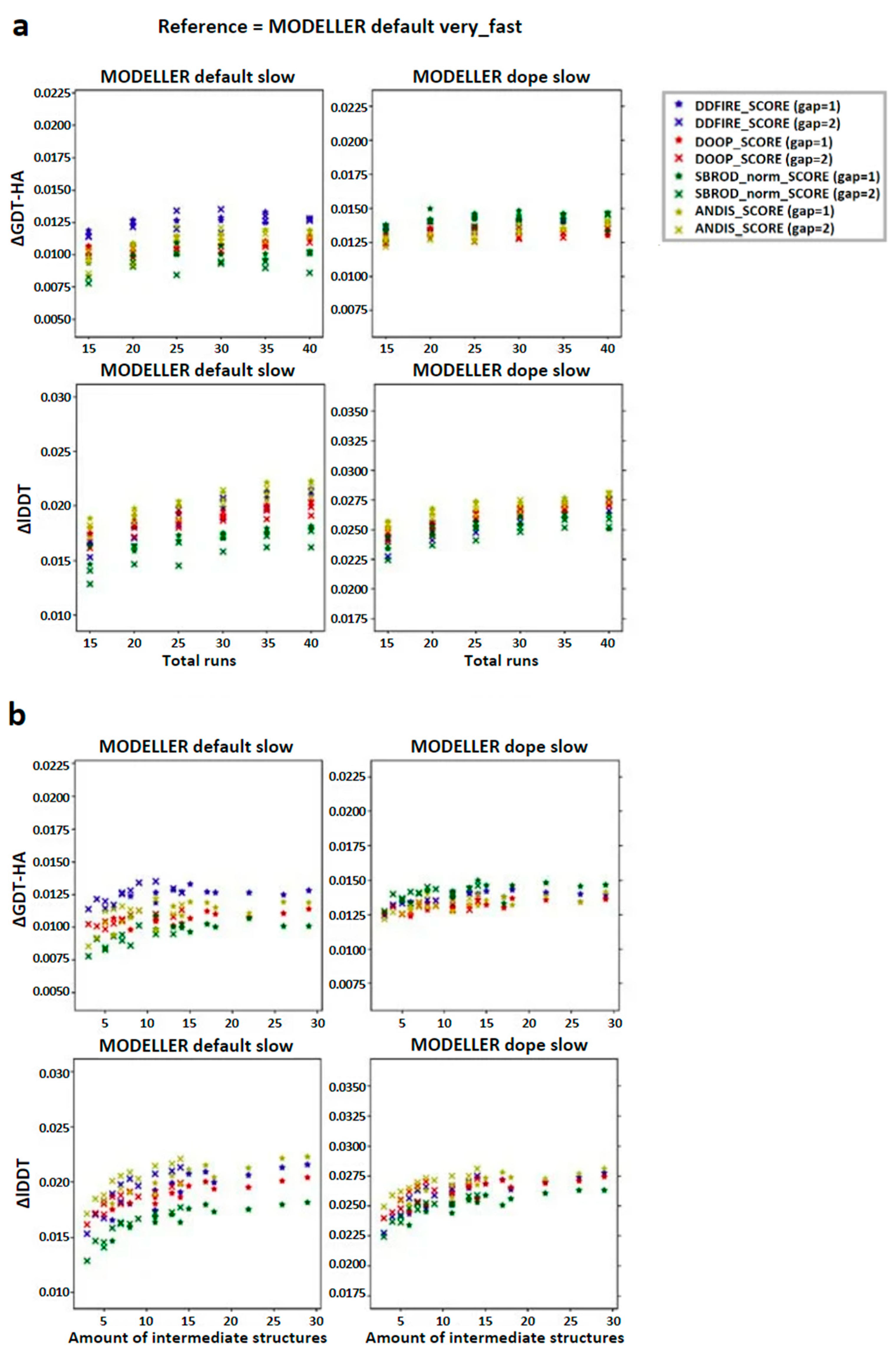
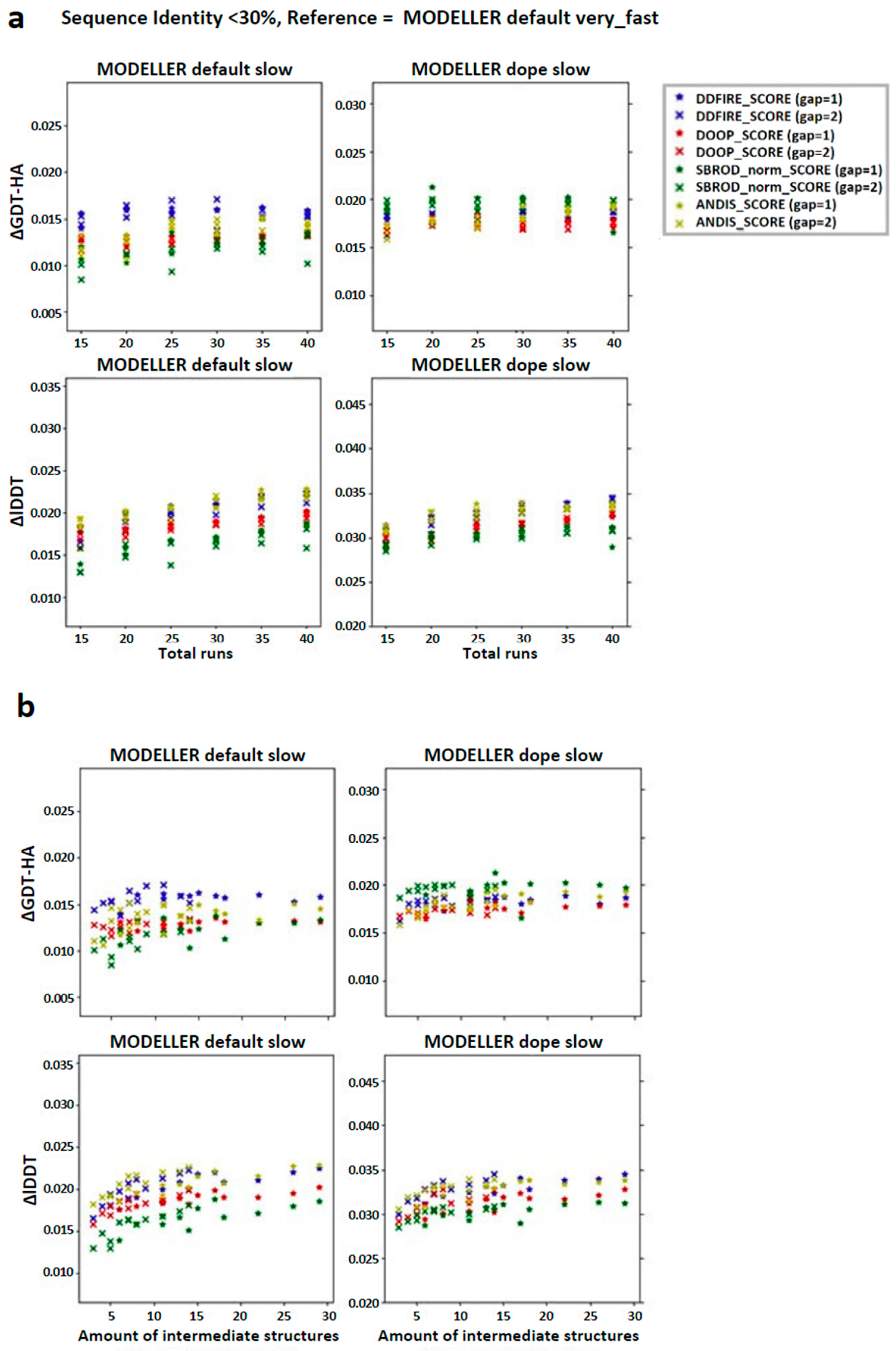
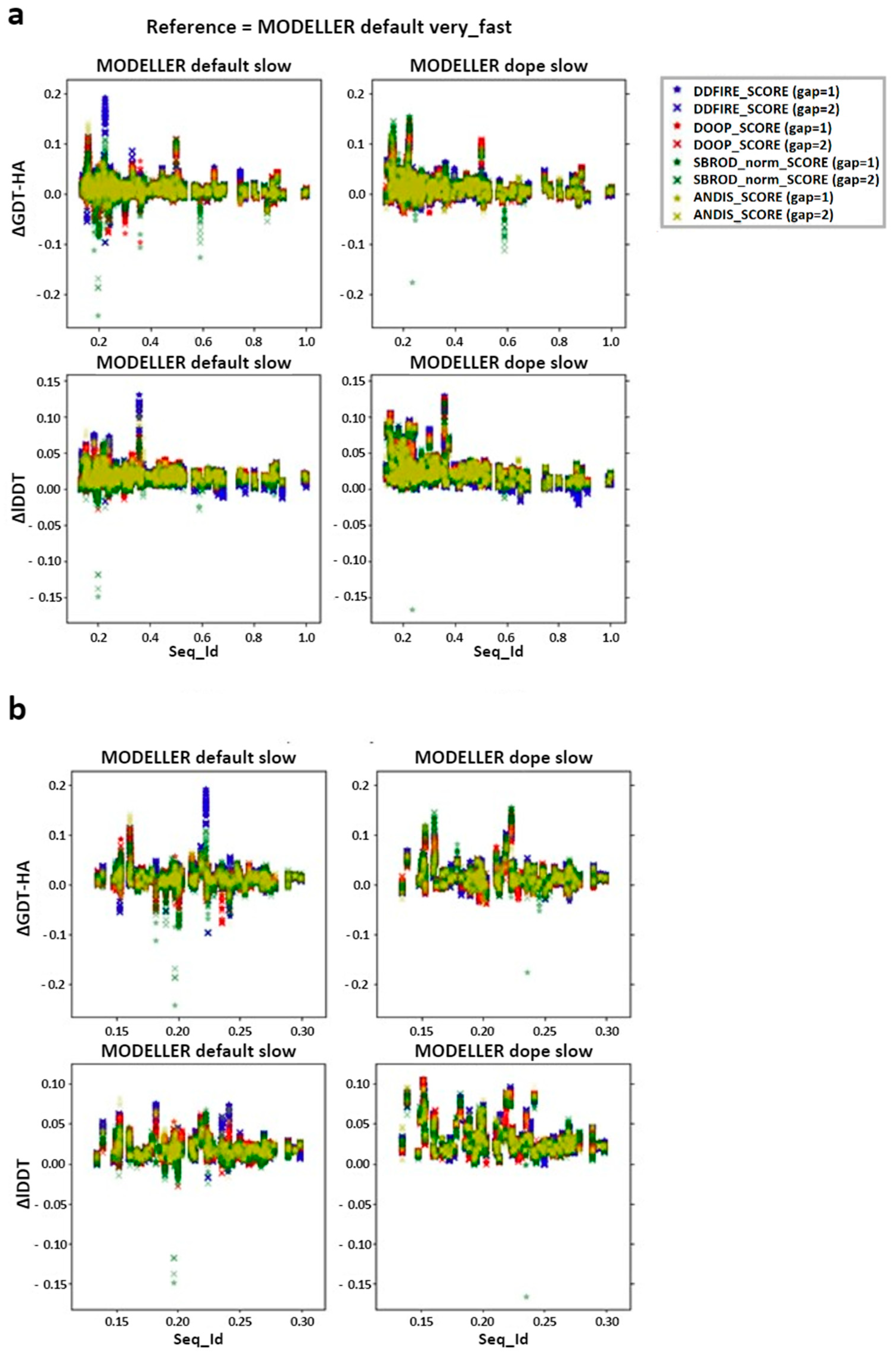
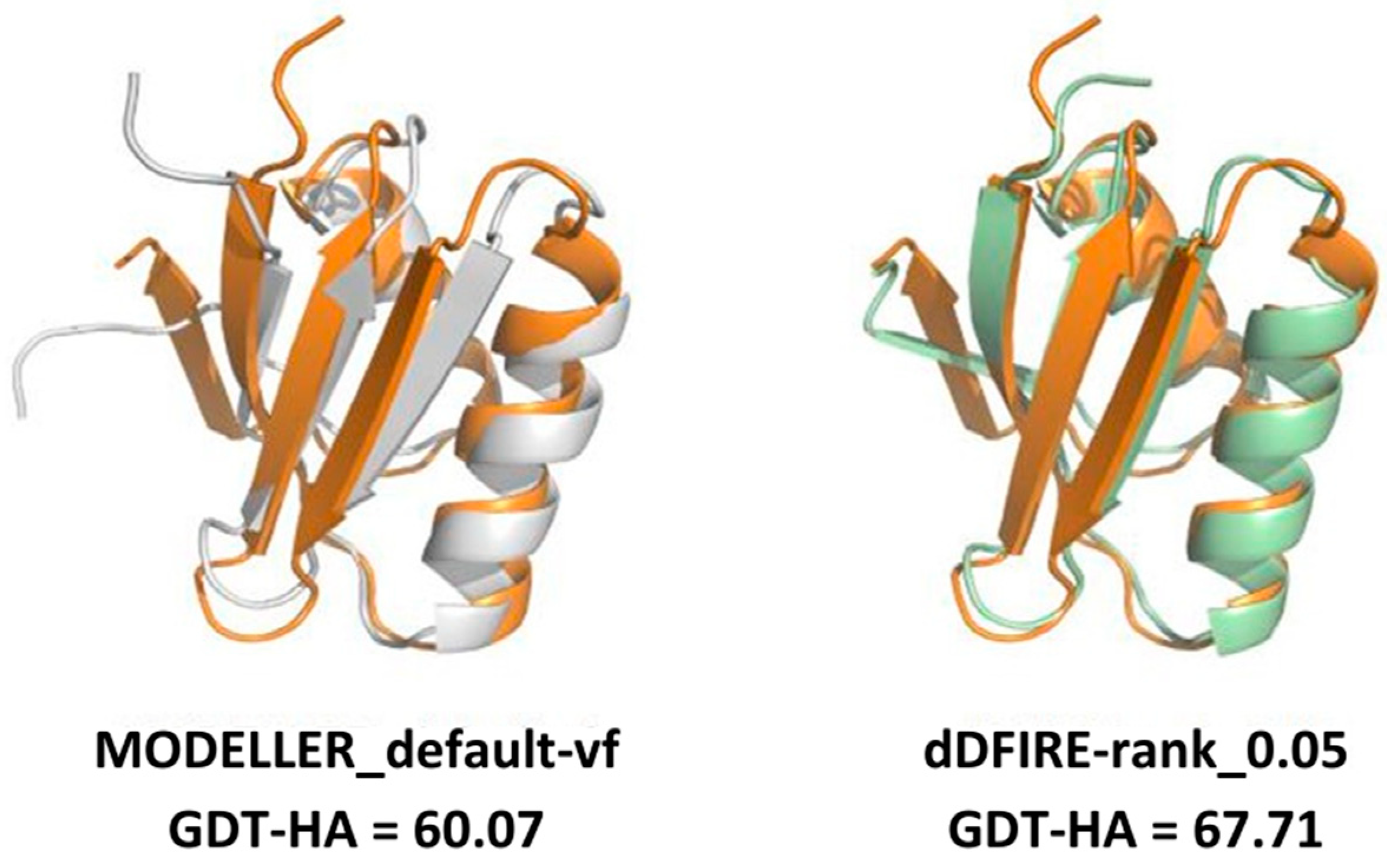
2.1. Comparing Single Template AVG Models with MODELLER Results
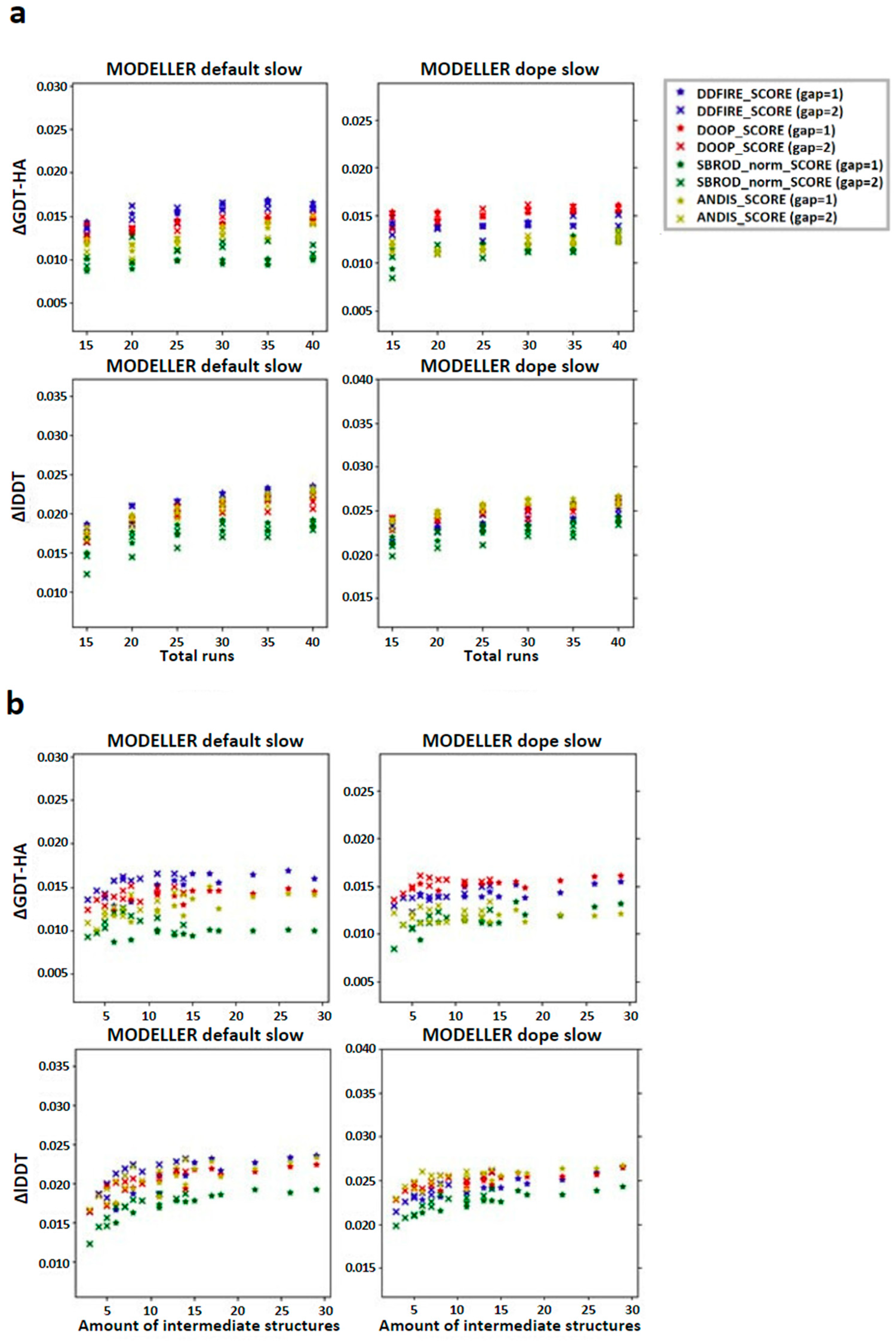
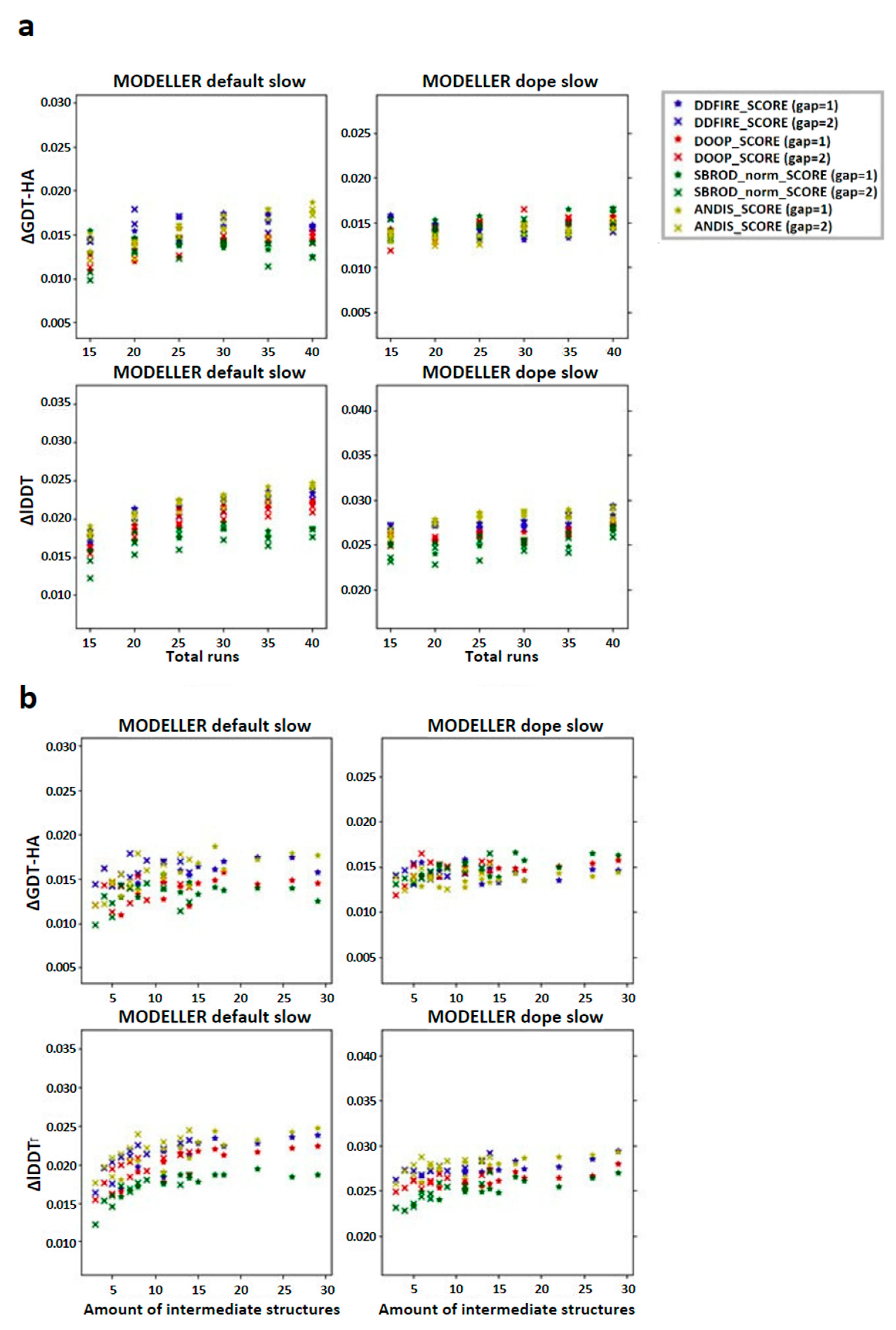
2.2. Comparing Multiple-Template AVG Models with MODELLER Results
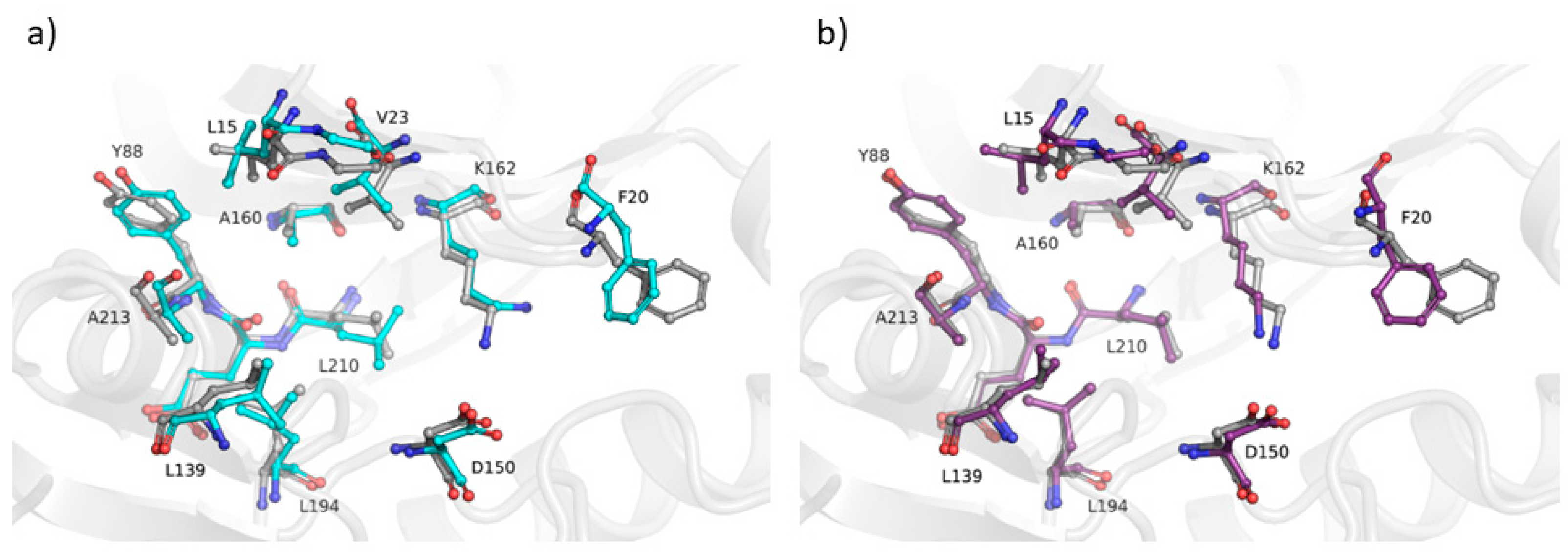
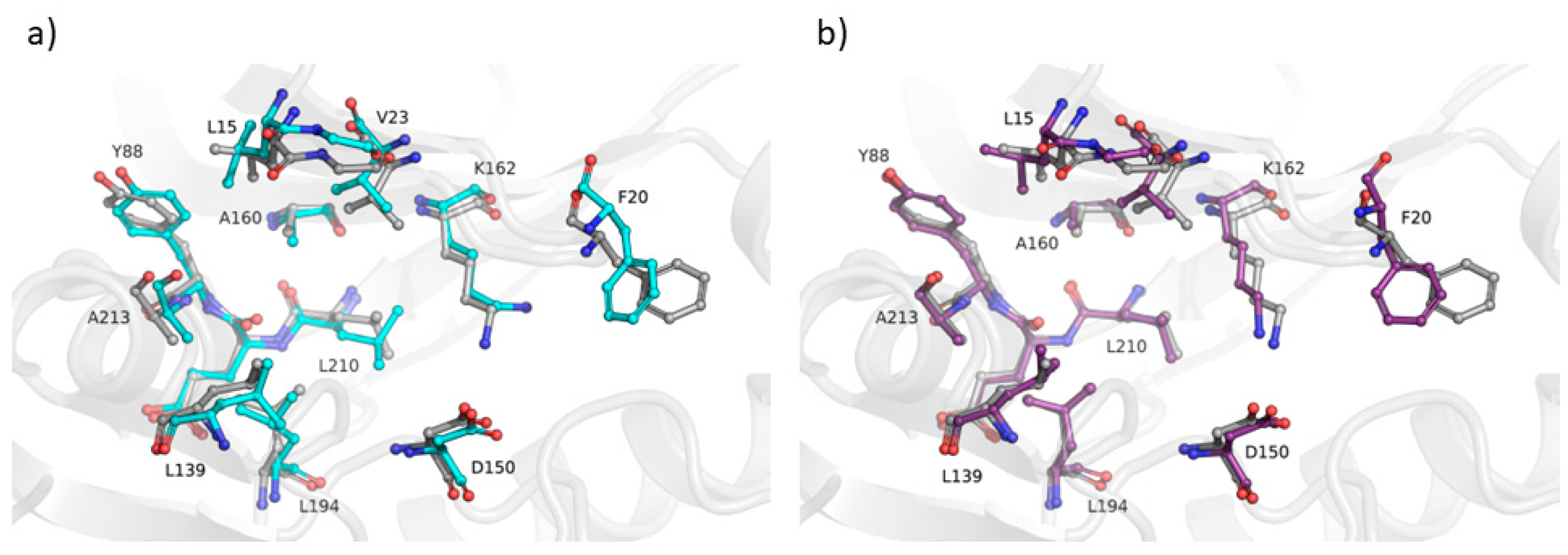
2.3. Assessing the Ability of the AVG Strategy to Predict Holo Protein Conformations
3. Discussion
4. Materials and Methods
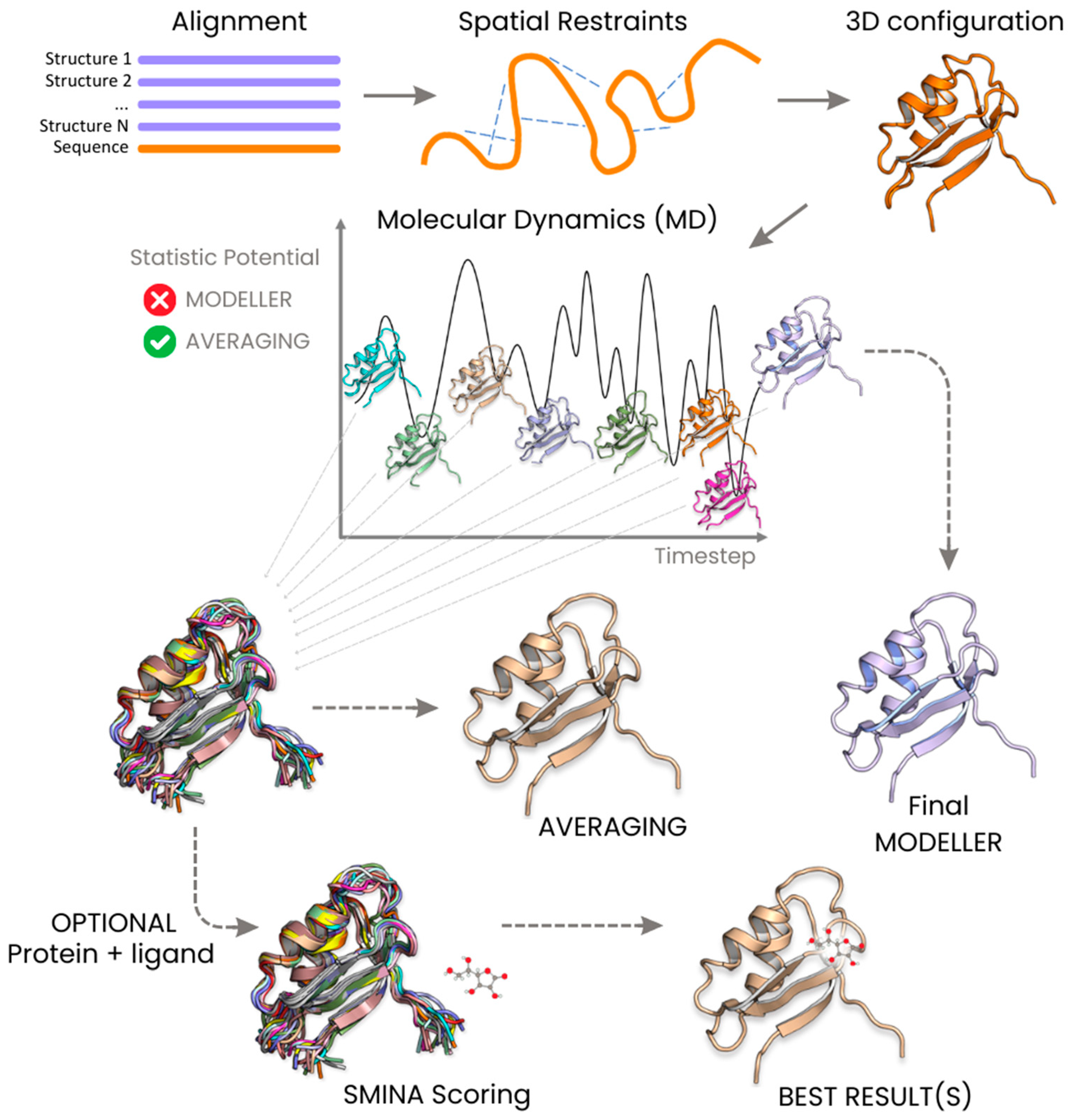
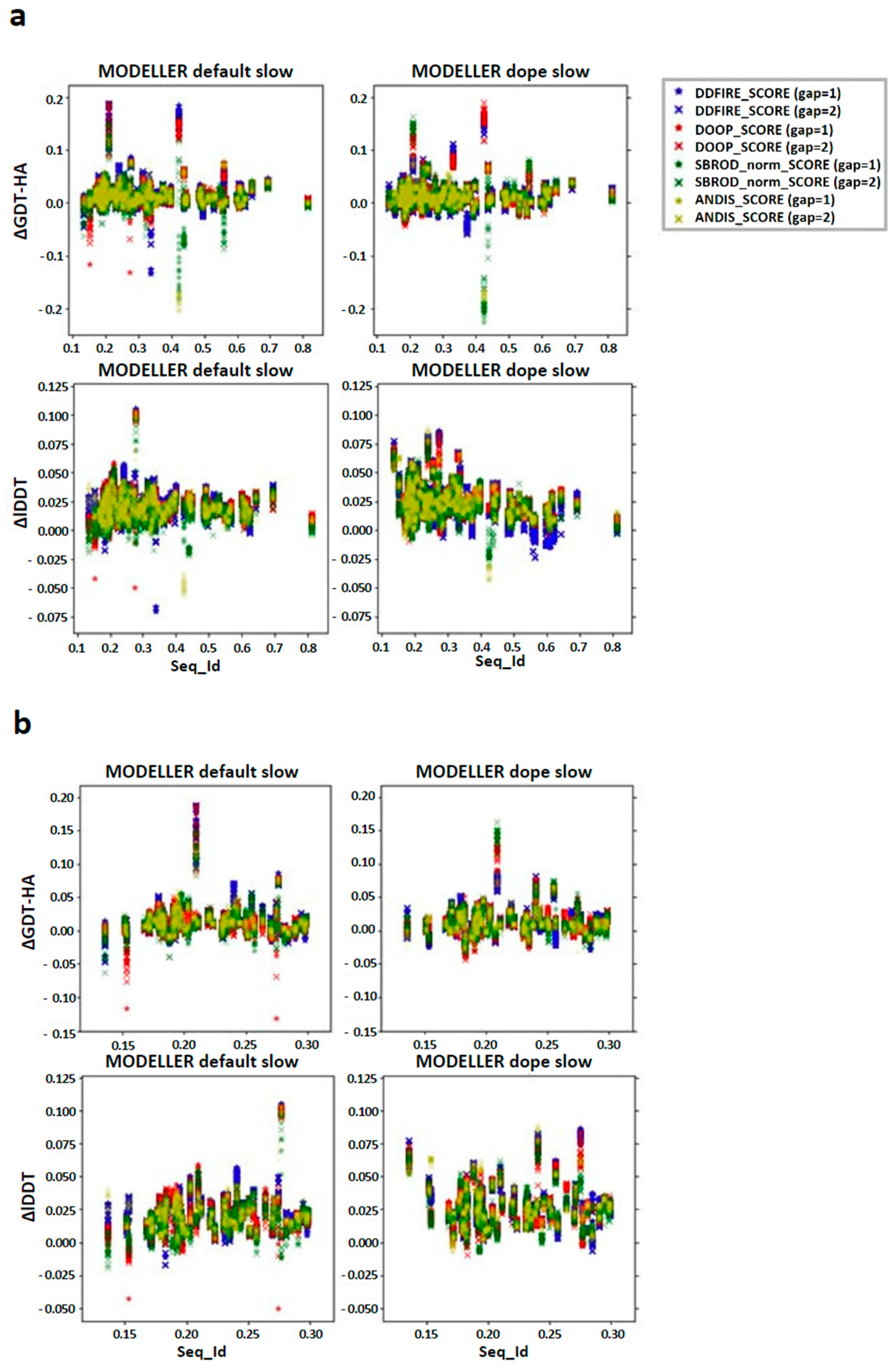
4.1. Statistical Potentials
4.2. MODELLER Algorithm and Its Variants
4.3. Analysis Set
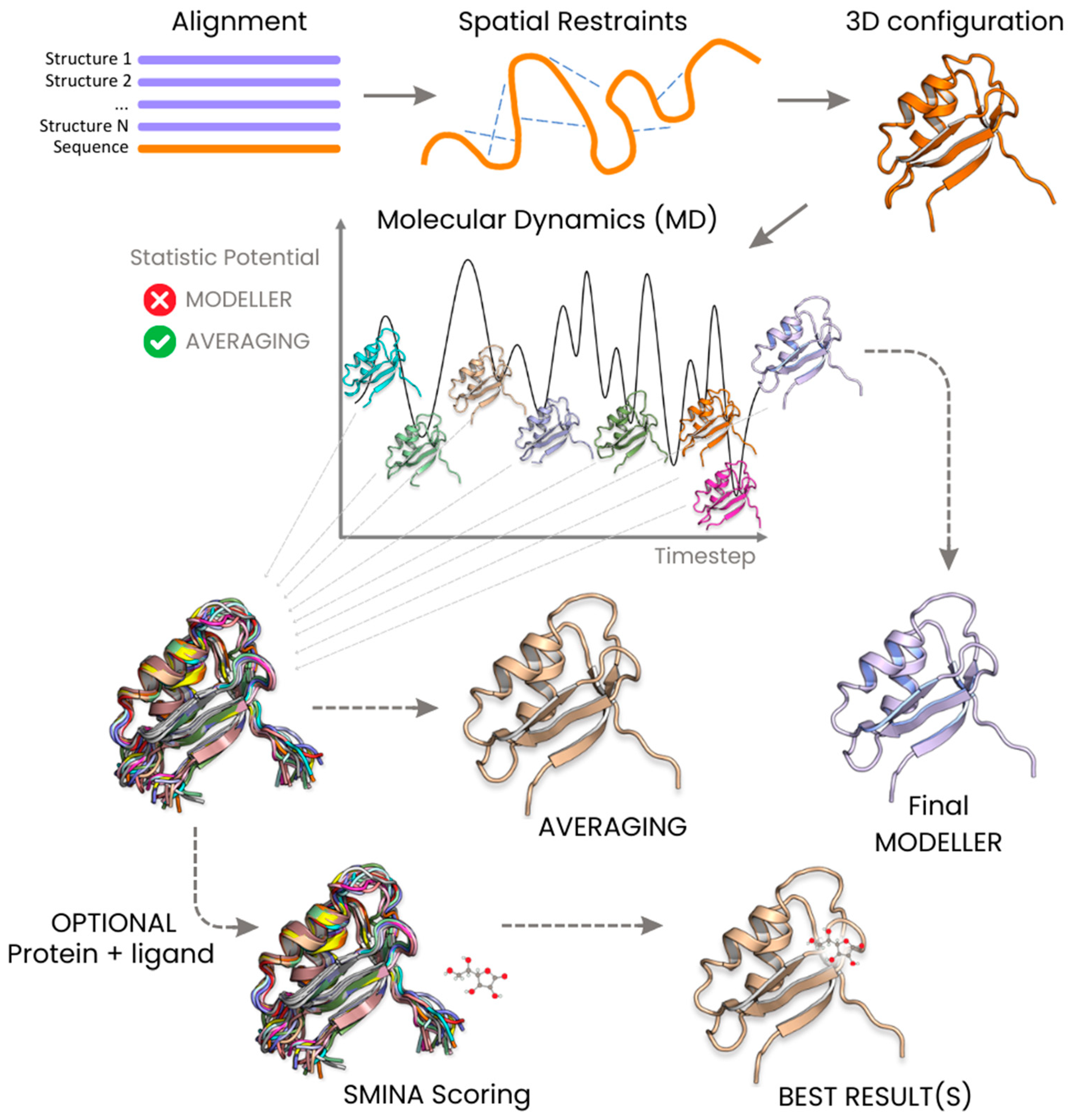
4.4. Building a Configurations Pool
4.5. GDT-HA and lDDT Metrics
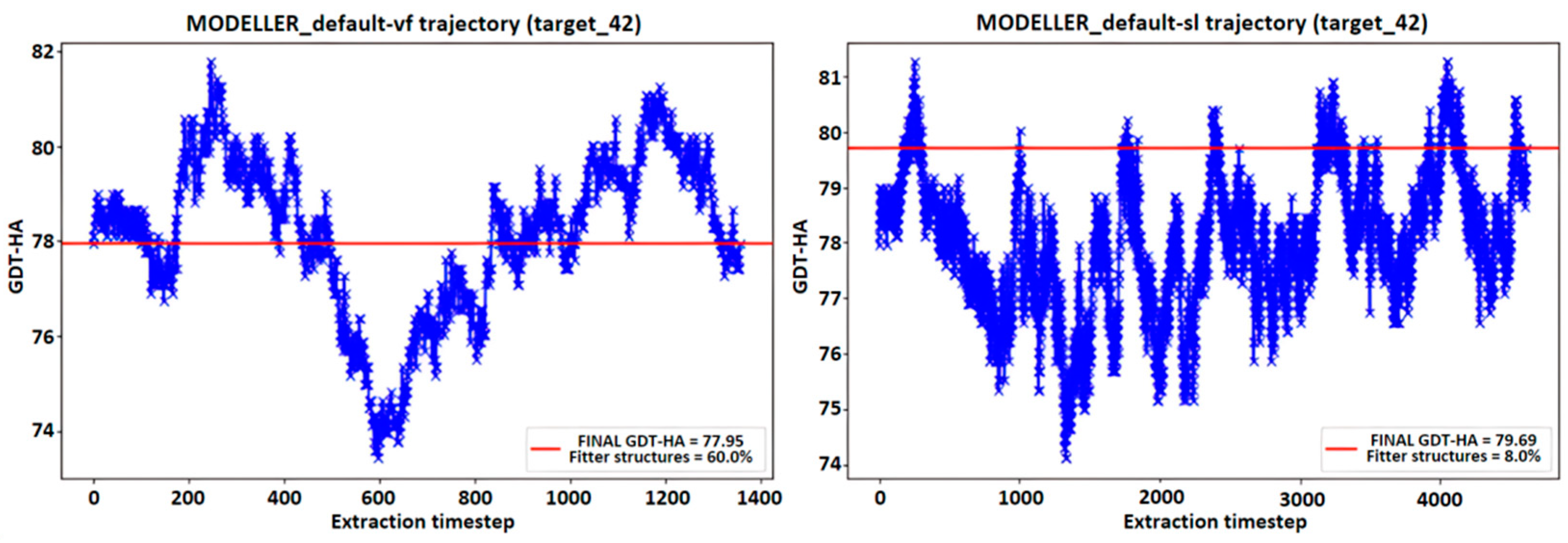
4.6. MODELLER Trajectories Analysis
4.7. Ranking by Means of Statistical Potentials
