
Traumatic Brain Injury Induces Nociceptin/Orphanin FQ and Nociceptin Opioid Peptide Receptor Expression within 24 Hours

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
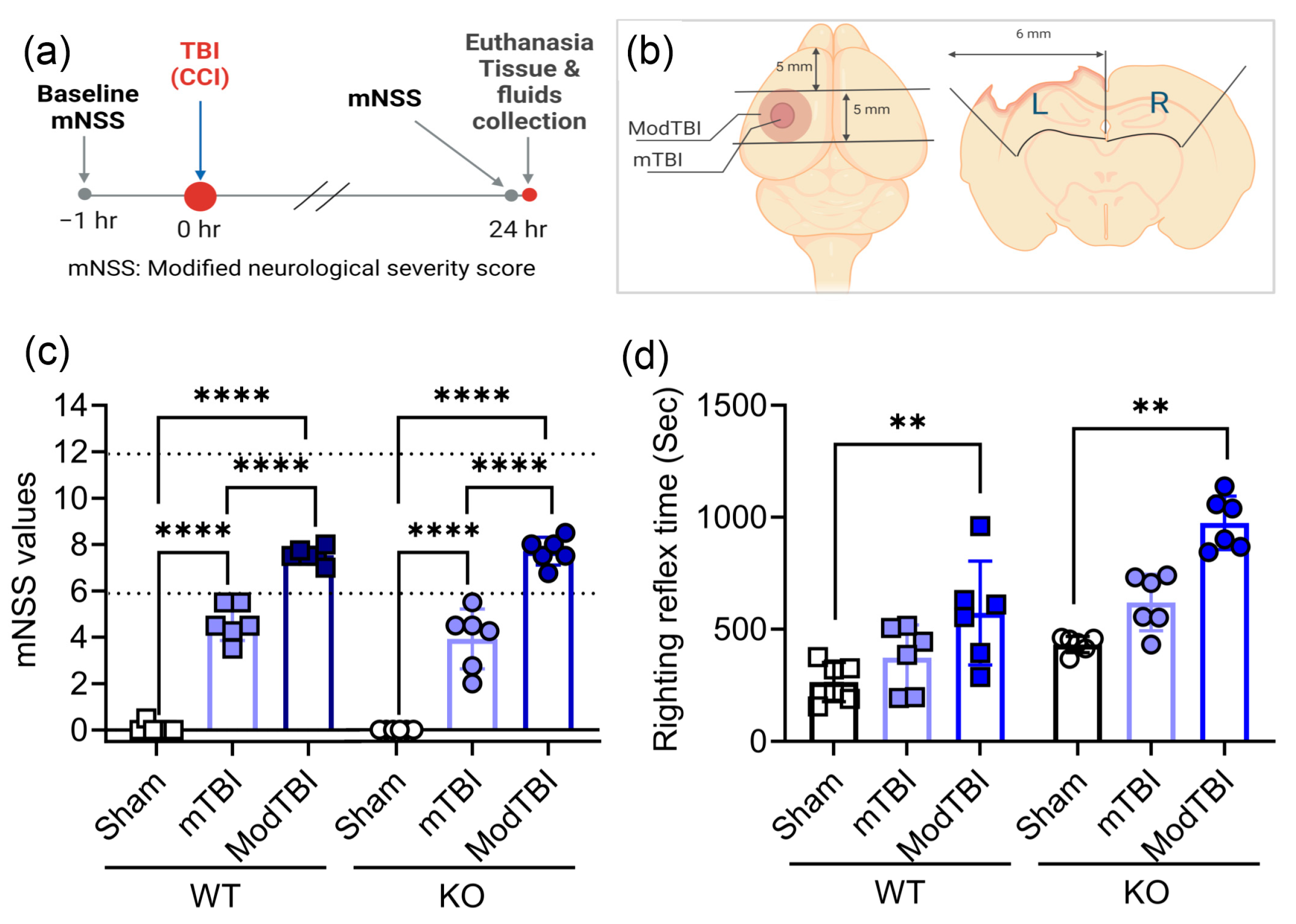
2.1. Neurological Scores on Day 1 Confirm Injury Severity; Moderate TBI Prolongs Righting Reflex Time in WT and KO Rats
2.2. N/OFQ Levels Increase on the TBI-Injured Side of the Brain and in CSF of WT Rats 24 h Post-TBI
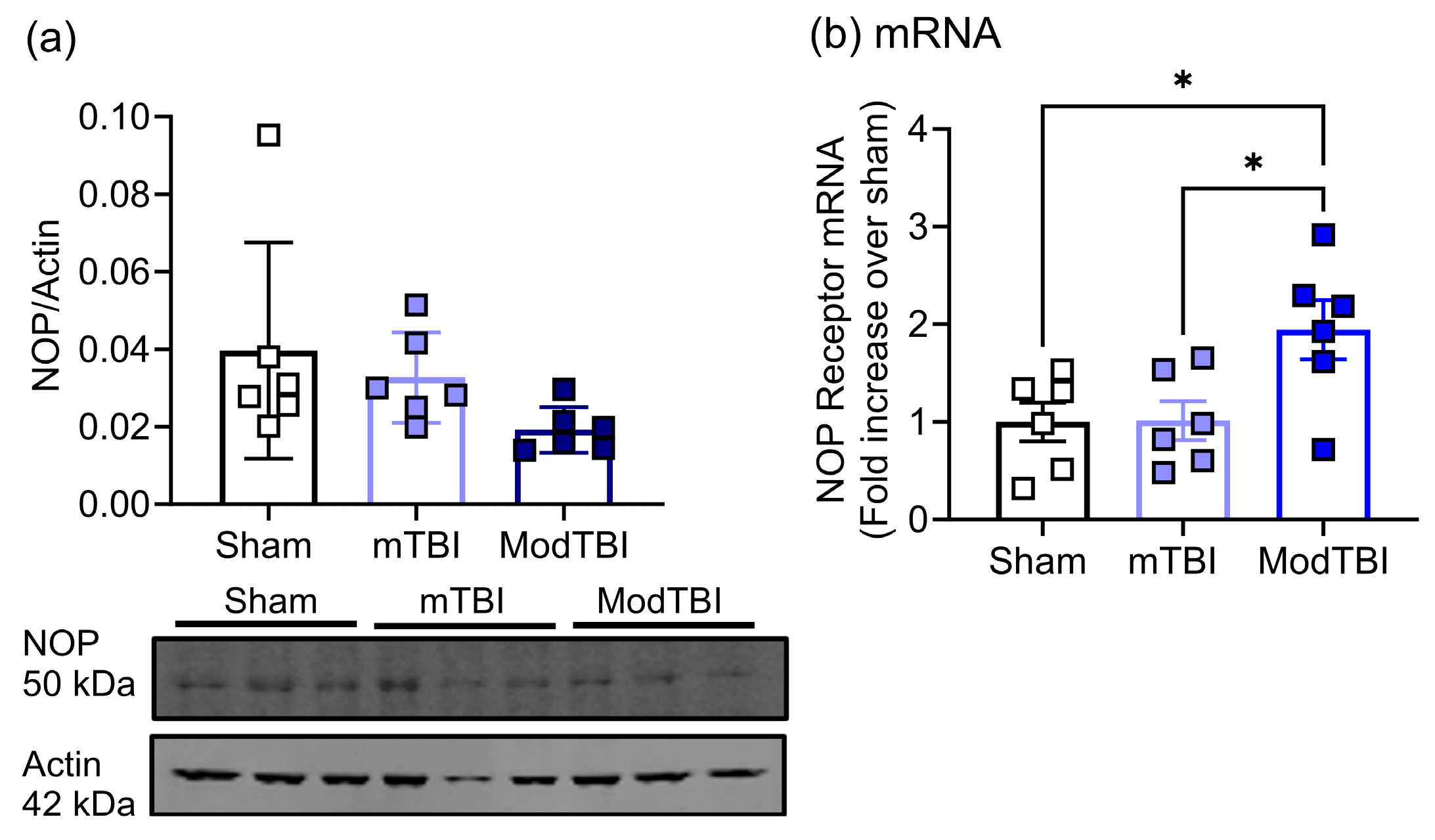
2.3. NOP Receptor Protein and mRNA Expression Changes in Injured Brain Tissue of WT Rats 24 h Post-TBI
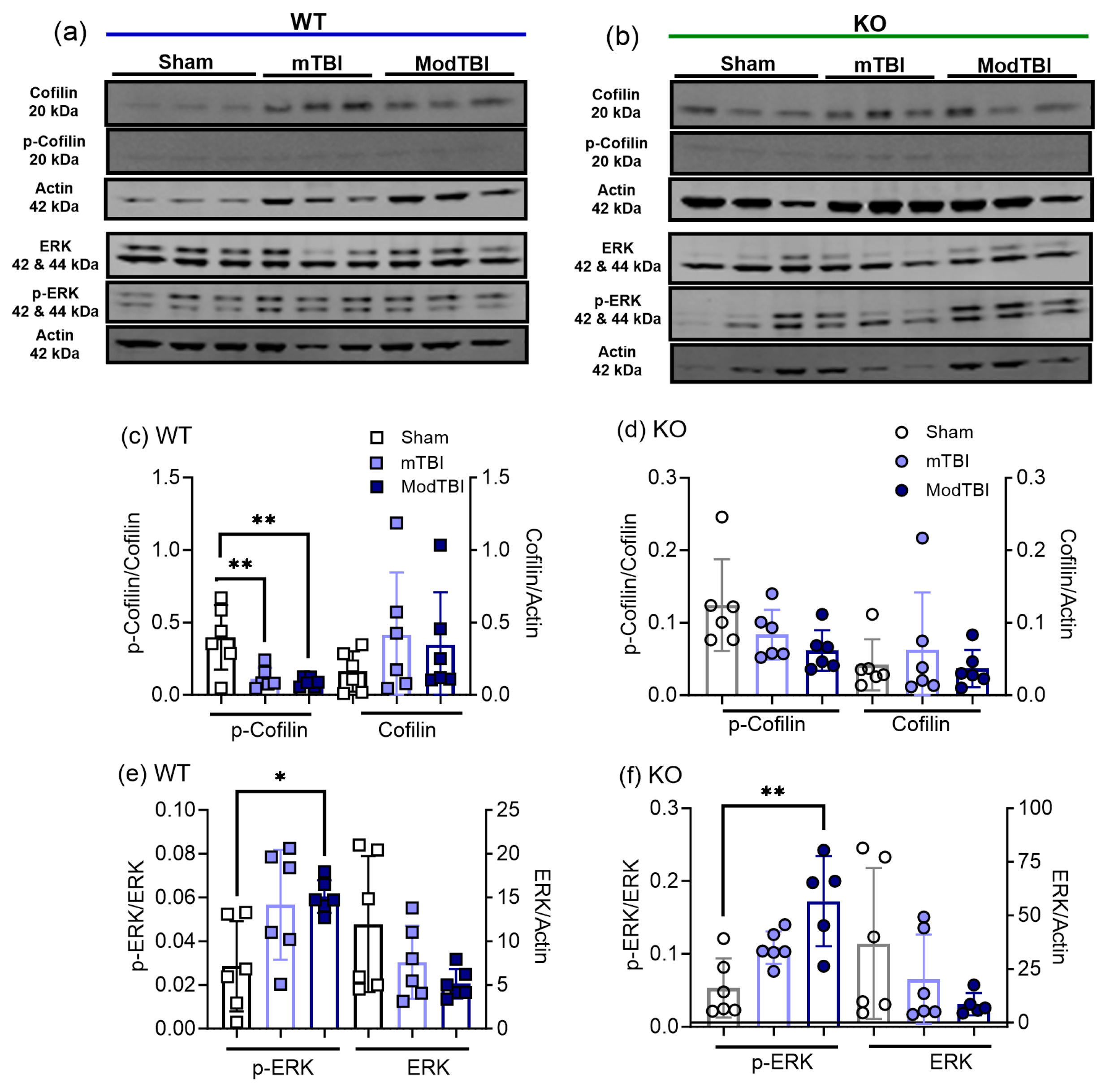
2.4. Cofilin-1 and Extracellular Regulated Kinase (ERK) Activation Increased following TBI Compared to Sham
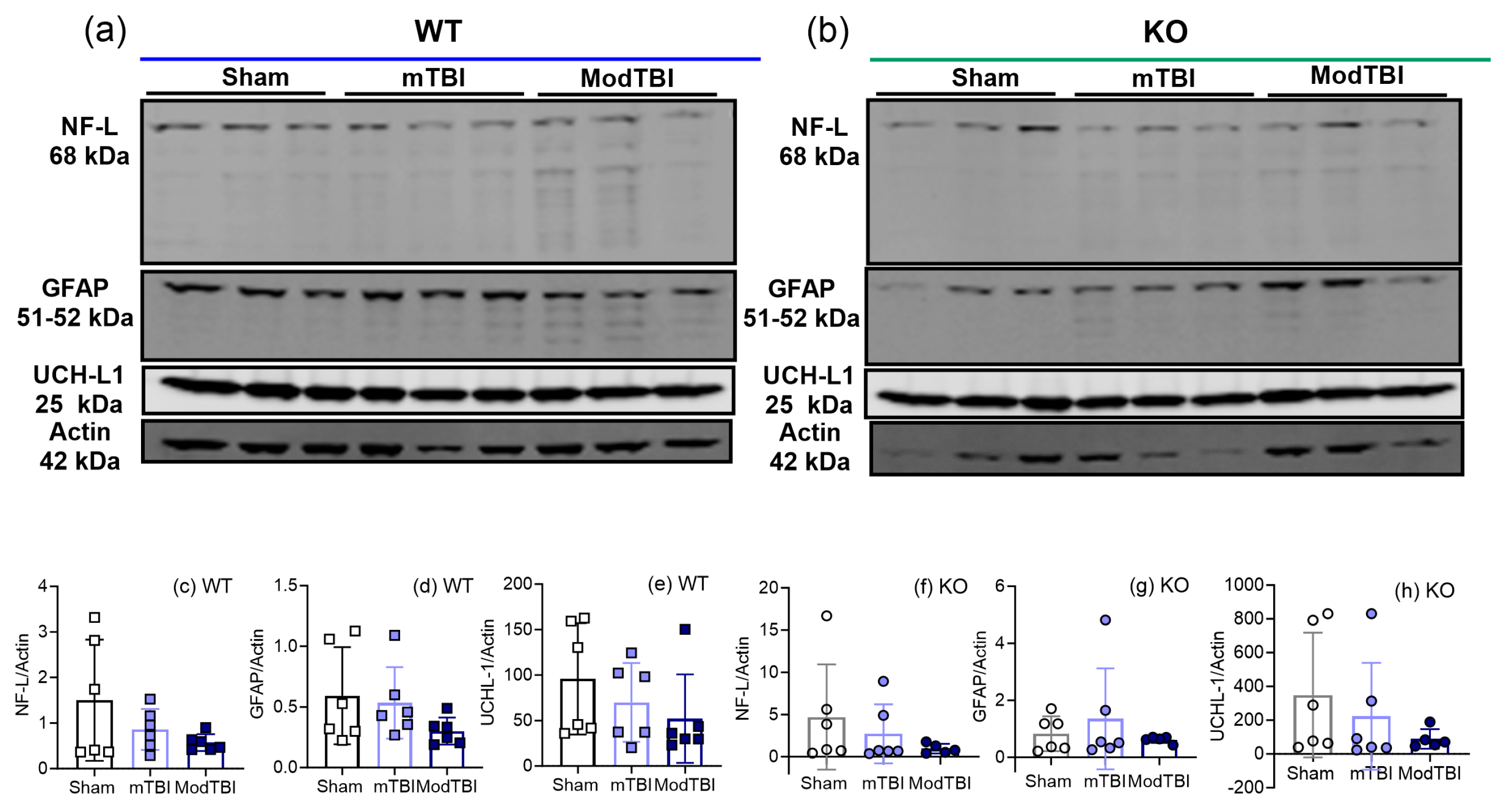
2.5. Injury Markers NF-L, GFAP, and UCH-L1 Remained Unchanged in Ipsilateral Brain Tissue 24 h post-TBI
