
Linking Gene Fusions to Bone Marrow Failure and Malignant Transformation in Dyskeratosis Congenita


Abstract
1. Introduction
2. Results
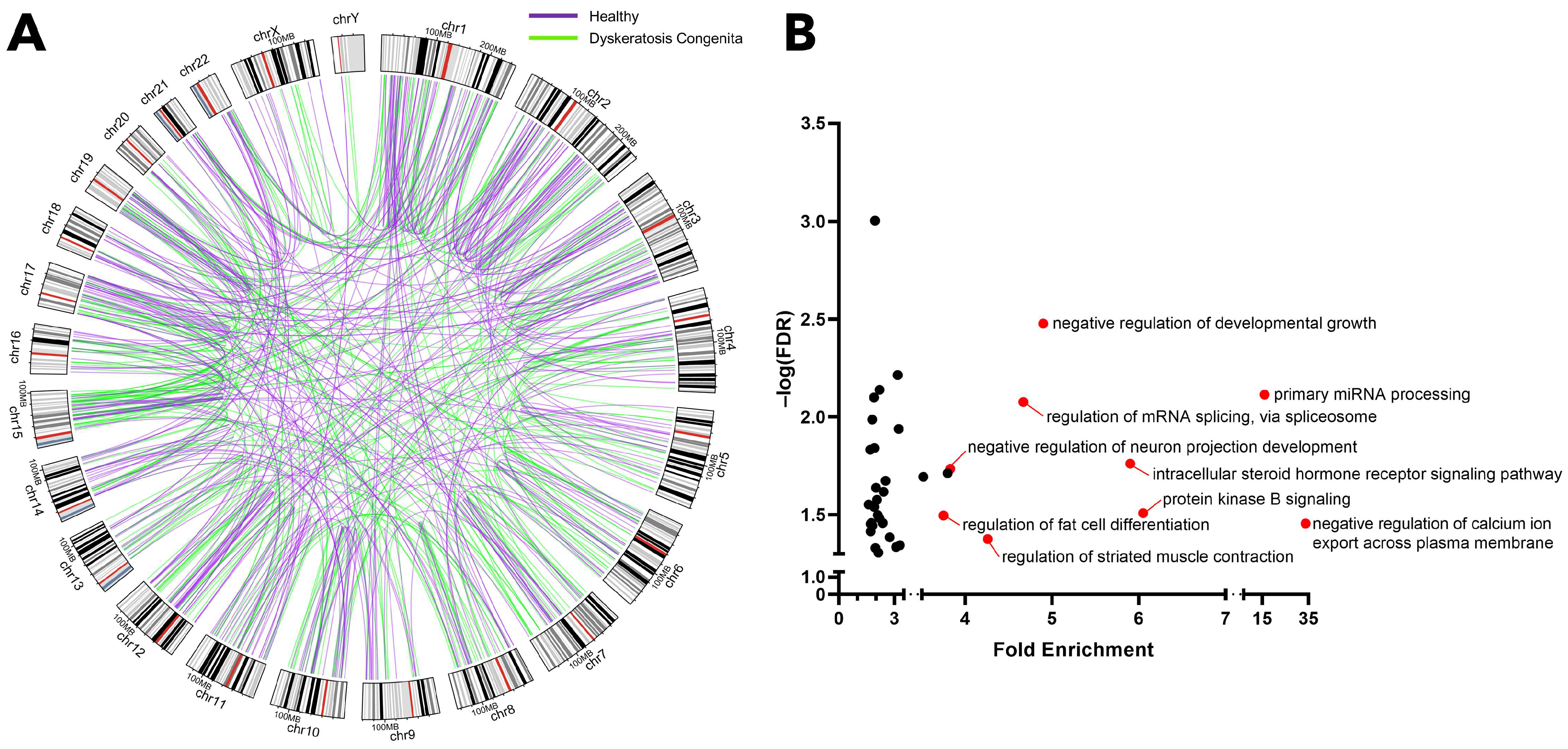
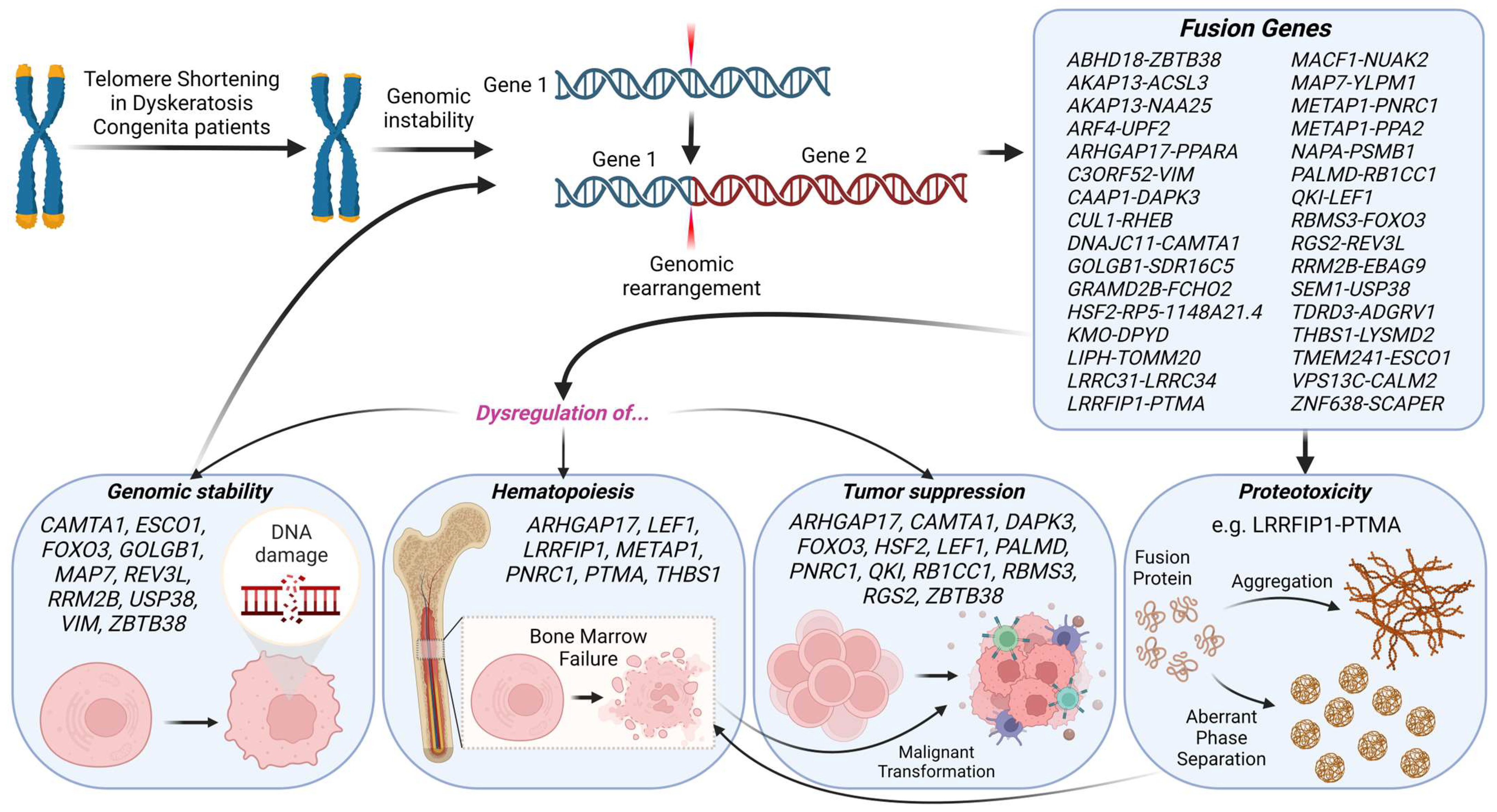
2.1. Identification of Gene Fusions Associated with Dyskeratosis Congenita
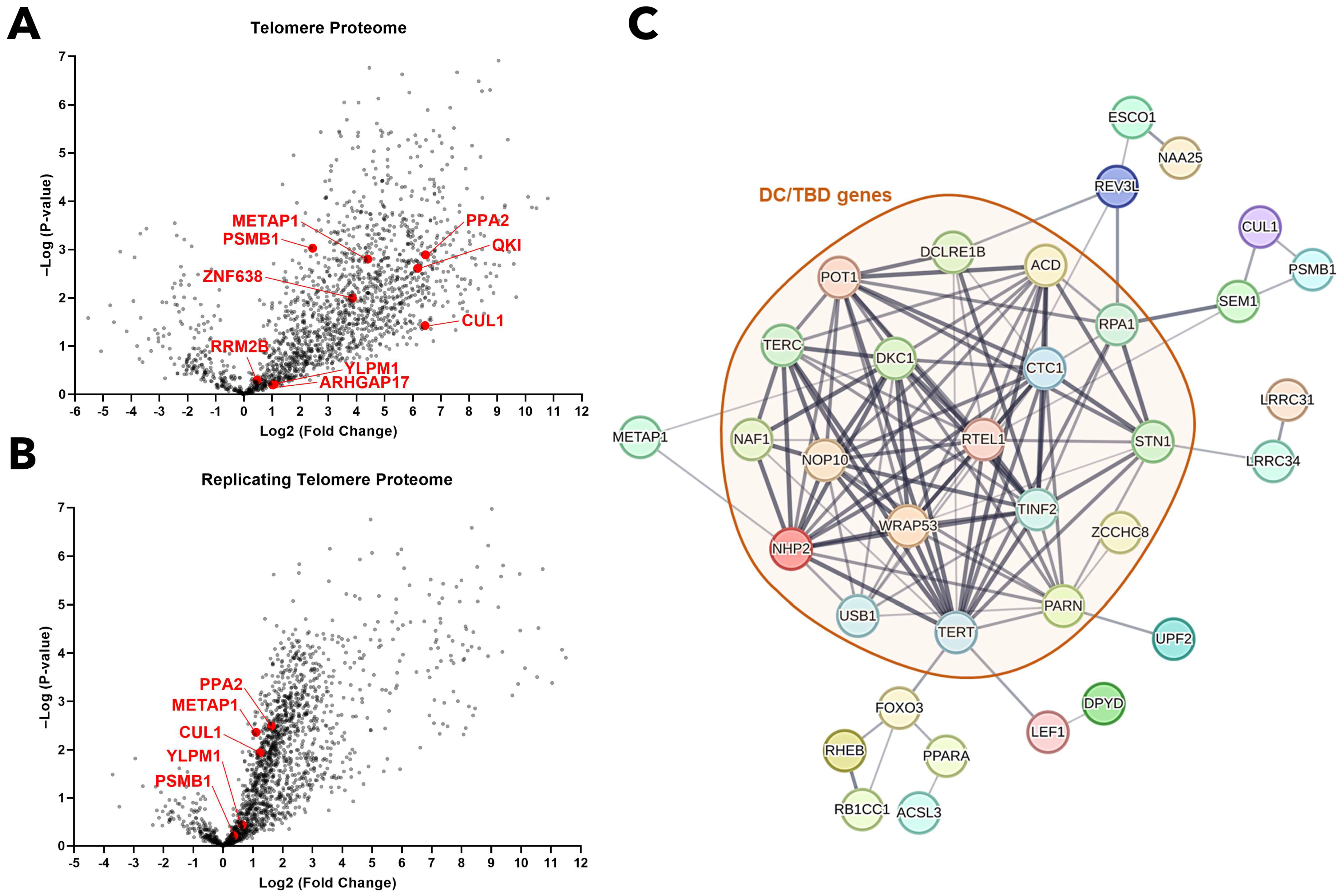
2.2. Genes Engaged in Fusion Events Predominantly Govern Genomic Integrity, Hematopoiesis, and Tumor Suppression
2.3. Chimeric Protein Proteotoxicity as a Potential Driver of BMF in DC
3. Materials and Methods
3.1. Dyskeratosis Congenita RNA-Seq Data
3.2. Gene Fusion Analysis
3.3. Gene Ontology Analysis
3.4. Overrepresentation and Network Analyses
3.5. Protein–Protein Association Network Analysis
3.6. Disorder, Polyampholyte, and Self-Association Propensity Analysis
3.7. Prediction of Fusion Protein Tertiary Structure
3.8. Molecular Dynamics Simulations and Analysis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fernandez Garcia, M.S.; Teruya-Feldstein, J. The diagnosis and treatment of dyskeratosis congenita: A review. J. Blood Med. 2014, 5, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Kam, M.L.W.; Nguyen, T.T.T.; Ngeow, J.Y.Y. Telomere biology disorders. NPJ Genom. Med. 2021, 6, 36. [Google Scholar] [CrossRef] [PubMed]
- Robledo Aguilar, A.; Gomez, F.; Tojo Sierra, R.; Larripa, P. Dyskeratosis congenita Zinsser-Cole-Engmann form with abnormal karyotype. Dermatologica 1974, 148, 98–103. [Google Scholar] [CrossRef] [PubMed]
- Dokal, I.; Bungey, J.; Williamson, P.; Oscier, D.; Hows, J.; Luzzatto, L. Dyskeratosis congenita fibroblasts are abnormal and have unbalanced chromosomal rearrangements. Blood 1992, 80, 3090–3096. [Google Scholar] [CrossRef] [PubMed]
- Kehrer, H.; Krone, W. Chromosome abnormalities in cell cultures derived from the leukoplakia of a female patient with dyskeratosis congenita. Am. J. Med. Genet. 1992, 42, 217–218. [Google Scholar] [CrossRef] [PubMed]
- Scappaticci, S.; Fraccaro, M.; Cerimele, D. Chromosome abnormalities in dyskeratosis congenita. Am. J. Med. Genet. 1989, 34, 609–610. [Google Scholar] [CrossRef] [PubMed]
- Gao, Q.; Liang, W.W.; Foltz, S.M.; Mutharasu, G.; Jayasinghe, R.G.; Cao, S.; Liao, W.W.; Reynolds, S.M.; Wyczalkowski, M.A.; Yao, L.; et al. Driver Fusions and Their Implications in the Development and Treatment of Human Cancers. Cell Rep. 2018, 23, 227–238.e3. [Google Scholar] [CrossRef]
- Uhrig, S.; Ellermann, J.; Walther, T.; Burkhardt, P.; Frohlich, M.; Hutter, B.; Toprak, U.H.; Neumann, O.; Stenzinger, A.; Scholl, C.; et al. Accurate and efficient detection of gene fusions from RNA sequencing data. Genome Res. 2021, 31, 448–460. [Google Scholar] [CrossRef]
- Emily, N.; Brian, R.; Valerie, K. The oncogenic fusion protein TAZ-CAMTA1 promotes genomic instability and senescence through hypertranscription. bioRxiv 2022. [Google Scholar] [CrossRef]
- Price, J.C.; Pollock, L.M.; Rudd, M.L.; Fogoros, S.K.; Mohamed, H.; Hanigan, C.L.; Le Gallo, M.; NIH Intramural Sequencing Center (NISC) Comparative Sequencing Program; Zhang, S.; Cruz, P.; et al. Sequencing of candidate chromosome instability genes in endometrial cancers reveals somatic mutations in ESCO1, CHTF18, and MRE11A. PLoS ONE 2014, 8, e63313. [Google Scholar] [CrossRef]
- Kawasumi, R.; Abe, T.; Arakawa, H.; Garre, M.; Hirota, K.; Branzei, D. ESCO1/2’s roles in chromosome structure and interphase chromatin organization. Genes. Dev. 2017, 31, 2136–2150. [Google Scholar] [CrossRef]
- Chung, Y.M.; Park, S.H.; Tsai, W.B.; Wang, S.Y.; Ikeda, M.A.; Berek, J.S.; Chen, D.J.; Hu, M.C. FOXO3 signalling links ATM to the p53 apoptotic pathway following DNA damage. Nat. Commun. 2012, 3, 1000. [Google Scholar] [CrossRef]