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Lee, C.; Su, B.-H.; Tseng, Y.J. Comparative studies of AlphaFold, RoseTTAFold and Modeller: A case study involving the use of G-protein-coupled receptors. Brief. Bioinform. 2022, 23, bbac308. [Google Scholar] [CrossRef]
- Zuluaga, F.H.G.; D’Arminio, N.; Bardozzo, F.; Tagliaferri, R.; Marabotti, A. An automated pipeline integrating AlphaFold 2 and MODELLER for protein structure prediction. Comput. Struct. Biotechnol. J. 2023, 21, 5620–5629. [Google Scholar] [CrossRef]
- Sala, D.; Engelberger, F.; Mchaourab, H.S.; Meiler, J. Modeling conformational states of proteins with AlphaFold. Curr. Opin. Struct. Biol. 2023, 81, 102645. [Google Scholar] [CrossRef]
- Heo, L.; Feig, M. Multi-state modeling of G-protein coupled receptors at experimental accuracy. Proteins 2022, 90, 1873–1885. [Google Scholar] [CrossRef]
- Janson, G.; Valdes-Garcia, G.; Heo, L.; Feig, M. Direct generation of protein conformational ensembles via machine learning. Nat. Commun. 2023, 14, 774. [Google Scholar] [CrossRef]
- Mirjalili, V.; Feig, M. Protein Structure Refinement through Structure Selection and Averaging from Molecular Dynamics Ensembles. J. Chem. Theory Comput. 2013, 9, 1294–1303. [Google Scholar] [CrossRef]