2.6. Pearson Correlation Analysis Supports the Relationship between Changes in N/OFQ/NOP and Other Outcomes following TBI
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Induction of TBI
4.3. mNSS
4.4. Processing and Collection of Biofluid and Brain Tissue Samples
4.5. Radioimmunoassay (RIA)
4.6. Immunoblotting
4.7. Real-Time PCR
4.8. Data Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Pervez, M.; Kitagawa, R.S.; Chang, T.R. Definition of Traumatic Brain Injury, Neurosurgery, Trauma Orthopedics, Neuroimaging, Psychology, and Psychiatry in Mild Traumatic Brain Injury. Neuroimaging Clin. N. Am. 2018, 28, 1–13. [Google Scholar] [CrossRef]
- Blennow, K.; DBrody, L.; Kochanek, P.M.; Levin, H.; McKee, A.; Ribbers, G.M.; Yaffe, K.; Zetterberg, H. Traumatic brain injuries. Nat. Rev. Dis. Primers 2016, 2, 16084. [Google Scholar] [CrossRef]
- Dixon, K.J. Pathophysiology of Traumatic Brain Injury. Phys. Med. Rehabil. Clin. N. Am. 2017, 28, 215–225. [Google Scholar] [CrossRef]
- Eapen, B.C.; Cifu, D.X. Rehabilitation after Traumatic Brain Injury; Elsevier Health Sciences: Amsterdam, The Netherlands, 2018. [Google Scholar]
- Ponsford, J.; Sloan, S.; Snow, P. Traumatic Brain Injury: Rehabilitation for Everyday Adaptive Living; Psychology Press: London, UK, 2012. [Google Scholar]
- Capizzi, A.; Woo, J.; Verduzco-Gutierrez, M. Traumatic Brain Injury: An Overview of Epidemiology, Pathophysiology, and Medical Management. Med. Clin. N. Am. 2020, 104, 213–238. [Google Scholar] [CrossRef]
- Picetti, E.; Rossi, S.; Abu-Zidan, F.M.; Ansaloni, L.; Armonda, R.; Baiocchi, G.L.; Bala, M.; Balogh, Z.J.; Berardino, M.; Biffl, W.L.; et al. WSES consensus conference guidelines: Monitoring and management of severe adult traumatic brain injury patients with polytrauma in the first 24 hours. World J. Emerg. Surg. 2019, 14, 53. [Google Scholar] [CrossRef] [PubMed]
- Bodien, Y.G.; Barra, A.; Temkin, N.R.; Barber, J.; Foreman, B.; Vassar, M.; Robertson, C.; Taylor, S.R.; Markowitz, A.J.; Manley, G.T.; et al. Diagnosing Level of Consciousness: The Limits of the Glasgow Coma Scale Total Score. J. Neurotrauma 2021, 38, 3295–3305. [Google Scholar] [CrossRef] [PubMed]
- Wiles, M.D. Management of traumatic brain injury: A narrative review of current evidence. Anaesthesia 2022, 77 (Suppl. S1), 102–112. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.K.W.; Sun, J. FDA Authorizes Marketing of First Blood Test to Aid in the Evaluation of Concussion in Adults; FDA: Silver Spring, MD, USA, 2018.
