
The ATM Ser49Cys Variant Effects ATM Function as a Regulator of Oncogene-Induced Senescence

 , ,
, , 
Abstract
1. Introduction
2. Results
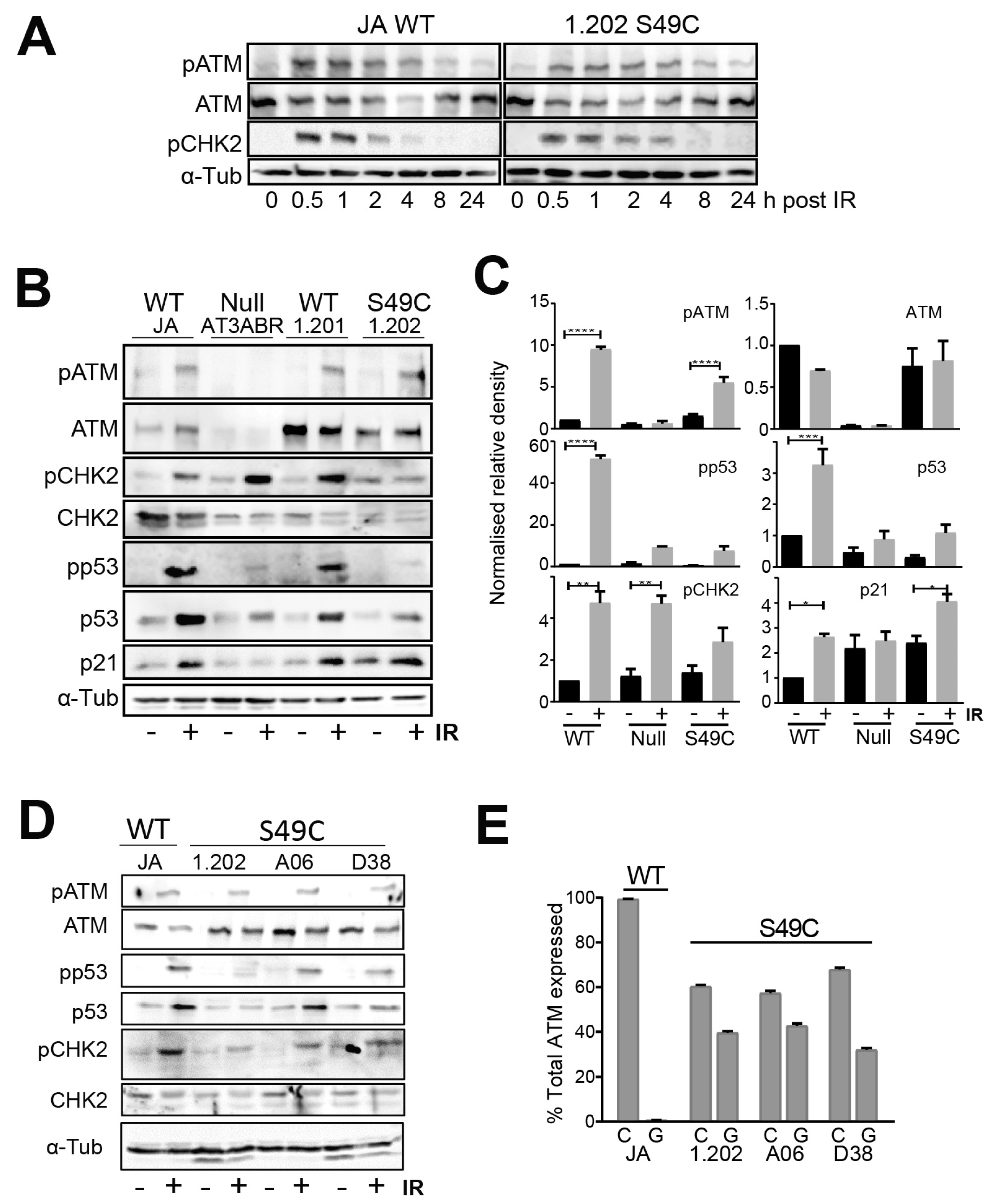
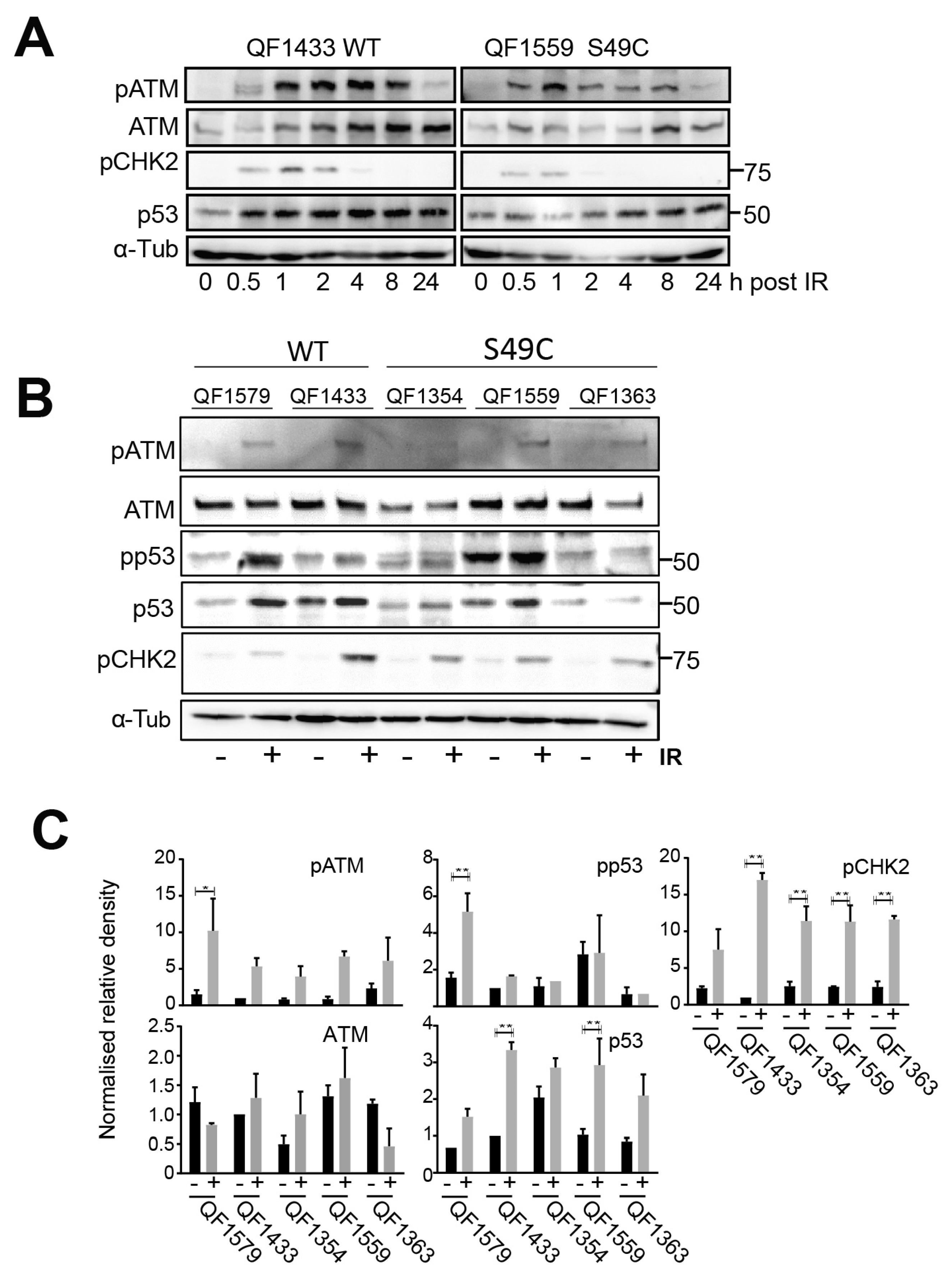
2.1. ATM Ser49Cys Variant Protein Is Functional for Acute p53 Activation by DNA Damage
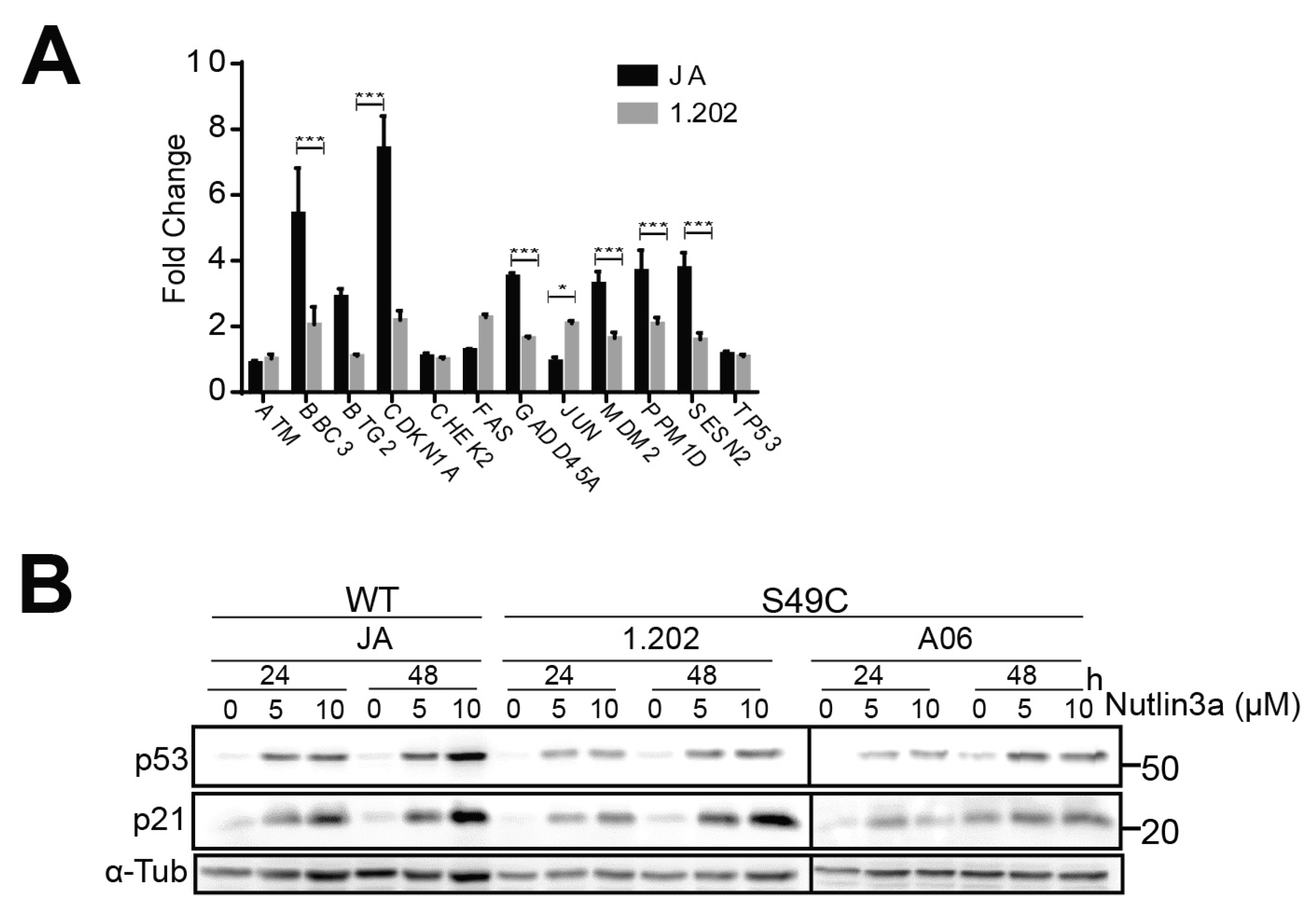
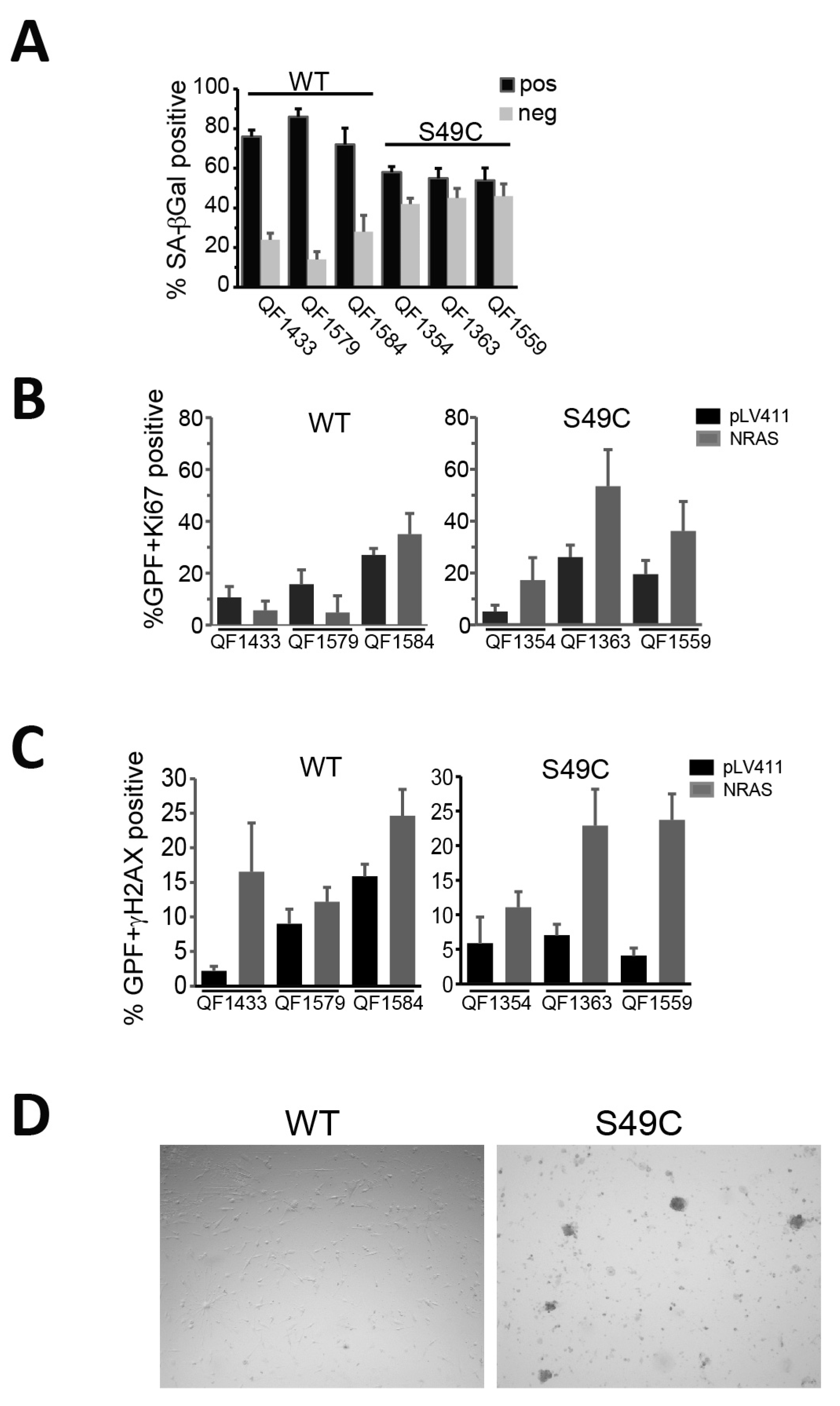
2.2. ATM Ser49Cys Variant Delays Senescence in Primary Human Melanoblasts
2.3. ATM Ser49Cys Is Not Associated with Increased Melanoma Risk
3. Discussion
4. Materials and Methods
4.1. Antibodies
4.2. Production of Patient-Derived LCLs
4.3. Cell Culture
4.4. Genotyping of LCLs and Primary Human Melanoblasts
4.5. Allelic Expression Variation
4.6. Immunofluorescence
4.7. Induction of Senescence
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bakkenist, C.J.; Kastan, M.B. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature 2003, 421, 499–506. [Google Scholar] [CrossRef] [PubMed]
- Thompson, L.H. Recognition, signaling, and repair of DNA double-strand breaks produced by ionizing radiation in mammalian cells: The molecular choreography. Mutat. Res. 2012, 751, 158–246. [Google Scholar] [CrossRef]
- Shiloh, Y.; Ziv, Y. The ATM protein kinase: Regulating the cellular response to genotoxic stress, and more. Nat. Rev. Mol. Cell Biol. 2013, 14, 197–210. [Google Scholar] [CrossRef] [PubMed]
- Bartkova, J.; Rezaei, N.; Liontos, M.; Karakaidos, P.; Kletsas, D.; Issaeva, N.; Vassiliou, L.V.; Kolettas, E.; Niforou, K.; Zoumpourlis, V.C.; et al. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature 2006, 444, 633–637. [Google Scholar] [CrossRef]