- Mirjalili, V.; Noyes, K.; Feig, M. Physics-based protein structure refinement through multiple molecular dynamics trajectories and structure averaging. Proteins Struct. Funct. Bioinform. 2014, 82, 196–207. [Google Scholar] [CrossRef]
- Park, H.; DiMaio, F.; Baker, D. The Origin of Consistent Protein Structure Refinement from Structural Averaging. Structure 2015, 23, 1123–1128. [Google Scholar] [CrossRef]
- Feig, M.; Mirjalili, V. Protein structure refinement via molecular-dynamics simulations: What works and what does not? Proteins Struct. Funct. Bioinform. 2016, 84, 282–292. [Google Scholar] [CrossRef]
- Sánchez, R.; Šali, A. Evaluation of comparative protein structure modeling by MODELLER-3. Proteins Struct. Funct. Bioinform. 1997, 29, 50–58. [Google Scholar] [CrossRef]
- Janson, G.; Grottesi, A.; Pietrosanto, M.; Ausiello, G.; Guarguaglini, G.; Paiardini, A. Revisiting the “satisfaction of spatial restraints” approach of MODELLER for protein homology modeling. PLoS Comput. Biol. 2019, 15, e1007219. [Google Scholar] [CrossRef]
- Narunsky, A.; Nepomnyachiy, S.; Ashkenazy, H.; Kolodny, R.; Ben-Tal, N. ConTemplate Suggests Possible Alternative Conformations for a Query Protein of Known Structure. Structure 2015, 23, 2162–2170. [Google Scholar] [CrossRef]
- Monzon, A.M.; Zea, D.J.; Marino-Buslje, C.; Parisi, G. Homology modeling in a dynamical world. Protein Sci. 2017, 26, 2195–2206. [Google Scholar] [CrossRef]
- Koes, D.R.; Baumgartner, M.P.; Camacho, C.J. Lessons learned in empirical scoring with smina from the CSAR 2011 benchmarking exercise. J. Chem. Inf. Model. 2013, 53, 1893–1904. [Google Scholar] [CrossRef]
- Zhang, J.; Li, H.; Zhao, X.; Wu, Q.; Huang, S.Y. Holo Protein Conformation Generation from Apo Structures by Ligand Binding Site Refinement. J. Chem. Inf. Model. 2022, 62, 5806–5820. [Google Scholar] [CrossRef]
- Kryshtafovych, A.; Monastyrskyy, B.; Fidelis, K.; Moult, J.; Schwede, T.; Tramontano, A. Evaluation of the template-based modeling in CASP12. Proteins 2018, 86, 321–334. [Google Scholar] [CrossRef]
- Karelina, M.; Noh, J.J.; Dror, R.O. How accurately can one predict drug binding modes using AlphaFold models? eLife 2023, 12, RP89386. [Google Scholar] [CrossRef]