- Castaño-Leon, A.M.; Carabias, C.S.; Hilario, A.; Ramos, A.; Navarro-Main, B.; Paredes, I.; Munarriz, P.M.; Panero, I.; Fernández, C.E.; García-Pérez, D.; et al. Serum assessment of traumatic axonal injury: The correlation of GFAP, t-Tau, UCH-L1, and NfL levels with diffusion tensor imaging metrics and its prognosis utility. J. Neurosurg. 2023, 138, 454–464. [Google Scholar] [CrossRef] [PubMed]
- Iverson, G.L.; Minkkinen, M.; Karr, J.E.; Berghem, K.; Zetterberg, H.; Blennow, K.; Posti, J.P.; Luoto, T.M. Examining four blood biomarkers for the detection of acute intracranial abnormalities following mild traumatic brain injury in older adults. Front. Neurol. 2022, 13, 960741. [Google Scholar] [CrossRef] [PubMed]
- Kochanek, P.M.; Dixon, C.E.; Mondello, S.; Wang, K.K.K.; Lafrenaye, A.; Bramlett, H.M.; Dietrich, W.D.; Hayes, R.L.; Shear, D.A.; Gilsdorf, J.S.; et al. Multi-Center Pre-clinical Consortia to Enhance Translation of Therapies and Biomarkers for Traumatic Brain Injury: Operation Brain Trauma Therapy and Beyond. Front. Neurol. 2018, 9, 640. [Google Scholar] [CrossRef]
- Liliang, P.C.; Liang, C.L.; Weng, H.C.; Lu, K.; Wang, K.W.; Chen, H.J.; Chuang, J.H. Tau proteins in serum predict outcome after severe traumatic brain injury. J. Surg. Res. 2010, 160, 302–307. [Google Scholar] [CrossRef] [PubMed]
- Pandey, S.; Singh, K.; Sharma, V.; Pandey, D.; Jha, R.P.; Rai, S.K.; Chauhan, R.S.; Singh, R. A prospective pilot study on serum cleaved tau protein as a neurological marker in severe traumatic brain injury. Br. J. Neurosurg. 2017, 31, 356–363. [Google Scholar] [CrossRef] [PubMed]
- Campbell, J.N.; Low, B.; Kurz, J.E.; Patel, S.S.; Young, M.T.; Churn, S.B. Mechanisms of dendritic spine remodeling in a rat model of traumatic brain injury. J. Neurotrauma 2012, 29, 218–234. [Google Scholar] [CrossRef] [PubMed]
- Bahader, G.A.; James, A.W.; Almarghalani, D.A.; Shah, Z.A. Cofilin Inhibitor Protects against Traumatic Brain Injury-Induced Oxidative Stress and Neuroinflammation. Biology 2023, 12, 630. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Aravind, A.; Pfister, B.J.; Chandra, N.; Haorah, J. Animal Models of Traumatic Brain Injury and Assessment of Injury Severity. Mol. Neurobiol. 2019, 56, 5332–5345. [Google Scholar] [CrossRef] [PubMed]
- Najem, D.; Rennie, K.; Ribecco-Lutkiewicz, M.; Ly, D.; Haukenfrers, J.; Liu, Q.; Nzau, M.; Fraser, D.D.; Bani-Yaghoub, M. Traumatic brain injury: Classification, models, and markers. Biochem. Cell Biol. 2018, 96, 391–406. [Google Scholar] [CrossRef] [PubMed]
- Dixon, C.E.; Clifton, G.L.; Lighthall, J.W.; Yaghmai, A.A.; Hayes, R.L. A controlled cortical impact model of traumatic brain injury in the rat. J. Neurosci. Methods 1991, 39, 253–262. [Google Scholar] [CrossRef]
- Bunzow, J.R.; Saez, C.; Mortrud, M.; Bouvier, C.; Williams, J.T.; Low, M.; Grandy, D.K. Molecular cloning and tissue distribution of a putative member of the rat opioid receptor gene family that is not a mu, delta or kappa opioid receptor type. FEBS Lett. 1994, 347, 284–288. [Google Scholar] [CrossRef]
- Chen, Y.; Fan, Y.; Liu, J.; Mestek, A.; Tian, M.; Kozak, C.A.; Yu, L. Molecular cloning, tissue distribution and chromosomal localization of a novel member of the opioid receptor gene family. FEBS Lett. 1994, 347, 279–283. [Google Scholar] [CrossRef]
- Fukuda, K.; Kato, S.; Mori, K.; Nishi, M.; Takeshima, H.; Iwabe, N.; Miyata, T.; Houtani, T.; Sugimoto, T. cDNA cloning and regional distribution of a novel member of the opioid receptor family. FEBS Lett. 1994, 343, 42–46. [Google Scholar] [CrossRef] [PubMed]