- Bigarella, C.L.; Li, J.; Rimmele, P.; Liang, R.; Sobol, R.W.; Ghaffari, S. FOXO3 Transcription Factor Is Essential for Protecting Hematopoietic Stem and Progenitor Cells from Oxidative DNA Damage. J. Biol. Chem. 2017, 292, 3005–3015. [Google Scholar] [CrossRef]
- White, R.R.; Maslov, A.Y.; Lee, M.; Wilner, S.E.; Levy, M.; Vijg, J. FOXO3a acts to suppress DNA double-strand break-induced mutations. Aging Cell 2020, 19, e13184. [Google Scholar] [CrossRef]
- George, G.; Karolina, K.; Muzamil, M.K.; Peter, T.; Beate, N.; Emma, L.; Rainer, P. The Golgi complex is a regulatory hub for homologous recombination-mediated DNA repair. bioRxiv 2022. [Google Scholar] [CrossRef]
- Dullovi, A.; Ozgencil, M.; Rajvee, V.; Tse, W.Y.; Cutillas, P.R.; Martin, S.A.; Horejsi, Z. Microtubule-associated proteins MAP7 and MAP7D1 promote DNA double-strand break repair in the G1 cell cycle phase. iScience 2023, 26, 106107. [Google Scholar] [CrossRef] [PubMed]
- Ben Yamin, B.; Ahmed-Seghir, S.; Tomida, J.; Despras, E.; Pouvelle, C.; Yurchenko, A.; Goulas, J.; Corre, R.; Delacour, Q.; Droin, N.; et al. DNA polymerase zeta contributes to heterochromatin replication to prevent genome instability. EMBO J. 2021, 40, e104543. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.; Guo, R.; Huang, Q.; Yen, Y. Chromosomal instability triggered by Rrm2b loss leads to IL-6 secretion and plasmacytic neoplasms. Cell Rep. 2013, 3, 1389–1397. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Yang, C.; Li, T.; Yu, S.; Gan, T.; Hu, J.; Cui, J.; Zheng, X. The Deubiquitinase USP38 Promotes NHEJ Repair through Regulation of HDAC1 Activity and Regulates Cancer Cell Response to Genotoxic Insults. Cancer Res. 2020, 80, 719–731. [Google Scholar] [CrossRef] [PubMed]
- Patteson, A.E.; Vahabikashi, A.; Pogoda, K.; Adam, S.A.; Mandal, K.; Kittisopikul, M.; Sivagurunathan, S.; Goldman, A.; Goldman, R.D.; Janmey, P.A. Vimentin protects cells against nuclear rupture and DNA damage during migration. J. Cell Biol. 2019, 218, 4079–4092. [Google Scholar] [CrossRef] [PubMed]
- Miotto, B.; Chibi, M.; Xie, P.; Koundrioukoff, S.; Moolman-Smook, H.; Pugh, D.; Debatisse, M.; He, F.; Zhang, L.; Defossez, P.-A. The RBBP6/ZBTB38/MCM10 Axis Regulates DNA Replication and Common Fragile Site Stability. Cell Rep. 2014, 7, 575–587. [Google Scholar] [CrossRef]
- Nagy, Z.; Wynne, K.; von Kriegsheim, A.; Gambaryan, S.; Smolenski, A. Cyclic Nucleotide-dependent Protein Kinases Target ARHGAP17 and ARHGEF6 Complexes in Platelets. J. Biol. Chem. 2015, 290, 29974–29983. [Google Scholar] [CrossRef]
- Gutierrez, A.; Sanda, T.; Ma, W.; Zhang, J.; Grebliunaite, R.; Dahlberg, S.; Neuberg, D.; Protopopov, A.; Winter, S.S.; Larson, R.S.; et al. Inactivation of LEF1 in T-cell acute lymphoblastic leukemia. Blood 2010, 115, 2845–2851. [Google Scholar] [CrossRef]
- Yin, X.; Liu, P.; Liu, Y.Y.; Liu, M.Y.; Fan, W.L.; Liu, B.Y.; Zhao, J.H. LRRFIP1 expression triggers platelet agglutination by enhancing alphaIIbbeta3 expression. Exp. Ther. Med. 2019, 18, 269–277. [Google Scholar] [CrossRef]
- Goodall, A.H.; Burns, P.; Salles, I.; Macaulay, I.C.; Jones, C.I.; Ardissino, D.; de Bono, B.; Bray, S.L.; Deckmyn, H.; Dudbridge, F.; et al. Transcription profiling in human platelets reveals LRRFIP1 as a novel protein regulating platelet function. Blood 2010, 116, 4646–4656. [Google Scholar] [CrossRef]
- Frobel, J.; Cadeddu, R.P.; Hartwig, S.; Bruns, I.; Wilk, C.M.; Kundgen, A.; Fischer, J.C.; Schroeder, T.; Steidl, U.G.; Germing, U.; et al. Platelet proteome analysis reveals integrin-dependent aggregation defects in patients with myelodysplastic syndromes. Mol. Cell Proteom. 2013, 12, 1272–1280. [Google Scholar] [CrossRef]
