
Re-Engineering Therapeutic Anti-Aβ Monoclonal Antibody to Target Amyloid Light Chain

 , ,
, , 
 , and
, and
Abstract
1. Introduction
2. Results
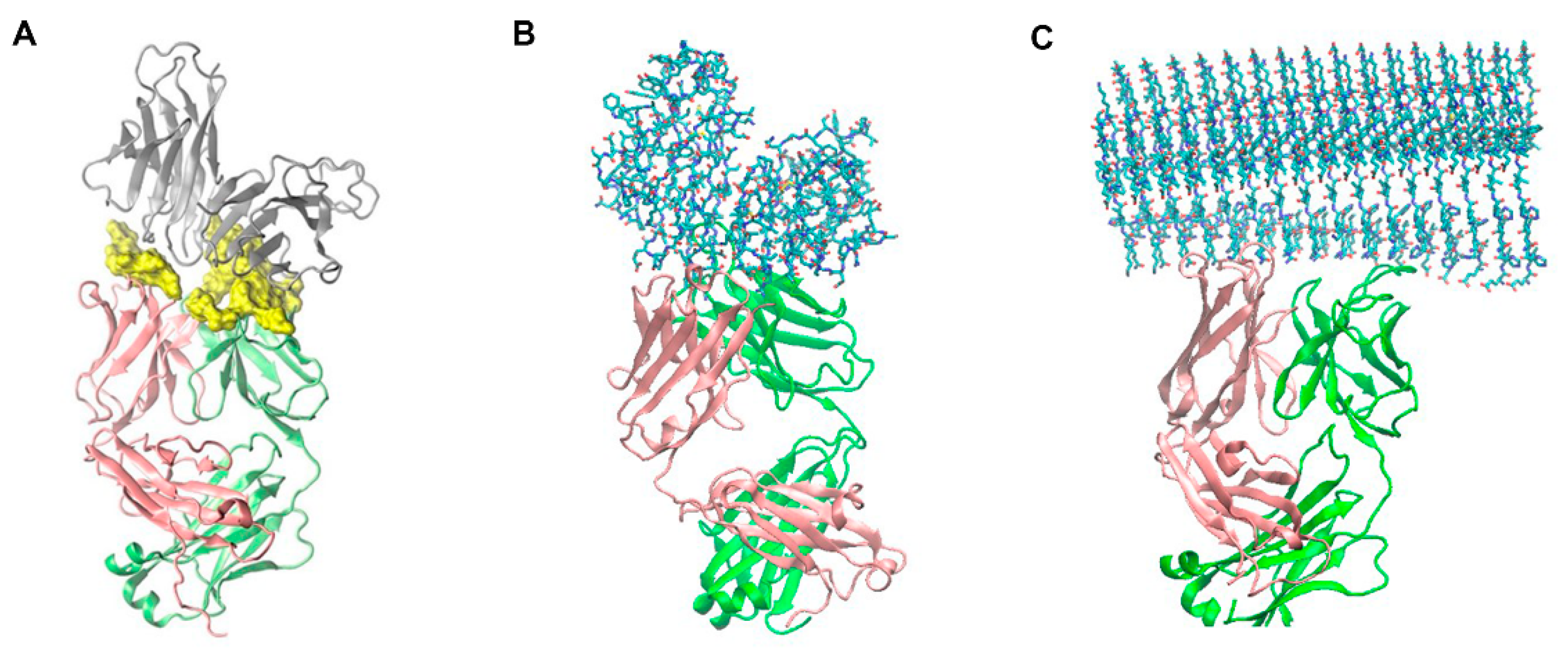
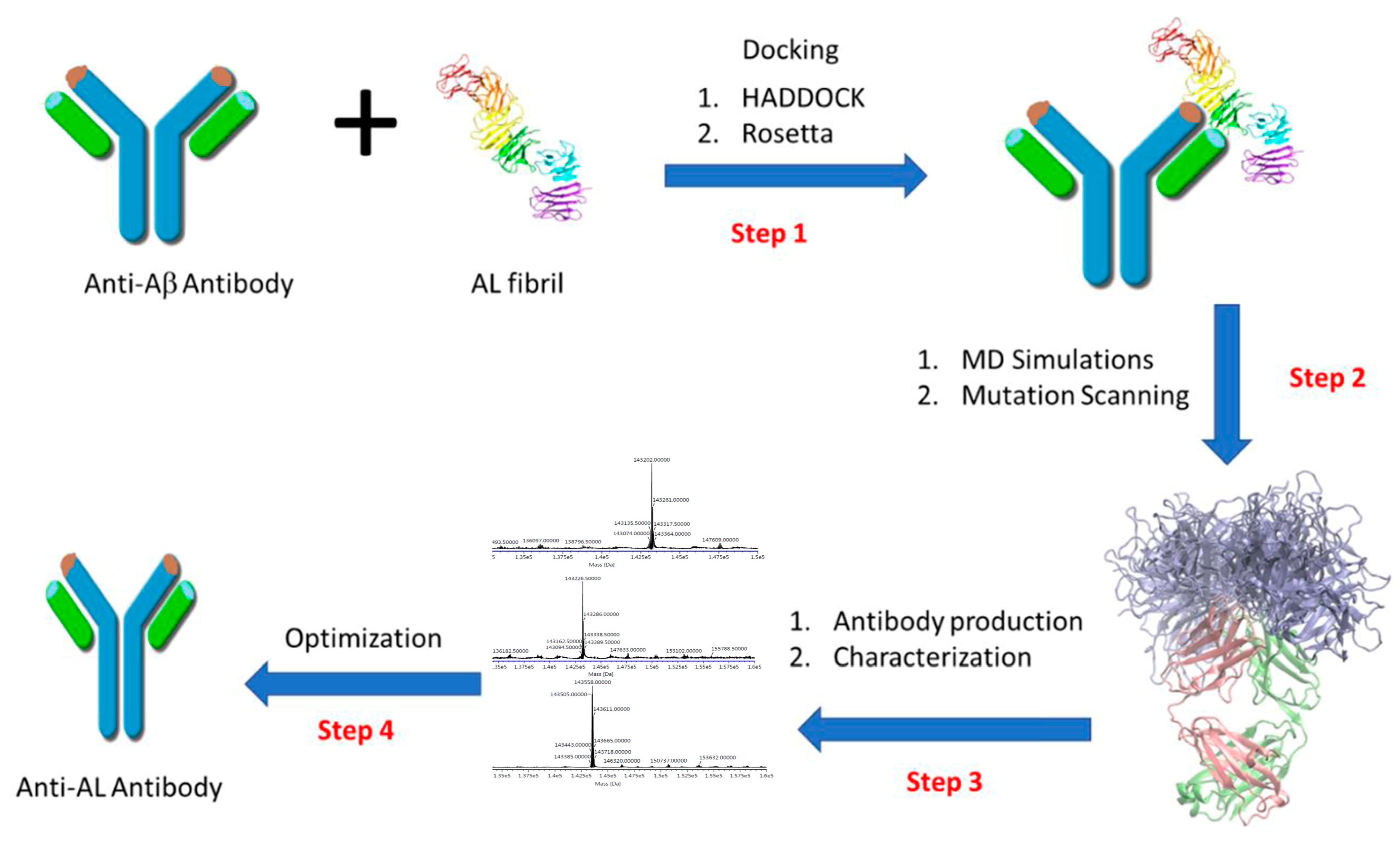
2.1. Molecular Modeling and Simulation of Antibody-AL Binding
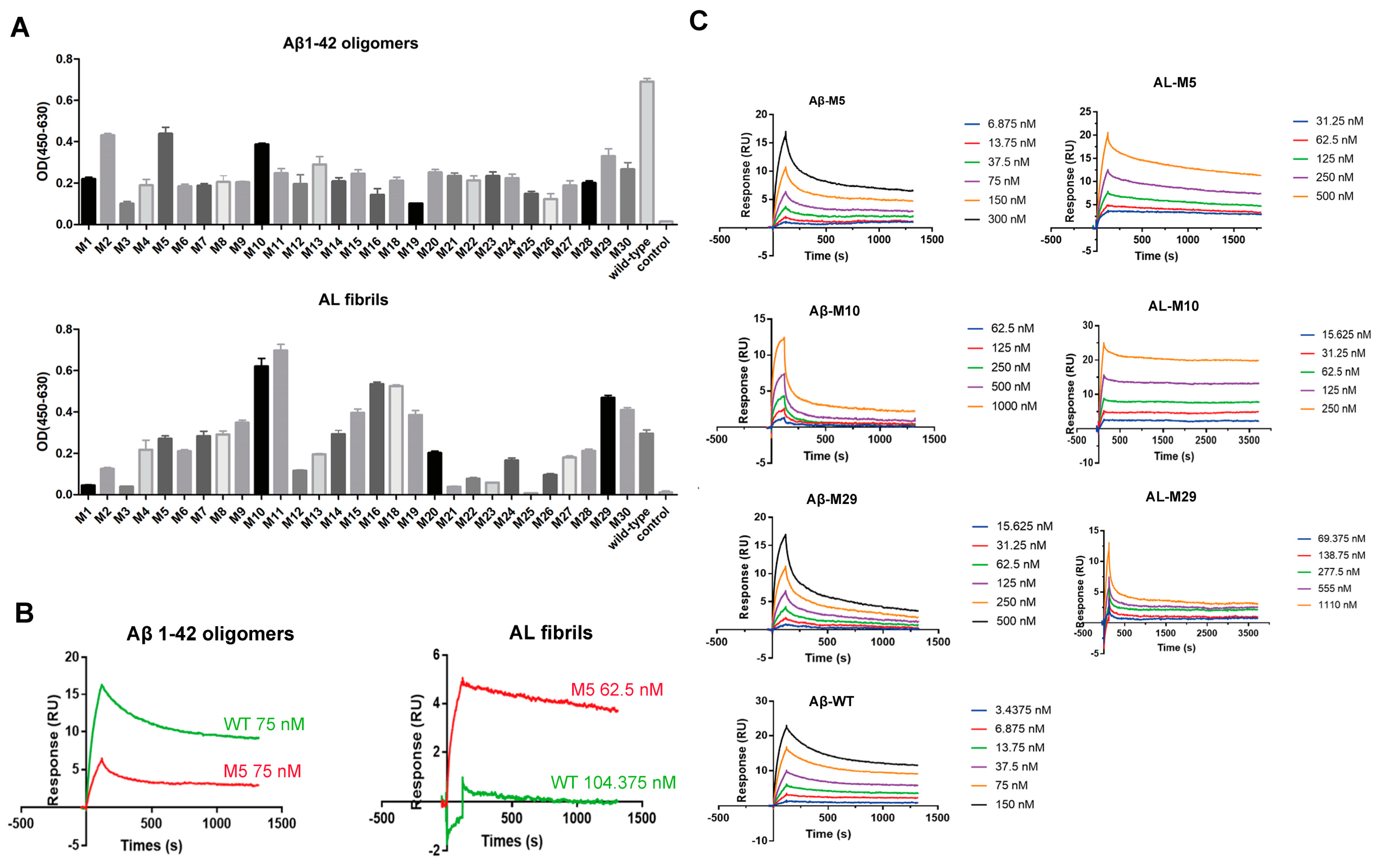
2.2. Engineered Antibodies Bind Both Aβ Oligomers and AL
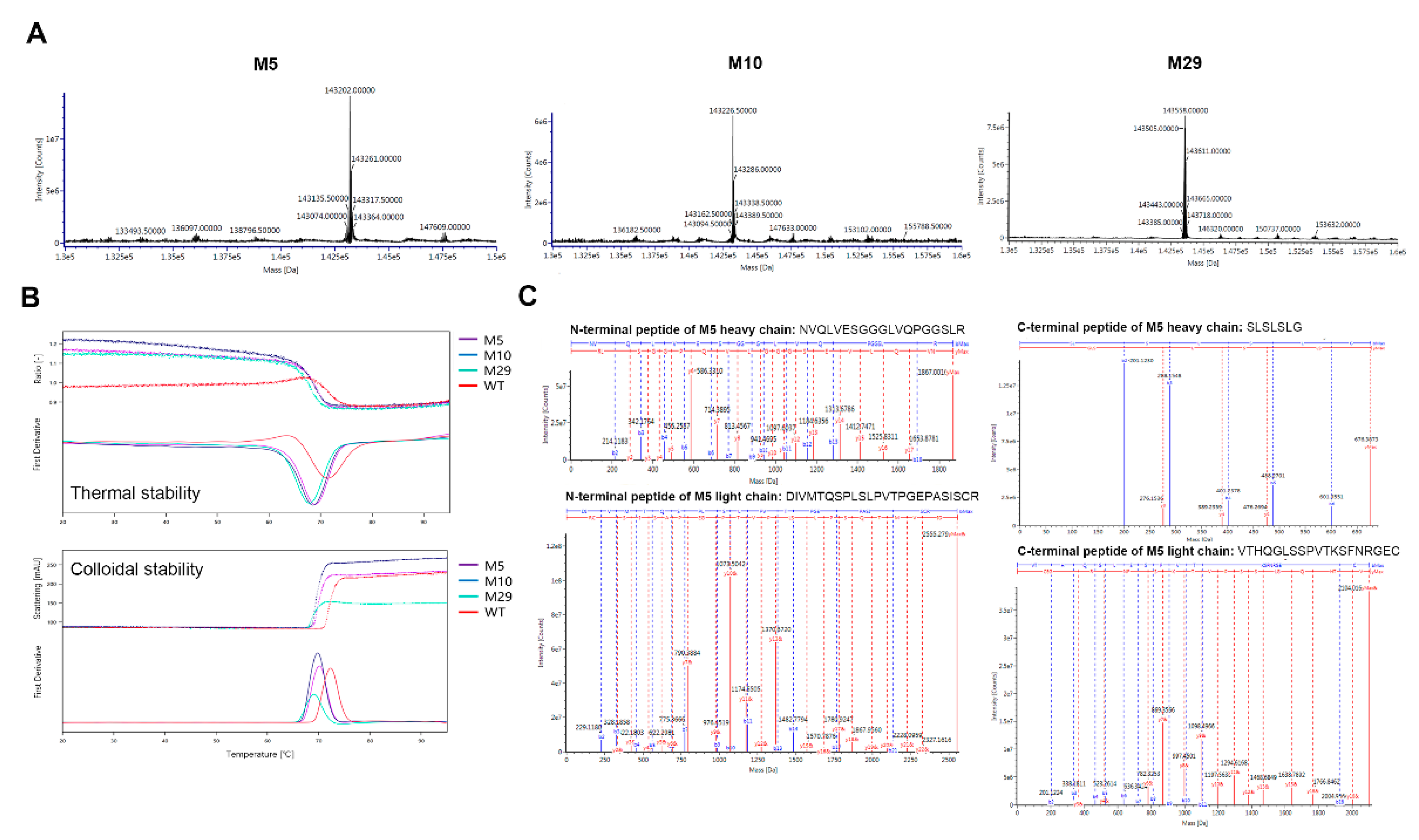
2.3. Characterization of Engineered Aβ and AL Binding Antibodies
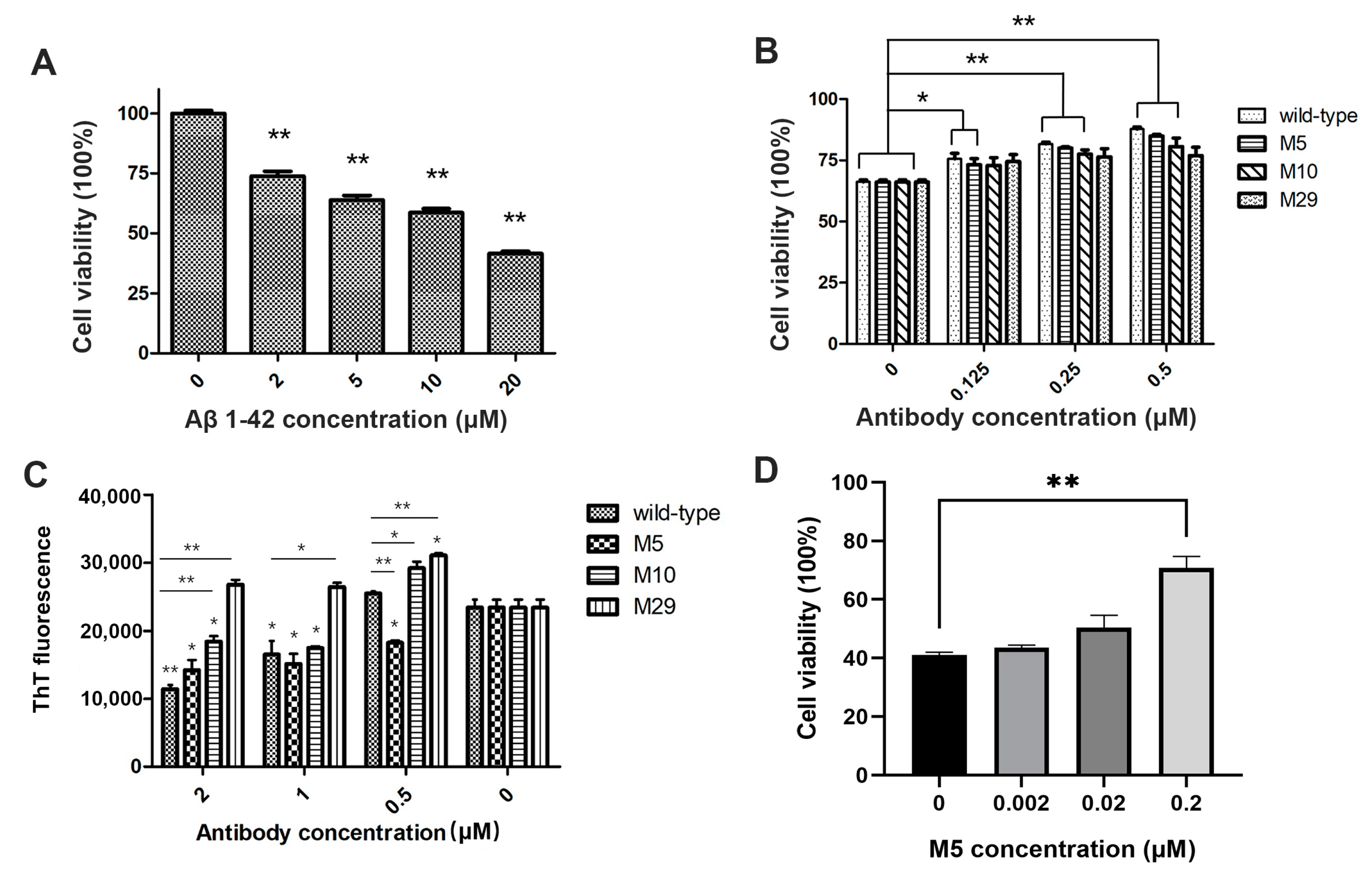