- Michaloglou, C.; Vredeveld, L.C.; Soengas, M.S.; Denoyelle, C.; Kuilman, T.; van der Horst, C.M.; Majoor, D.M.; Shay, J.W.; Mooi, W.J.; Peeper, D.S. BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature 2005, 436, 720–724. [Google Scholar] [CrossRef]
- Shain, A.H.; Yeh, I.; Kovalyshyn, I.; Sriharan, A.; Talevich, E.; Gagnon, A.; Dummer, R.; North, J.; Pincus, L.; Ruben, B.; et al. The Genetic Evolution of Melanoma from Precursor Lesions. N. Engl. J. Med. 2015, 373, 1926–1936. [Google Scholar] [CrossRef]
- Ambrose, M.; Gatti, R.A. Pathogenesis of ataxia-telangiectasia: The next generation of ATM functions. Blood 2013, 121, 4036–4045. [Google Scholar] [CrossRef]
- Choi, M.; Kipps, T.; Kurzrock, R. ATM Mutations in Cancer: Therapeutic Implications. Mol. Cancer Ther. 2016, 15, 1781–1791. [Google Scholar] [CrossRef]
- Barrett, J.H.; Iles, M.M.; Harland, M.; Taylor, J.C.; Aitken, J.F.; Andresen, P.A.; Bishop, D.T. Genome-wide association study identifies three new melanoma susceptibility loci. Nat. Genet. 2011, 43, 1108–1113. [Google Scholar] [CrossRef]
- Baughan, S.L.; Darwiche, F.; Tainsky, M.A. Functional analysis of ATM variants in a high risk cohort provides insight into missing heritability. Cancer Genet. 2022, 264–265, 40–49. [Google Scholar] [CrossRef]