- Chen, X.; Wang, F.; Zhang, Y.; Ma, X.; Cao, P.; Yuan, L.; Wang, L.; Chen, J.; Zhou, X.; Wu, Q.; et al. Fusion gene map of acute leukemia revealed by transcriptome sequencing of a consecutive cohort of 1000 cases in a single center. Blood Cancer J. 2021, 11, 112. [Google Scholar] [CrossRef]
- Freire, M.; Barbeito, P.; Sarandeses, C.S.; Díaz-Jullien, C.; Muras, J.; Covelo, G.; Moreira, D.; Freire-Cobo, C. Prothymosin α, a protein implicated in the proliferation and survival of lymphocytes. J. Immunol. Sci. 2018, 2, 19–25. [Google Scholar] [CrossRef]
- Aburima, A.; Berger, M.; Spurgeon, B.E.J.; Webb, B.A.; Wraith, K.S.; Febbraio, M.; Poole, A.W.; Naseem, K.M. Thrombospondin-1 promotes hemostasis through modulation of cAMP signaling in blood platelets. Blood 2021, 137, 678–689. [Google Scholar] [CrossRef] [PubMed]
- Mertens, F.; Johansson, B.; Fioretos, T.; Mitelman, F. The emerging complexity of gene fusions in cancer. Nat. Rev. Cancer 2015, 15, 371–381. [Google Scholar] [CrossRef] [PubMed]
- Pan, S.; Deng, Y.; Fu, J.; Zhang, Y.; Zhang, Z.; Ru, X.; Qin, X. Tumor Suppressive Role of ARHGAP17 in Colon Cancer Through Wnt/beta-Catenin Signaling. Cell Physiol. Biochem. 2018, 46, 2138–2148. [Google Scholar] [CrossRef]
- He, Z.; Yang, C.; He, Y.; Gong, B.; Yin, C.; Feng, J.; Chen, L.; Tang, J.; Chen, Y. CAMTA1, a novel antitumor gene, regulates proliferation and the cell cycle in glioma by inhibiting AKT phosphorylation. Cell Signal 2021, 79, 109882. [Google Scholar] [CrossRef]
- Henrich, K.O.; Bauer, T.; Schulte, J.; Ehemann, V.; Deubzer, H.; Gogolin, S.; Muth, D.; Fischer, M.; Benner, A.; Konig, R.; et al. CAMTA1, a 1p36 tumor suppressor candidate, inhibits growth and activates differentiation programs in neuroblastoma cells. Cancer Res. 2011, 71, 3142–3151. [Google Scholar] [CrossRef]
- Takahashi, M.; Lio, C.J.; Campeau, A.; Steger, M.; Ay, F.; Mann, M.; Gonzalez, D.J.; Jain, M.; Sharma, S. The tumor suppressor kinase DAPK3 drives tumor-intrinsic immunity through the STING-IFN-beta pathway. Nat. Immunol. 2021, 22, 485–496. [Google Scholar] [CrossRef] [PubMed]
- Dansen, T.B.; Burgering, B.M. Unravelling the tumor-suppressive functions of FOXO proteins. Trends Cell Biol. 2008, 18, 421–429. [Google Scholar] [CrossRef]
- Bjork, J.K.; Akerfelt, M.; Joutsen, J.; Puustinen, M.C.; Cheng, F.; Sistonen, L.; Nees, M. Heat-shock factor 2 is a suppressor of prostate cancer invasion. Oncogene 2016, 35, 1770–1784. [Google Scholar] [CrossRef] [PubMed]
- Carr, T.; McGregor, S.; Dias, S.; Verykokakis, M.; Le Beau, M.M.; Xue, H.H.; Sigvardsson, M.; Bartom, E.T.; Kee, B.L. Oncogenic and Tumor Suppressor Functions for Lymphoid Enhancer Factor 1 in E2a(-/-) T Acute Lymphoblastic Leukemia. Front. Immunol. 2022, 13, 845488. [Google Scholar] [CrossRef] [PubMed]
- Cai, M.Y.; Xu, Y.L.; Rong, H.; Yang, H. Low Level of PALMD Contributes to the Metastasis of Uveal Melanoma. Front. Oncol. 2022, 12, 802941. [Google Scholar] [CrossRef]
- Gaviraghi, M.; Vivori, C.; Pareja Sanchez, Y.; Invernizzi, F.; Cattaneo, A.; Santoliquido, B.M.; Frenquelli, M.; Segalla, S.; Bachi, A.; Doglioni, C.; et al. Tumor suppressor PNRC1 blocks rRNA maturation by recruiting the decapping complex to the nucleolus. EMBO J. 2018, 37, e99179. [Google Scholar] [CrossRef]
- Bian, Y.; Wang, L.; Lu, H.; Yang, G.; Zhang, Z.; Fu, H.; Lu, X.; Wei, M.; Sun, J.; Zhao, Q.; et al. Downregulation of tumor suppressor QKI in gastric cancer and its implication in cancer prognosis. Biochem. Biophys. Res. Commun. 2012, 422, 187–193. [Google Scholar] [CrossRef]
- Chen, P.; Duan, Y.; Lu, X.; Chen, L.; Zhang, W.; Wang, H.; Hu, R.; Liu, S. RB1CC1 functions as a tumor-suppressing gene in renal cell carcinoma via suppression of PYK2 activity and disruption of TAZ-mediated PDL1 transcription activation. Cancer Immunol. Immunother. 2021, 70, 3261–3275. [Google Scholar] [CrossRef]