- Tanaka, S.; Scheraga, H.A. Medium- and Long-Range Interaction Parameters between Amino Acids for Predicting Three-Dimensional Structures of Proteins. Macromolecules 1976, 9, 945–950. [Google Scholar] [CrossRef]
- Sippl, M.J. Calculation of conformational ensembles from potentials of mean force: An approach to the knowledge-based prediction of local structures in globular proteins. J. Mol. Biol. 1990, 213, 859–883. [Google Scholar] [CrossRef]
- Yang, Y.; Zhou, Y. Specific interactions for ab initio folding of protein terminal regions with secondary structures. Proteins Struct. Funct. Bioinform. 2008, 72, 793–803. [Google Scholar] [CrossRef]
- Karasikov, M.; Pagès, G.; Grudinin, S. Smooth orientation-dependent scoring function for coarse-grained protein quality assessment. Bioinformatics 2019, 35, 2801–2808. [Google Scholar] [CrossRef]
- Chae, M.H.; Krull, F.; Knapp, E.W. Optimized distance-dependent atom-pair-based potential DOOP for protein structure prediction. Proteins Struct. Funct. Bioinform. 2015, 83, 881–890. [Google Scholar] [CrossRef]
- Yu, Z.; Yao, Y.; Deng, H.; Yi, M. ANDIS: An atomic angle- and distance-dependent statistical potential for protein structure quality assessment. BMC Bioinform. 2019, 20, 299. [Google Scholar] [CrossRef]
- Shen, M.; Sali, A. Statistical potential for assessment and prediction of protein structures. Protein Sci. 2006, 15, 2507–2524. [Google Scholar] [CrossRef]
- Zemla, A. LGA: A method for finding 3D similarities in protein structures. Nucleic Acids Res. 2003, 31, 3370–3374. [Google Scholar] [CrossRef]
- Mariani, V.; Biasini, M.; Barbato, A.; Schwede, T. lDDT: A local superposition-free score for comparing protein structures and models using distance difference tests. Bioinformatics 2013, 29, 2722–2728. [Google Scholar] [CrossRef]
- Wu, T.; Guo, Z.; Cheng, J. Atomic protein structure refinement using all-atom graph representations and SE(3)-equivariant graph transformer. Bioinformatics 2023, 39, btad298. [Google Scholar] [CrossRef]
- Huang, Y.J.; Mao, B.; Aramini, J.M.; Montelione, G.T. Assessment of template-based protein structure predictions in CASP10. Proteins 2014, 82, 43–56. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | PDB Apo | PDB Holo | Holo Ligand | PDB Template | Template Ligand | %id | RMSD Default Modeller | RMSD Averaging Method |