- Fletcher, O.; Johnson, N.; dos Santos Silva, I.; Orr, N.; Ashworth, A.; Nevanlinna, H.; Heikkinen, T.; Aittomaki, K.; Blomqvist, C.; Burwinkel, B.; et al. Missense variants in ATM in 26,101 breast cancer cases and 29,842 controls. Cancer Epidemiol. Biomark. Prev. 2010, 19, 2143–2151. [Google Scholar] [CrossRef]
- Dombernowsky, S.L.; Weischer, M.; Allin, K.H.; Bojesen, S.E.; Tybjjrg-Hansen, A.; Nordestgaard, B.G. Risk of cancer by ATM missense mutations in the general population. J. Clin. Oncol. 2008, 26, 3057–3062. [Google Scholar] [CrossRef] [PubMed]
- Law, M.H.; Bishop, D.T.; Lee, J.E.; Brossard, M.; Martin, N.G.; Moses, E.K.; Song, F.; Barrett, J.H.; Kumar, R.; Easton, D.F.; et al. Genome-wide meta-analysis identifies five new susceptibility loci for cutaneous malignant melanoma. Nat. Genet. 2015, 47, 987–995. [Google Scholar] [CrossRef] [PubMed]
- Khanna, K.K.; Keating, K.E.; Kozlov, S.; Scott, S.; Gatei, M.; Hobson, K.; Taya, Y.; Gabrielli, B.; Chan, D.; Lees-Miller, S.P.; et al. ATM associates with and phosphorylates p53: Mapping the region of interaction. Nat. Genet. 1998, 20, 398–400. [Google Scholar] [CrossRef]
- McInerney-Leo, A.M.; Wheeler, L.; Sturm, R.A.; Tan, J.M.; Harris, J.E.; Anderson, L.; Jagirdar, K.; Brown, M.A.; Leo, P.J.; Soyer, H.P.; et al. Point mutation in p14ARF-specific exon 1β of CDKN2A causing familial melanoma and astrocytoma. Br. J. Dermatol. 2018, 178, e263–e264. [Google Scholar] [CrossRef]
- Goldstein, A.M.; Chan, M.; Harland, M.; Gillanders, E.M.; Hayward, N.K.; Avril, M.F.; Azizi, E.; Bianchi-Scarra, G.; Bishop, D.T.; Bressac-de Paillerets, B.; et al. High-risk melanoma susceptibility genes and pancreatic cancer, neural system tumors, and uveal melanoma across GenoMEL. Cancer Res. 2006, 66, 9818–9828. [Google Scholar] [CrossRef]