- Zhu, L.; Xi, P.W.; Li, X.X.; Sun, X.; Zhou, W.B.; Xia, T.S.; Shi, L.; Hu, Y.; Ding, Q.; Wei, J.F. The RNA binding protein RBMS3 inhibits the metastasis of breast cancer by regulating Twist1 expression. J. Exp. Clin. Cancer Res. 2019, 38, 105. [Google Scholar] [CrossRef] [PubMed]
- Lyu, J.H.; Park, D.W.; Huang, B.; Kang, S.H.; Lee, S.J.; Lee, C.; Bae, Y.S.; Lee, J.G.; Baek, S.H. RGS2 suppresses breast cancer cell growth via a MCPIP1-dependent pathway. J. Cell Biochem. 2015, 116, 260–267. [Google Scholar] [CrossRef] [PubMed]
- Ding, G.; Lu, W.; Zhang, Q.; Li, K.; Zhou, H.; Wang, F.; Zhao, C.; Fan, C.; Wang, J. ZBTB38 suppresses prostate cancer cell proliferation and migration via directly promoting DKK1 expression. Cell Death Dis. 2021, 12, 998. [Google Scholar] [CrossRef] [PubMed]
- Alter, B.P.; Giri, N.; Savage, S.A.; Rosenberg, P.S. Cancer in dyskeratosis congenita. Blood 2009, 113, 6549–6557. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.G.; Nager, A.C.; Lunardi, T.; Vancevska, A.; Lossaint, G.; Lingner, J. The human telomeric proteome during telomere replication. Nucleic Acids Res. 2021, 49, 12119–12135. [Google Scholar] [CrossRef] [PubMed]
- Latysheva, N.S.; Oates, M.E.; Maddox, L.; Flock, T.; Gough, J.; Buljan, M.; Weatheritt, R.J.; Babu, M.M. Molecular Principles of Gene Fusion Mediated Rewiring of Protein Interaction Networks in Cancer. Mol. Cell 2016, 63, 579–592. [Google Scholar] [CrossRef] [PubMed]
- Chua, B.A.; Signer, R.A.J. Hematopoietic stem cell regulation by the proteostasis network. Curr. Opin. Hematol. 2020, 27, 254–263. [Google Scholar] [CrossRef]
- Mathangasinghe, Y.; Fauvet, B.; Jane, S.M.; Goloubinoff, P.; Nillegoda, N.B. The Hsp70 chaperone system: Distinct roles in erythrocyte formation and maintenance. Haematologica 2021, 106, 1519–1534. [Google Scholar] [CrossRef]
- Das, R.K.; Pappu, R.V. Conformations of intrinsically disordered proteins are influenced by linear sequence distributions of oppositely charged residues. Proc. Natl. Acad. Sci. USA 2013, 110, 13392–13397. [Google Scholar] [CrossRef]
- Holehouse, A.S.; Das, R.K.; Ahad, J.N.; Richardson, M.O.; Pappu, R.V. CIDER: Resources to Analyze Sequence-Ensemble Relationships of Intrinsically Disordered Proteins. Biophys. J. 2017, 112, 16–21. [Google Scholar] [CrossRef]
- Bagashev, A.; Fitzgerald, M.C.; Larosa, D.F.; Rose, P.P.; Cherry, S.; Johnson, A.C.; Sullivan, K.E. Leucine-rich repeat (in Flightless I) interacting protein-1 regulates a rapid type I interferon response. J. Interferon Cytokine Res. 2010, 30, 843–852. [Google Scholar] [CrossRef]
- Samara, P.; Ioannou, K.; Tsitsilonis, O.E. Prothymosin Alpha and Immune Responses: Are We Close to Potential Clinical Applications? Vitam. Horm. 2016, 102, 179–207. [Google Scholar] [CrossRef]
- Akdel, M.; Pires, D.E.V.; Pardo, E.P.; Jänes, J.; Zalevsky, A.O.; Mészáros, B.; Bryant, P.; Good, L.L.; Laskowski, R.A.; Pozzati, G.; et al. A structural biology community assessment of AlphaFold2 applications. Nat. Struct. Mol. Biol. 2022, 29, 1056–1067. [Google Scholar] [CrossRef]
- Radom, F.; Plückthun, A.; Paci, E. Assessment of ab initio models of protein complexes by molecular dynamics. PLoS Comput. Biol. 2018, 14, e1006182. [Google Scholar] [CrossRef]
- Mensah, M.A.; Niskanen, H.; Magalhaes, A.P.; Basu, S.; Kircher, M.; Sczakiel, H.L.; Reiter, A.M.V.; Elsner, J.; Meinecke, P.; Biskup, S.; et al. Aberrant phase separation and nucleolar dysfunction in rare genetic diseases. Nature 2023, 614, 564–571. [Google Scholar] [CrossRef]