|---|---|---|---|---|---|---|---|---|
| Carbonic anhydrase II | 2CBE | 1BCD | FMS | 4YGF | AZM | 29.3 | 0.5 | 0.2 |
| Peroxisome proliferator-activated receptor γ | 1PRG | 2GTK | 208 | 6NWS | L77 | 28.0 | 1.7 | 0.5 |
| Vascular endothelial growth factor receptor kinase | 1VR2 | 2P2I | 608 | 6P3D | 0LI | 29.1 | 0.8 | 0.7 |
| Rac-beta serine/threonine protein kinase | 1GZK | 3D0E | G93 | 6BXI | ANP | 29.6 | 0.9 | 0.9 |
| AmpC β-Lactamase | 2BLS | 1L2S | STC | 4E6W | APB | 26.1 | 0.9 | 0.5 |
| Catechol O-methyltransferase | 4PYI | 3BWM | SAM | 5ZW4 | SAM | 25.2 | 3.2 | 0.8 |
| Beta-secretase 1 | 2ZHV | 3L5D | BDV | 1PPM | 0P1 | 27.5 | 2.2 | 2.0 |
| Fatty acid-binding protein, adipocyte | 3RZY | 2NNQ | T4B | 2IFB | PLM | 29.2 | 0.6 | 0.3 |
| Peroxisome proliferator-activated receptor δ | 2GWX | 2ZNP | K55 | 7KXD | Z7G | 27.4 | 1.5 | 0.6 |
| Blood coagulation factor Xa | 1HCG | 3KL6 | 443 | 6ZOV | GBS | 27.4 | 0.5 | 0.3 |