- Duffy, D.L.; Jagirdar, K.; Lee, K.J.; McWhirter, S.R.; McMeniman, E.K.; De’Ambrosis, B.; Pflugfelder, A.; Rayner, J.E.; Whiteman, D.C.; Brown, M.A.; et al. Genes Determining Nevus Count and Dermoscopic Appearance in Australian Melanoma Cases and Controls. J. Investig. Dermatol. 2020, 140, 498–501.e17. [Google Scholar] [CrossRef]
- Rayner, J.E.; Duffy, D.L.; Smit, D.J.; Jagirdar, K.; Lee, K.J.; De’Ambrosis, B.; Smithers, B.M.; McMeniman, E.K.; McInerney-Leo, A.M.; Schaider, H.; et al. Germline and somatic albinism variants in amelanotic/hypomelanotic melanoma: Increased carriage of TYR and OCA2 variants. PLoS ONE 2020, 15, e0238529. [Google Scholar] [CrossRef]
- Canela-Xandri, O.; Rawlik, K.; Tenesa, A. An atlas of genetic associations in UK Biobank. Nat. Genet. 2018, 50, 1593–1599. [Google Scholar] [CrossRef]
- Pandita, T.K.; Lieberman, H.B.; Lim, D.S.; Dhar, S.; Zheng, W.; Taya, Y.; Kastan, M.B. Ionizing radiation activates the ATM kinase throughout the cell cycle. Oncogene 2000, 19, 1386–1391. [Google Scholar] [CrossRef]
- Pomerantz, J.; Schreiber-Agus, N.; Liegeois, N.J.; Silverman, A.; Alland, L.; Chin, L.; Potes, J.; Chen, K.; Orlow, I.; Lee, H.W.; et al. The Ink4a tumor suppressor gene product, p19Arf, interacts with MDM2 and neutralizes MDM2’s inhibition of p53. Cell 1998, 92, 713–723. [Google Scholar] [CrossRef]