- Pavlov, N.A.; Cherny, D.I.; Heim, G.; Jovin, T.M.; Subramaniam, V. Amyloid fibrils from the mammalian protein prothymosin alpha. FEBS Lett. 2002, 517, 37–40. [Google Scholar] [CrossRef] [PubMed]
- Gough, S.M.; Slape, C.I.; Aplan, P.D. NUP98 gene fusions and hematopoietic malignancies: Common themes and new biologic insights. Blood 2011, 118, 6247–6257. [Google Scholar] [CrossRef] [PubMed]
- Du, H.; Guo, Y.; Ma, D.; Tang, K.; Cai, D.; Luo, Y.; Xie, C. A case report of heterozygous TINF2 gene mutation associated with pulmonary fibrosis in a patient with dyskeratosis congenita. Medicine 2018, 97, e0724. [Google Scholar] [CrossRef] [PubMed]
- Fukuhara, A.; Tanino, Y.; Ishii, T.; Inokoshi, Y.; Saito, K.; Fukuhara, N.; Sato, S.; Saito, J.; Ishida, T.; Yamaguchi, H.; et al. Pulmonary fibrosis in dyskeratosis congenita with TINF2 gene mutation. Eur. Respir. J. 2013, 42, 1757–1759. [Google Scholar] [CrossRef] [PubMed]
- Dvorak, L.A.; Vassallo, R.; Kirmani, S.; Johnson, G.; Hartman, T.E.; Tazelaar, H.D.; Leslie, K.O.; Colby, T.V.; Cockcroft, D.W.; Churg, A.M.; et al. Pulmonary fibrosis in dyskeratosis congenita: Report of 2 cases. Hum. Pathol. 2015, 46, 147–152. [Google Scholar] [CrossRef] [PubMed]
- Giri, N.; Lee, R.; Faro, A.; Huddleston, C.B.; White, F.V.; Alter, B.P.; Savage, S.A. Lung transplantation for pulmonary fibrosis in dyskeratosis congenita: Case Report and systematic literature review. BMC Blood Disord. 2011, 11, 3. [Google Scholar] [CrossRef]
- Xi, Y.; Kim, T.; Brumwell, A.N.; Driver, I.H.; Wei, Y.; Tan, V.; Jackson, J.R.; Xu, J.; Lee, D.K.; Gotts, J.E.; et al. Local lung hypoxia determines epithelial fate decisions during alveolar regeneration. Nat. Cell Biol. 2017, 19, 904–914. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Jia, L.; Tian, G.; Dong, Y.; Zhang, X.; Zhou, Z.; Luo, X.; Li, Y.; Yao, W. shinyCircos-V2.0: Leveraging the creation of Circos plot with enhanced usability and advanced features. iMeta 2023, 2, e109. [Google Scholar] [CrossRef]
- Thomas, P.D.; Ebert, D.; Muruganujan, A.; Mushayahama, T.; Albou, L.P.; Mi, H. PANTHER: Making genome-scale phylogenetics accessible to all. Protein Sci. 2022, 31, 8–22. [Google Scholar] [CrossRef]
- Liao, Y.; Wang, J.; Jaehnig, E.J.; Shi, Z.; Zhang, B. WebGestalt 2019: Gene set analysis toolkit with revamped UIs and APIs. Nucleic Acids Res. 2019, 47, W199–W205. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef]
- Grill, S.; Nandakumar, J. Molecular mechanisms of telomere biology disorders. J. Biol. Chem. 2021, 296, 100064. [Google Scholar] [CrossRef]
- Dinic, J.; Marciel, A.B.; Tirrell, M.V. Polyampholyte physics: Liquid–liquid phase separation and biological condensates. Curr. Opin. Colloid Interface Sci. 2021, 54, 101457. [Google Scholar] [CrossRef]
- Hatos, A.; Tosatto, S.C.E.; Vendruscolo, M.; Fuxreiter, M. FuzDrop on AlphaFold: Visualizing the sequence-dependent propensity of liquid-liquid phase separation and aggregation of proteins. Nucleic Acids Res. 2022, 50, W337–W344. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.H.; Forman-Kay, J.D.; Chan, H.S. Sequence-Specific Polyampholyte Phase Separation in Membraneless Organelles. Phys. Rev. Lett. 2016, 117, 178101. [Google Scholar] [CrossRef] [PubMed]
- Saar, K.L.; Morgunov, A.S.; Qi, R.; Arter, W.E.; Krainer, G.; Lee, A.A.; Knowles, T.P.J. Learning the molecular grammar of protein condensates from sequence determinants and embeddings. Proc. Natl. Acad. Sci. USA 2021, 118, e2019053118. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Zidek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Schrodinger, LLC. The PyMOL Molecular Graphics System; Version 2.5.0; 2023; (Unpublished). [Google Scholar]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- Páll, S.; Abraham, M.J.; Kutzner, C.; Hess, B.; Lindahl, E. Tackling Exascale Software Challenges in Molecular Dynamics Simulations with GROMACS. In Proceedings of the International Conference on Exascale Applications and Software, Stockholm, Sweden, 2–3 April 2014; pp. 3–27. [Google Scholar]