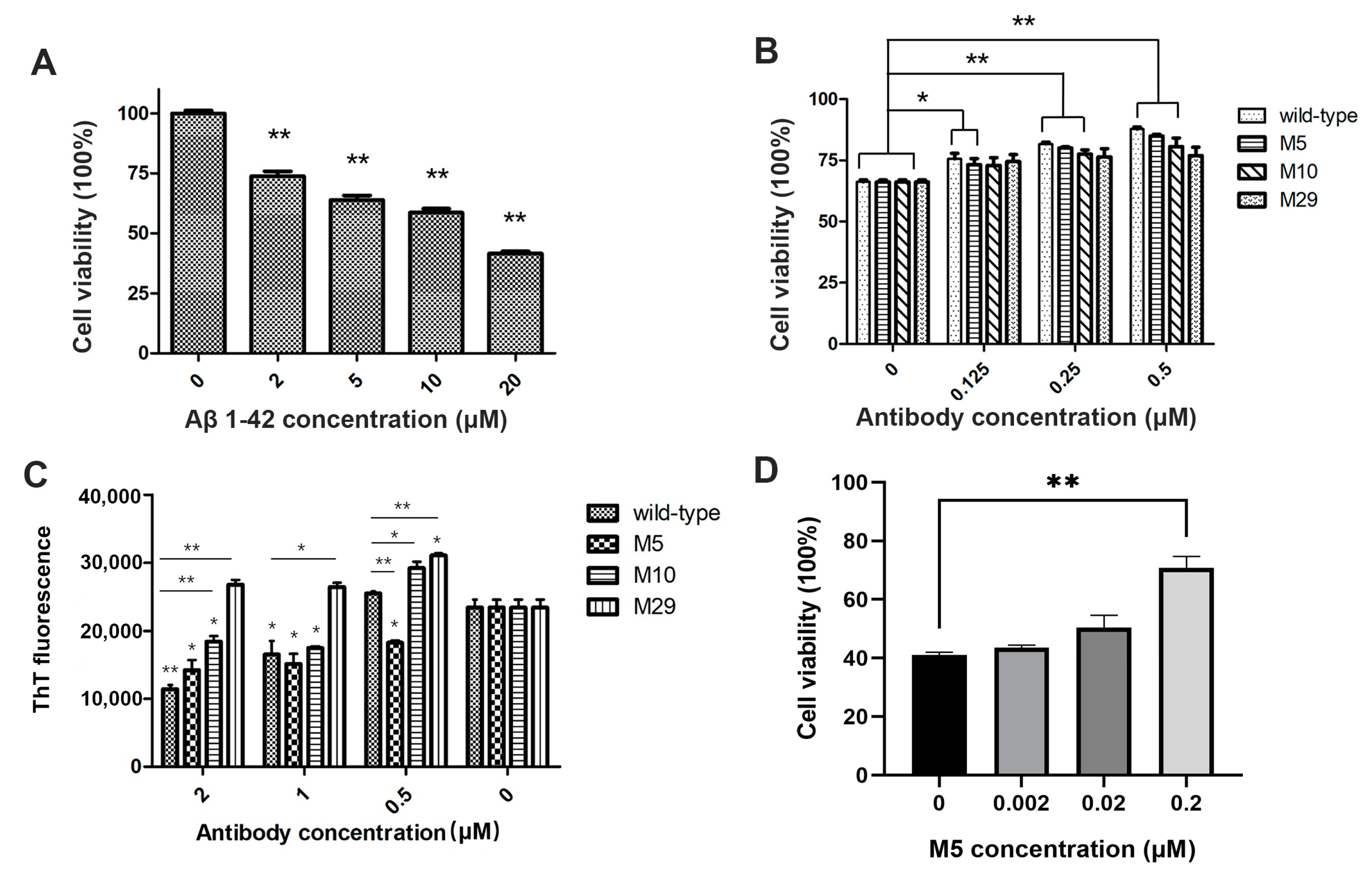
2.4. Bioactivity of Engineered Antibodies
3. Discussion
4. Materials and Methods
4.1. Computational Antibody Re-Engineering
4.2. Genes Cloning and Expression Vector Construction
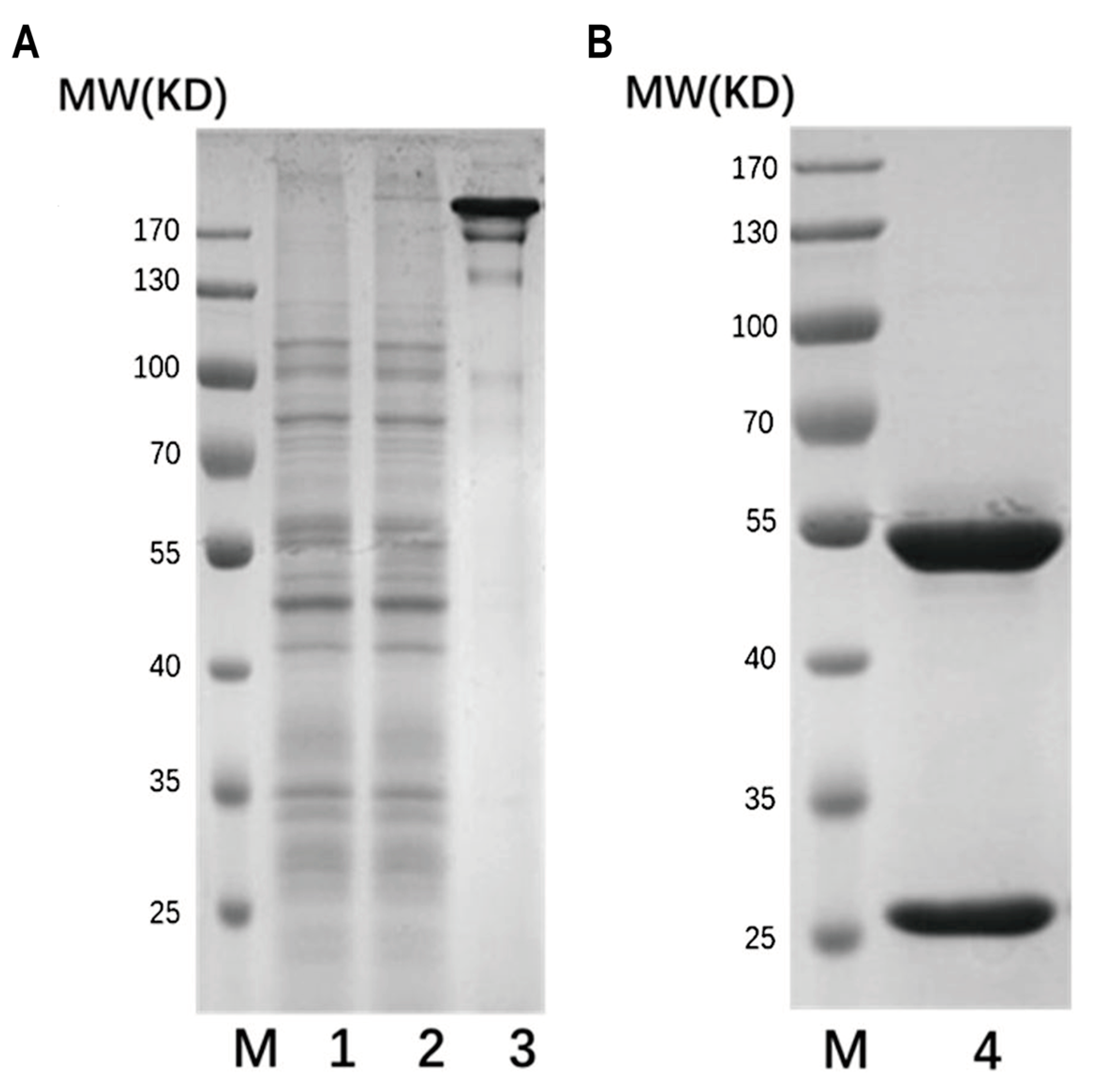
4.3. Expression and Purification of Wild and Mutant Types
4.4. Preparation of Aβ Oligomers and AL Amyloid Fibrils
4.5. Affinity Screening by ELISA
4.6. Antibody Affinity Measurement by SPR
4.7. Thermal Stability Analysis
4.8. Molecular Weight Analysis and Peptide Mapping