| Aurora kinase A | 4J8N | 5OSD | ADP | 3D5W | ADP | 34.1 | 0.6 | 0.3 |
| Mean RMSD | 1.2 | 0.8 | ||||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rosignoli, S.; Lustrino, E.; Di Silverio, I.; Paiardini, A. Making Use of Averaging Methods in MODELLER for Protein Structure Prediction. Int. J. Mol. Sci. 2024, 25, 1731. https://doi.org/10.3390/ijms25031731
Rosignoli S, Lustrino E, Di Silverio I, Paiardini A. Making Use of Averaging Methods in MODELLER for Protein Structure Prediction. International Journal of Molecular Sciences. 2024; 25(3):1731. https://doi.org/10.3390/ijms25031731
Chicago/Turabian StyleRosignoli, Serena, Elisa Lustrino, Iris Di Silverio, and Alessandro Paiardini. 2024. "Making Use of Averaging Methods in MODELLER for Protein Structure Prediction" International Journal of Molecular Sciences 25, no. 3: 1731. https://doi.org/10.3390/ijms25031731
APA StyleRosignoli, S., Lustrino, E., Di Silverio, I., & Paiardini, A. (2024). Making Use of Averaging Methods in MODELLER for Protein Structure Prediction. International Journal of Molecular Sciences, 25(3), 1731. https://doi.org/10.3390/ijms25031731