- Pronk, S.; Páll, S.; Schulz, R.; Larsson, P.; Bjelkmar, P.; Apostolov, R.; Shirts, M.R.; Smith, J.C.; Kasson, P.M.; van der Spoel, D.; et al. GROMACS 4.5: A high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 2013, 29, 845–854. [Google Scholar] [CrossRef]
- Hess, B.; Kutzner, C.; van der Spoel, D.; Lindahl, E. GROMACS 4: Algorithms for Highly Efficient, Load-Balanced, and Scalable Molecular Simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J.C. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef]
- Lindahl, E.; Hess, B.; van der Spoel, D. GROMACS 3.0: A package for molecular simulation and trajectory analysis. Mol. Model. Annu. 2001, 7, 306–317. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; van der Spoel, D.; van Drunen, R. GROMACS: A message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 1995, 91, 43–56. [Google Scholar] [CrossRef]
- Lindahl, E.; Abraham, M.J.; Hess, B.; van der Spoel, D. GROMACS 2021.5 Source Code; 2021.5; Zenodo: Genève, Switzerland, 2022. [Google Scholar]
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, M.; de Groot, B.L.; Grubmuller, H.; MacKerell, A.D., Jr. CHARMM36m: An improved force field for folded and intrinsically disordered proteins. Nat. Methods 2017, 14, 71–73. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Byrd, R.H.; Lu, P.; Nocedal, J. Algorithm 778: L-BFGS-B: Fortran subroutines for large-scale bound-constrained optimization. ACM Trans. Math. Softw. 1997, 23, 550–560. [Google Scholar] [CrossRef]
- Byrd, R.H.; Lu, P.; Nocedal, J.; Zhu, C. A Limited Memory Algorithm for Bound Constrained Optimization. SIAM J. Sci. Comput. 1995, 16, 1190–1208. [Google Scholar] [CrossRef]
- Bernetti, M.; Bussi, G. Pressure control using stochastic cell rescaling. J. Chem. Phys. 2020, 153, 114107. [Google Scholar] [CrossRef] [PubMed]
- Kunzmann, P.; Hamacher, K. Biotite: A unifying open source computational biology framework in Python. BMC Bioinform. 2018, 19, 346. [Google Scholar] [CrossRef]
- Kunzmann, P.; Müller, T.D.; Greil, M.; Krumbach, J.H.; Anter, J.M.; Bauer, D.; Islam, F.; Hamacher, K. Biotite: New tools for a versatile Python bioinformatics library. BMC Bioinform. 2023, 24, 236. [Google Scholar] [CrossRef]
- Virtanen, P.; Gommers, R.; Oliphant, T.E.; Haberland, M.; Reddy, T.; Cournapeau, D.; Burovski, E.; Peterson, P.; Weckesser, W.; Bright, J.; et al. SciPy 1.0: Fundamental algorithms for scientific computing in Python. Nat. Methods 2020, 17, 261–272. [Google Scholar] [CrossRef] [PubMed]
- Harris, C.R.; Millman, K.J.; van der Walt, S.J.; Gommers, R.; Virtanen, P.; Cournapeau, D.; Wieser, E.; Taylor, J.; Berg, S.; Smith, N.J.; et al. Array programming with NumPy. Nature 2020, 585, 357–362. [Google Scholar] [CrossRef]
- Hunter, J.D. Matplotlib: A 2D Graphics Environment. Comput. Sci. Eng. 2007, 9, 90–95. [Google Scholar] [CrossRef]
- Nelson, N.D.; Bertuch, A.A. Dyskeratosis congenita as a disorder of telomere maintenance. Mutat. Res. 2012, 730, 43–51. [Google Scholar] [CrossRef]
- Ikonnikova, A.Y.; Ammour, Y.I.; Snezhkina, A.V.; Krasnov, G.S.; Kudryavtseva, A.V.; Nasedkina, T.V. Identification of Fusion Transcripts in Leukemic Cells by Whole-Transcriptome Sequencing. Mol. Biol. 2018, 52, 200–205. [Google Scholar] [CrossRef]
- Liu, W.; Duan, Q.; Gong, L.; Yang, Y.; Huang, Z.; Guo, H.; Niu, X. A novel LRRFIP1-ALK fusion in inflammatory myofibroblastic tumor of hip and response to crizotinib. Investig. New Drugs 2021, 39, 278–282. [Google Scholar] [CrossRef]