4.9. Aβ42 Oligomers Bioactivity Measured by Toxicity on SH-SY5Y Cells
4.10. AL Fibrils Formation Measured by Thioflavin T Fluorescence
4.11. AL Fibrils Bioactivity Measured by Toxicity on AC16 Cells
4.12. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Aguzzi, A.; O’Connor, T. Protein aggregation diseases: Pathogenicity and therapeutic perspectives. Nat. Rev. Drug Discov. 2010, 9, 237–248. [Google Scholar] [CrossRef]
- Sanchorawala, V. Light-chain (AL) amyloidosis: Diagnosis and treatment. Clin. J. Am. Soc. Nephrol. 2006, 1, 1331–1341. [Google Scholar] [CrossRef]
- Kelly, J.W. Does protein aggregation drive postmitotic tissue degeneration? Sci. Transl. Med. 2021, 13, eaax0914. [Google Scholar] [CrossRef]
- Ballard, C.; Gauthier, S.; Corbett, A.; Brayne, C.; Aarsland, D.; Jones, E. Alzheimer’s disease. Lancet 2011, 377, 1019–1031. [Google Scholar] [CrossRef] [PubMed]
- De Genst, E.; Messer, A.; Dobson, C.M. Antibodies and protein misfolding: From structural research tools to therapeutic strategies. Biochim. Biophys. Acta 2014, 1844, 1907–1919. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, A.; Spires-Jones, T. Clearing the way for tau immunotherapy in Alzheimer’s disease. J. Neurochem. 2015, 132, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.; Bogdanovic, N.; Winblad, B.; Portelius, E.; Andreasen, N.; Cedazo-Minguez, A.; Zetterberg, H. Pathways to Alzheimer’s disease. J. Intern. Med. 2014, 275, 296–303. [Google Scholar] [CrossRef] [PubMed]
- Salloway, S.; Honigberg, L.A.; Cho, W.; Ward, M.; Friesenhahn, M.; Brunstein, F.; Quartino, A.; Clayton, D.; Mortensen, D.; Bittner, T.; et al. Amyloid positron emission tomography and cerebrospinal fluid results from a crenezumab anti-amyloid-beta antibody double-blind, placebo-controlled, randomized phase II study in mild-to-moderate Alzheimer’s disease (BLAZE). Alzheimers Res. Ther. 2018, 10, 96. [Google Scholar] [CrossRef] [PubMed]
- Adolfsson, O.; Pihlgren, M.; Toni, N.; Varisco, Y.; Buccarello, A.L.; Antoniello, K.; Lohmann, S.; Piorkowska, K.; Gafner, V.; Atwal, J.K.; et al. An effector-reduced anti-β-amyloid (Aβ) antibody with unique aβ binding properties promotes neuroprotection and glial engulfment of Aβ. J. Neurosci. 