- Goldstein, A.M.; Xiao, Y.; Sampson, J.; Zhu, B.; Rotunno, M.; Bennett, H.; Wen, Y.; Jones, K.; Vogt, A.; Burdette, L.; et al. Rare germline variants in known melanoma susceptibility genes in familial melanoma. Hum. Mol. Genet. 2017, 26, 4886–4895. [Google Scholar] [CrossRef] [PubMed]
- Duffy, D.L.; Zhu, G.; Li, X.; Sanna, M.; Iles, M.M.; Jacobs, L.C.; Evans, D.M.; Yazar, S.; Beesley, J.; Law, M.H.; et al. Novel pleiotropic risk loci for melanoma and nevus density implicate multiple biological pathways. Nat. Commun. 2018, 9, 4774. [Google Scholar] [CrossRef] [PubMed]
- Landi, M.T.; Bishop, D.T.; MacGregor, S.; Machiela, M.J.; Stratigos, A.J.; Ghiorzo, P.; Brossard, M.; Calista, D.; Choi, J.; Fargnoli, M.C.; et al. Genome-wide association meta-analyses combining multiple risk phenotypes provide insights into the genetic architecture of cutaneous melanoma susceptibility. Nat. Genet. 2020, 52, 494–504. [Google Scholar] [CrossRef] [PubMed]
- Potrony, M.; Badenas, C.; Aguilera, P.; Puig-Butille, J.A.; Carrera, C.; Malvehy, J.; Puig, S. Update in genetic susceptibility in melanoma. Ann. Transl. Med. 2015, 3, 210. [Google Scholar] [CrossRef] [PubMed]
- Maas, E.J.; Wallingford, C.K.; DeBortoli, E.; Smit, D.J.; Betz-Stablein, B.; Aoude, L.G.; Stark, M.S.; Sturm, R.A.; Soyer, H.P.; McInerney-Leo, A.M. GOLM1: Expanding our understanding of melanoma susceptibility. J. Med. Genet. 2023, 60, 835–837. [Google Scholar] [CrossRef] [PubMed]