- Yeh, I.; Botton, T.; Talevich, E.; Shain, A.H.; Sparatta, A.J.; de la Fouchardiere, A.; Mully, T.W.; North, J.P.; Garrido, M.C.; Gagnon, A.; et al. Activating MET kinase rearrangements in melanoma and Spitz tumours. Nat. Commun. 2015, 6, 7174. [Google Scholar] [CrossRef]
- Soler, G.; Nusbaum, S.; Varet, B.; Macintyre, E.A.; Vekemans, M.; Romana, S.P.; Radford-Weiss, I. LRRFIP1, a new FGFR1 partner gene associated with 8p11 myeloproliferative syndrome. Leukemia 2009, 23, 1359–1361. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N.; Gillespie, J.R.; Millett, I.S.; Khodyakova, A.V.; Vasiliev, A.M.; Chernovskaya, T.V.; Vasilenko, R.N.; Kozlovskaya, G.D.; Dolgikh, D.A.; Fink, A.L.; et al. Natively unfolded human prothymosin alpha adopts partially folded collapsed conformation at acidic pH. Biochemistry 1999, 38, 15009–15016. [Google Scholar] [CrossRef] [PubMed]
- Baidya, L.; Reddy, G. pH Induced Switch in the Conformational Ensemble of Intrinsically Disordered Protein Prothymosin-alpha and Its Implications for Amyloid Fibril Formation. J. Phys. Chem. Lett. 2022, 13, 9589–9598. [Google Scholar] [CrossRef] [PubMed]
- Haas, B.J.; Dobin, A.; Li, B.; Stransky, N.; Pochet, N.; Regev, A. Accuracy assessment of fusion transcript detection via read-mapping and de novo fusion transcript assembly-based methods. Genome Biol. 2019, 20, 213. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene 1 | Gene 2 | Breakpoint 1 | Breakpoint 2 | Type | Split Reads 1 | Split Reads 2 | Discordant Mates |
|---|---|---|---|---|---|---|---|
| ABHD18 | ZBTB38 | 4:128011700 | 3:141445718 | translocation | 15 | 22 | 19 |
| AKAP13 | ACSL3 | 15:85669830 | 2:222922708 | translocation | 4 | 2 | 1 |
| AKAP13 | NAA25 | 15:85645954 | 12:112039339 | translocation | 12 | 22 | 11 |
| ARF4 | UPF2 | 3:57575548 | 10:12035441 | translocation | 3 | 2 | 7 |
| ARHGAP17 | PPARA | 16:24964197 | 22:46218263 | translocation | 2 | 9 | 4 |
| C3ORF52 | VIM | 3:112093489 | 10:17236294 | translocation | 8 | 8 | 7 |
| CAAP1 | DAPK3 | 9:26884810 | 19:3969829 | translocation | 5 | 20 | 15 |
| CUL1 | RHEB | 7:148767749 | 7:151467211 | inversion | 2 | 4 | 7 |
| DNAJC11 | CAMTA1 | 1:6701729 | 1:6820181 | inversion | 4 | 5 | 2 |
| GOLGB1 | SDR16C5 | 3:121722262 | 8:56309027 | translocation | 12 | 14 | 11 |
| GRAMD2B | FCHO2 | 5:126371567 | 5:72989427 | duplication | 8 | 8 | 14 |
| HSF2 | RP5-1148A21.4 | 6:122423686 | 6:63576436 | duplication | 1 | 3 | 13 |
| KMO | DPYD | 1:241532498 | 1:97193248 | inversion | 1 | 4 | 4 |
| LIPH | TOMM20 | 3:185552423 | 1:235122372 | translocation | 3 | 6 | 6 |
| LRRC31 | LRRC34 | 3:169854813 | 3:169808745 | deletion/read-through | 17 | 11 | 16 |
| LRRFIP1 | PTMA | 2:237627740 | 2:231711348 | duplication | 2 | 6 | 5 |
| MACF1 | NUAK2 | 1:39388658 | 1:205311825 | inversion | 14 | 11 | 31 |
| MAP7 | YLPM1 | 6:136550342 | 14:74829213 | translocation | 5 | 9 | 5 |
| METAP1 | PNRC1 | 4:98995867 | 6:89083753 | translocation | 6 | 8 | 11 |
| METAP1 | PPA2 | 4:99043387 | 4:105399164 | inversion | 6 | 1 | 5 |
| NAPA | PSMB1 | 19:47514843 | 6:170549113 | translocation | 1 | 2 | 4 |
| PALMD | RB1CC1 | 1:99646362 | 8:52658976 | translocation | 2 | 1 | 3 |
| QKI | LEF1 | 6:163535125 | 4:108089257 | translocation | 8 | 14 | 15 |
| RBMS3 | FOXO3 | 3:29281756 | 6:108663455 | translocation | 7 | 11 | 6 |
| RGS2 | REV3L | 1:192809181 | 6:111405630 | translocation | 5 | 1 | 2 |
| RRM2B | EBAG9 | 8:102212776 | 8:109550810 | inversion | 2 | 1 | 1 |
| SEM1 | USP38 | 7:96694798 | 4:143195716 | translocation | 2 | 2 | 1 |
| TDRD3 | ADGRV1 | 13:60467379 | 5:90756979 | translocation | 7 | 9 | 19 |
| THBS1 | LYSMD2 | 15:39587520 | 15:51725121 | inversion | 20 | 17 | 29 |