- Mollereau, C.; Parmentier, M.; Mailleux, P.; Butour, J.L.; Moisand, C.; Chalon, P.; Caput, D.; Vassart, G.; Meunier, J.C. ORL1, a novel member of the opioid receptor family. Cloning, functional expression and localization. FEBS Lett. 1994, 341, 33–38. [Google Scholar] [CrossRef]
- Pan, Y.X.; Cheng, J.; Xu, J.; Rossi, G.; Jacobson, E.; Ryan-Moro, J.; Brooks, A.I.; Dean, G.E.; Standifer, K.M.; Pasternak, G.W. Cloning and functional characterization through antisense mapping of a kappa 3-related opioid receptor. Mol. Pharmacol. 1995, 47, 1180–1188. [Google Scholar]
- Wang, J.B.; PJohnson, S.; Imai, Y.; Persico, A.M.; Ozenberger, B.A.; Eppler, C.M.; Uhl, G.R. cDNA cloning of an orphan opiate receptor gene family member and its splice variant. FEBS Lett. 1994, 348, 75–79. [Google Scholar] [CrossRef]
- Wick, M.J.; Minnerath, S.R.; Lin, X.; Elde, R.; Law, P.Y.; Loh, H.H. Isolation of a novel cDNA encoding a putative membrane receptor with high homology to the cloned mu, delta, and kappa opioid receptors. Brain Res. Mol. Brain Res. 1994, 27, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Mollereau, C.; Simons, M.J.; Soularue, P.; Liners, F.; Vassart, G.; Meunier, J.C.; Parmentier, M. Structure, tissue distribution, and chromosomal localization of the prepronociceptin gene. Proc. Natl. Acad. Sci. USA 1996, 93, 8666–8670. [Google Scholar] [CrossRef]
- Al Yacoub, O.N.; Awwad, H.O.; Zhang, Y.; Standifer, K.M. Therapeutic potential of nociceptin/orphanin FQ peptide (NOP) receptor modulators for treatment of traumatic brain injury, traumatic stress, and their co-morbidities. Pharmacol. Ther. 2022, 231, 107982. [Google Scholar] [CrossRef] [PubMed]
- Armstead, W.M. Role of altered cyclooxygenase metabolism in impaired cerebrovasodilation to nociceptin/orphanin FQ following brain injury. Brain Res. Bull. 2000, 53, 807–812. [Google Scholar] [CrossRef] [PubMed]
- Armstead, W.M. NOC/oFQ contributes to age-dependent impairment of NMDA-induced cerebrovasodilation after brain injury. Am. J. Physiol. Heart Circ. Physiol. 2000, 279, H2188–H2195. [Google Scholar] [CrossRef]
- Armstead, W.M. Role of NOC/oFQ in impaired opioid-induced pial artery dilation following brain injury. Brain Res. 2000, 869, 231–235. [Google Scholar] [CrossRef]
- Al Yacoub, O.N.; Tarantini, S.; Zhang, Y.; Csiszar, A.; Standifer, K.M. The Nocicieptin/Orphanin FQ peptide receptor antagonist, SB-612111, improves cerebral blood flow in a rat model of traumatic brain injury. Front. Pharmacol. 2023, 14, 1272969. [Google Scholar] [CrossRef]
- Armstead, W.M. Nociceptin/orphanin FQ dilates pial arteries by K(ATP) and K(ca) channel activation. Brain Res. 1999, 835, 315–323. [Google Scholar] [CrossRef]
- Armstead, W.M. Role of nociceptin/orphanin FQ in age-dependent cerebral hemodynamic effects of brain injury. J. Neurotrauma 2000, 17, 751–764. [Google Scholar] [CrossRef]
- Witta, J.; Buzas, B.; Cox, B.M. Traumatic brain injury induces nociceptin/orphanin FQ expression in neurons of the rat cerebral cortex. J. Neurotrauma 2003, 20, 523–532. [Google Scholar] [CrossRef] [PubMed]
- Awwad, H.O.; Durand, C.D.; Gonzalez, L.P.; Tompkins, P.; Zhang, Y.; Lerner, M.R.; Brackett, D.J.; Sherry, D.M.; Awasthi, V.; Standifer, K.M. Post-blast treatment with Nociceptin/Orphanin FQ peptide (NOP) receptor antagonist reduces brain injury-induced hypoxia and signaling proteins in vestibulomotor-related brain regions. Behav. Brain Res. 2018, 340, 183–194. [Google Scholar] [CrossRef] [PubMed]