- Cook, A.L.; Donatien, P.D.; Smith, A.G.; Murphy, M.; Jones, M.K.; Herlyn, M.; Bennett, D.C.; Leonard, J.H.; Sturm, R.A. Human melanoblasts in culture: Expression of BRN2 and synergistic regulation by fibroblast growth factor-2, stem cell factor, and endothelin-3. J. Investig. Dermatol. 2003, 121, 1150–1159. [Google Scholar] [CrossRef] [PubMed]
- Daley, G.M.; Duffy, D.L.; Pflugfelder, A.; Jagirdar, K.; Lee, K.J.; Yong, X.L.H.; Eigentler, T.K.; Weide, B.; Smithers, B.M.; Martin, N.G.; et al. GSTP1 does not modify MC1R effects on melanoma risk. Exp. Dermatol. 2017, 26, 730–733. [Google Scholar] [CrossRef] [PubMed]
- Cook, A.L.; Chen, W.; Thurber, A.E.; Smit, D.J.; Smith, A.G.; Bladen, T.G.; Brown, D.L.; Duffy, D.L.; Pastorino, L.; Bianchi-Scarra, G.; et al. Analysis of cultured human melanocytes based on polymorphisms within the SLC45A2/MATP, SLC24A5/NCKX5, and OCA2/P loci. J. Investig. Dermatol. 2009, 129, 392–405. [Google Scholar] [CrossRef]
- Jones, T.R.; Kang, I.H.; Wheeler, D.B.; Lindquist, R.A.; Papallo, A.; Sabatini, D.M.; Golland, P.; Carpenter, A.E. CellProfiler Analyst: Data exploration and analysis software for complex image-based screens. BMC Bioinform. 2008, 9, 482. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Trait | Eff. Allele | Beta | pv | MAF | HWE | ORbeta * |
|---|---|---|---|---|---|---|
| C43 Malignant melanoma of skin | C | 0.0003 | 0.6618 | 0.0147 | 0.1305 | 1.05 |
| D03 Melanoma in situ | C | −0.0004 | 0.3289 | 0.0147 | 0.1305 | 0.83 |
| C43-C44 Melanoma and other malignant neoplasms of skin | C | 0.0019 | 0.2599 | 0.0147 | 0.1305 | 1.05 |