| TMEM241 | ESCO1 | 18:23437781 | 18:21540700 | deletion | 3 | 5 | 3 |
| VPS13C | CALM2 | 15:62044212 | 2:47170764 | translocation | 1 | 4 | 6 |
| ZNF638 | SCAPER | 2:71350271 | 15:76665789 | translocation | 2 | 2 | 2 |
| Gene Fusion | Length (aa) | κ | FCR (≥0.35) | NCPR | Hydropathy | Disorder Promoting | Plot Region(≥3) | FuzDrop PLLPS | deePhase(≥0.5) |
|---|---|---|---|---|---|---|---|---|---|
| ABHD18-ZBTB38 | 242 | 0.162 | 0.273 | 0.033 | 3.867 | 0.599 | 2 | 0.2472 | 0.37 |
| AKAP13-ACSL3 | 1714 | 0.162 | 0.239 | −0.077 | 3.973 | 0.714 | 1 | 1.0000 | 0.83 |
| AKAP13-NAA25 | 1584 | 0.163 | 0.238 | −0.07 | 4.032 | 0.704 | 1 | 1.0000 | 0.84 |
| ARHGAP17-PPARA | 536 | 0.208 | 0.276 | 0.011 | 4.146 | 0.597 | 2 | 0.2019 | 0.31 |
| C3ORF52-VIM | 131 | 0.196 | 0.244 | −0.092 | 4.292 | 0.603 | 1 | 0.9178 | 0.26 |
| CUL1-RHEB | 391 | 0.141 | 0.274 | −0.008 | 3.925 | 0.614 | 2 | 0.2704 | 0.24 |
| DNAJC11-CAMTA1 | 1682 | 0.197 | 0.205 | −0.006 | 4.014 | 0.672 | 1 | 0.9994 | 0.86 |
| GOLGB1-SDR16C5 | 370 | 0.192 | 0.259 | −0.043 | 4.144 | 0.643 | 2 | 0.6604 | 0.53 |
| HSF2-RP5-1148A21.4 | 423 | 0.223 | 0.267 | −0.054 | 3.884 | 0.619 | 2 | 0.5350 | 0.67 |
| KMO-DPYD | 229 | 0.239 | 0.227 | 0.009 | 4.381 | 0.581 | 1 | 0.1621 | 0.16 |
| LIPH-TOMM20 | 120 | 0.261 | 0.242 | −0.008 | 4.413 | 0.617 | 1 | 0.1939 | 0.19 |
| LRRC31-LRRC34 | 747 | 0.203 | 0.216 | −0.017 | 4.326 | 0.564 | 1 | 0.1573 | 0.57 |
| LRRFIP1-PTMA | 128 | 0.417 | 0.562 | −0.328 | 2.505 | 0.852 | 3 | 0.9949 | 0.66 |
| MACF1-NUAK2 | 5828 | 0.148 | 0.276 | −0.036 | 3.945 | 0.654 | 2 | 0.9924 | 0.81 |
| MAP7-YLPM1 | 114 | 0.164 | 0.412 | −0.009 | 3.366 | 0.772 | 3 | 0.3332 | 0.35 |
| METAP1-PNRC1 | 185 | 0.152 | 0.238 | 0.065 | 3.76 | 0.649 | 1 | 0.5152 | 0.52 |
| METAP1-PPA2 | 334 | 0.195 | 0.254 | −0.003 | 3.996 | 0.626 | 2 | 0.2324 | 0.38 |
| NAPA-PSMB1 | 236 | 0.128 | 0.246 | 0 | 4.363 | 0.619 | 1 | 0.2091 | 0.089 |
| PALMD-RB1CC1 | 1046 | 0.117 | 0.324 | −0.056 | 3.81 | 0.649 | 2 | 0.7123 | 0.81 |
| QKI-LEF1 | 443 | 0.16 | 0.26 | 0.029 | 3.718 | 0.677 | 2 | 0.9907 | 0.73 |
| RBMS3-FOXO3 | 491 | 0.277 | 0.167 | −0.012 | 3.879 | 0.676 | 1 | 0.9996 | 0.85 |
| RGS2-REV3L | 3032 | 0.197 | 0.254 | 0.012 | 3.847 | 0.647 | 2 | 0.9999 | 0.84 |
| SEM1-USP38 | 826 | 0.213 | 0.215 | −0.044 | 4.185 | 0.61 | 1 | 0.6913 | 0.8 |
| TDRD3-ADGRV1 | 2459 | 0.199 | 0.193 | −0.053 | 4.562 | 0.593 | 1 | 0.6100 | 0.57 |
| TMEM241-ESCO1 | 74 | 0.31 | 0.203 | 0.014 | 4.526 | 0.5 | 1 | 0.1198 | 0.17 |
| VPS13C-CALM2 | 196 | 0.179 | 0.316 | −0.133 | 4.032 | 0.612 | 2 | 0.1353 | 0.1 |
| ZNF638-SCAPER | 1003 | 0.198 | 0.2 | 0.011 | 4.086 | 0.616 | 1 | 0.8597 | 0.77 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Güllülü, Ö.; Mayer, B.E.; Toplek, F.B. Linking Gene Fusions to Bone Marrow Failure and Malignant Transformation in Dyskeratosis Congenita. Int. J. Mol. Sci. 2024, 25, 1606. https://doi.org/10.3390/ijms25031606
Güllülü Ö, Mayer BE, Toplek FB. Linking Gene Fusions to Bone Marrow Failure and Malignant Transformation in Dyskeratosis Congenita. International Journal of Molecular Sciences. 2024; 25(3):1606. https://doi.org/10.3390/ijms25031606
Chicago/Turabian StyleGüllülü, Ömer, Benjamin E. Mayer, and Fran Bačić Toplek. 2024. "Linking Gene Fusions to Bone Marrow Failure and Malignant Transformation in Dyskeratosis Congenita" International Journal of Molecular Sciences 25, no. 3: 1606. https://doi.org/10.3390/ijms25031606
APA StyleGüllülü, Ö., Mayer, B. E., & Toplek, F. B. (2024). Linking Gene Fusions to Bone Marrow Failure and Malignant Transformation in Dyskeratosis Congenita. International Journal of Molecular Sciences, 25(3), 1606. https://doi.org/10.3390/ijms25031606