- Al Yacoub, O.N.; Awwad, H.O.; Standifer, K.M. Recovery from traumatic brain injury (TBI) is Nociceptin/Orphanin FQ peptide (NOP) receptor genotype-, sex-, and injury severity-dependent. J. Pharmacol. Exp. Ther. 2023, 388, 1–48. [Google Scholar] [CrossRef] [PubMed]
- Armstead, W.M. PTK, ERK and p38 MAPK contribute to impaired NMDA-induced vasodilation after brain injury. Eur. J. Pharmacol. 2003, 474, 249–254. [Google Scholar] [CrossRef] [PubMed]
- Armstead, W.M. Differential activation of ERK, p38, and JNK MAPK by nociceptin/orphanin FQ in the potentiation of prostaglandin cerebrovasoconstriction after brain injury. Eur. J. Pharmacol. 2006, 529, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Meberg, P.J.; Ono, S.; Minamide, L.S.; Takahashi, M.; Bamburg, J.R. Actin depolymerizing factor and cofilin phosphorylation dynamics: Response to signals that regulate neurite extension. Cell Motil. Cytoskeleton. 1998, 39, 172–190. [Google Scholar] [CrossRef]
- Wang, Y.; Shibasaki, F.; Mizuno, K. Calcium signal-induced cofilin dephosphorylation is mediated by Slingshot via calcineurin. J. Biol. Chem. 2005, 280, 12683–12689. [Google Scholar] [CrossRef]
- Xiong, Y.; Mahmood, A.; Chopp, M. Animal models of traumatic brain injury. Nat. Rev. Neurosci. 2013, 14, 128–142. [Google Scholar] [CrossRef]
- Rosenberger, J.; Petrovics, G.; Buzas, B. Oxidative stress induces proorphanin FQ and proenkephalin gene expression in astrocytes through p38- and ERK-MAP kinases and NF-kappaB. J. Neurochem. 2001, 79, 35–44. [Google Scholar] [CrossRef]
- Caputi, F.F.; Romualdi, P.; Candeletti, S. Regulation of the Genes Encoding the ppN/OFQ and NOP Receptor. Handb. Exp. Pharmacol. 2019, 254, 141–162. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Simon, F.M.; Rodriguez, R.E. Expression of the nociceptin receptor during zebrafish development: Influence of morphine and nociception. Int. J. Devl. Neurosci. 2016, 27, 315–320. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.C.; Akinyi, L.; Scharf, D.; Mo, J.; Larner, S.F.; Muller, U.; Oli, M.W.; Zheng, W.; Kobeissy, F.; Papa, L.; et al. Ubiquitin C-terminal hydrolase-L1 as a biomarker for ischemic and traumatic brain injury in rats. Eur. J. Neurosci. 2010, 31, 722–732. [Google Scholar] [CrossRef] [PubMed]
- Osier, N.D.; Bramlett, H.M.; Shear, D.A.; Mondello, S.; Carlson, S.W.; Dietrich, W.D.; Deng-Bryant, Y.; Wang, K.K.W.; Hayes, R.L.; Yang, Z.; et al. Kollidon VA64 Treatment in Traumatic Brain Injury: Operation Brain Trauma Therapy. J. Neurotrauma 2021, 38, 2454–2472. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, M.; Yang, Z.; Chawla, S.; Delso, N.; Pukenas, B.; Elmér, E.; Hugerth, M.; Margulies, S.S.; Ehinger, J.; Hansson, M.J.; et al. Evaluation of Diffusion Tensor Imaging and Fluid Based Biomarkers in a Large Animal Trial of Cyclosporine in Focal Traumatic Brain Injury. J. Neurotrauma 2021, 38, 1870–1878. [Google Scholar] [CrossRef] [PubMed]
- Korley, F.K.; Nikolian, V.C.; Williams, A.M.; Dennahy, I.S.; Weykamp, M.; Alam, H.B. Valproic Acid Treatment Decreases Serum Glial Fibrillary Acidic Protein and Neurofilament Light Chain Levels in Swine Subjected to Traumatic Brain Injury. J. Neurotrauma 2018, 35, 1185–1191. [Google Scholar] [CrossRef] [PubMed]