| Skin colour | C | 0.0007 | 0.8603 | 0.0147 | 0.1305 | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Atkinson, C.; McInerney-Leo, A.M.; Proctor, M.; Lanagan, C.; Stevenson, A.J.; Dehkhoda, F.; Caole, M.; Maas, E.; Ainger, S.; Pritchard, A.L.; et al. The ATM Ser49Cys Variant Effects ATM Function as a Regulator of Oncogene-Induced Senescence. Int. J. Mol. Sci. 2024, 25, 1664. https://doi.org/10.3390/ijms25031664
Atkinson C, McInerney-Leo AM, Proctor M, Lanagan C, Stevenson AJ, Dehkhoda F, Caole M, Maas E, Ainger S, Pritchard AL, et al. The ATM Ser49Cys Variant Effects ATM Function as a Regulator of Oncogene-Induced Senescence. International Journal of Molecular Sciences. 2024; 25(3):1664. https://doi.org/10.3390/ijms25031664
Chicago/Turabian StyleAtkinson, Caroline, Aideen M. McInerney-Leo, Martina Proctor, Catherine Lanagan, Alexander J. Stevenson, Farhad Dehkhoda, Mary Caole, Ellie Maas, Stephen Ainger, Antonia L. Pritchard, and et al. 2024. "The ATM Ser49Cys Variant Effects ATM Function as a Regulator of Oncogene-Induced Senescence" International Journal of Molecular Sciences 25, no. 3: 1664. https://doi.org/10.3390/ijms25031664
APA StyleAtkinson, C., McInerney-Leo, A. M., Proctor, M., Lanagan, C., Stevenson, A. J., Dehkhoda, F., Caole, M., Maas, E., Ainger, S., Pritchard, A. L., Johansson, P. A., Leo, P., Hayward, N. K., Sturm, R. A., Duncan, E. L., & Gabrielli, B. (2024). The ATM Ser49Cys Variant Effects ATM Function as a Regulator of Oncogene-Induced Senescence. International Journal of Molecular Sciences, 25(3), 1664. https://doi.org/10.3390/ijms25031664