- Niu, F.; Qian, K.; Qi, H.; Zhao, Y.; Jiang, Y.; Sun, M. Antiapoptotic and Anti-Inflammatory Effects of CPCGI in Rats with Traumatic Brain Injury. Neuropsychiatr. Dis. Treat. 2020, 16, 2975–2987. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.L.; Yu, M.M.; Yang, D.X.; Liu, Y.L.; Wei, X.E.; Li, W.B. Longitudinal Microstructural Changes in Traumatic Brain Injury in Rats: A Diffusional Kurtosis Imaging, Histology, and Behavior Study. AJNR Am. J. Neuroradiol. 2018, 39, 1650–1656. [Google Scholar] [CrossRef]
- Homberg, J.R.; Mul, J.D.; de Wit, E.; Cuppen, E. Complete knockout of the nociceptin/orphanin FQ receptor in the rat does not induce compensatory changes in mu, delta and kappa opioid receptors. Neuroscience 2009, 163, 308–315. [Google Scholar] [CrossRef]
- Rizzi, A.; Molinari, S.; Marti, M.; Marzola, G.; Calo, G. Nociceptin/orphanin FQ receptor knockout rats: In vitro and in vivo studies. Neuropharmacology 2011, 60, 572–579. [Google Scholar] [CrossRef] [PubMed]
- Percie du Sert, N.; Hurst, V.; Ahluwalia, A.; Alam, S.; Avey, M.T.; Baker, M.; Browne, W.J.; Clark, A.; Cuthill, I.C.; Dirnagl, U.; et al. The ARRIVE guidelines 2.0: Updated guidelines for reporting animal research. Br. J. Pharmacol. 2020, 177, 3617–3624. [Google Scholar] [CrossRef] [PubMed]
- Osier, N.D.; Dixon, C.E. The Controlled Cortical Impact Model: Applications, Considerations for Researchers, and Future Directions. Front. Neurol. 2016, 7, 134. [Google Scholar] [CrossRef] [PubMed]
- Brody, D.L.; Donald, C.M.; Kessens, C.C.; Yuede, C.; Parsadanian, M.; Spinner, M.; Kim, E.; Schwetye, K.E.; Holtzman, D.M.; Bayly, P.V. Electromagnetic controlled cortical impact device for precise, graded experimental traumatic brain injury. J. Neurotrauma 2007, 24, 657–673. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Sanberg, P.R.; Li, Y.; Wang, L.; Lu, M.; Willing, A.E.; Sanchez-Ramos, J.; Chopp, M. Intravenous administration of human umbilical cord blood reduces behavioral deficits after stroke in rats. Stroke 2001, 32, 2682–2688. [Google Scholar] [CrossRef]
- Shear, D.A.; Dixon, C.E.; Bramlett, H.M.; Mondello, S.; Dietrich, W.D.; Deng-Bryant, Y.; Schmid, K.E.; Wang, K.K.; Hayes, R.L.; Povlishock, J.T.; et al. Nicotinamide Treatment in Traumatic Brain Injury: Operation Brain Trauma Therapy. J. Neurotrauma 2016, 33, 523–537. [Google Scholar] [CrossRef]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative C(T) method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al Yacoub, O.N.; Zhang, Y.; Patankar, P.S.; Standifer, K.M. Traumatic Brain Injury Induces Nociceptin/Orphanin FQ and Nociceptin Opioid Peptide Receptor Expression within 24 Hours. Int. J. Mol. Sci. 2024, 25, 1658. https://doi.org/10.3390/ijms25031658
Al Yacoub ON, Zhang Y, Patankar PS, Standifer KM. Traumatic Brain Injury Induces Nociceptin/Orphanin FQ and Nociceptin Opioid Peptide Receptor Expression within 24 Hours. International Journal of Molecular Sciences. 2024; 25(3):1658. https://doi.org/10.3390/ijms25031658
Chicago/Turabian StyleAl Yacoub, Omar N., Yong Zhang, Panini S. Patankar, and Kelly M. Standifer. 2024. "Traumatic Brain Injury Induces Nociceptin/Orphanin FQ and Nociceptin Opioid Peptide Receptor Expression within 24 Hours" International Journal of Molecular Sciences 25, no. 3: 1658. https://doi.org/10.3390/ijms25031658
APA StyleAl Yacoub, O. N., Zhang, Y., Patankar, P. S., & Standifer, K. M. (2024). Traumatic Brain Injury Induces Nociceptin/Orphanin FQ and Nociceptin Opioid Peptide Receptor Expression within 24 Hours. International Journal of Molecular Sciences, 25(3), 1658. https://doi.org/10.3390/ijms25031658