2012, 32, 9677–9789. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Wang, Q.; Jiang, S.; Chen, W.; Zeng, C.; Liu, Z. The clinical features and outcomes of systemic AL amyloidosis: A cohort of 231 Chinese patients. Clin. Kidney J. 2015, 8, 120–126. [Google Scholar] [CrossRef]
- Huang, X.H.; Liu, Z.H. The Clinical Presentation and Management of Systemic Light-Chain Amyloidosis in China. Kidney Dis. 2016, 2, 1–9. [Google Scholar] [CrossRef]
- Zhao, Q.; Li, F.; Song, P.; Zhou, X.; Wang, L.; Yu, Y.; An, Z.; Wang, X.; Zhai, Y. Clinical Characteristics and Treatment Outcome of Chinese Patients With Systemic Amyloid Light-Chain Amyloidosis: A Retrospective Single-Center Analysis. Clin. Lymphoma Myeloma Leuk. 2016, 16, 104–110. [Google Scholar] [CrossRef]
- Kyle, R.A.; Gertz, M.A. Primary systemic amyloidosis: Clinical and laboratory features in 474 cases. Semin. Hematol. 1995, 32, 45–59. [Google Scholar]
- Merlini, G.; Dispenzieri, A.; Sanchorawala, V.; Schönland, S.O.; Palladini, G.; Hawkins, P.N.; Gertz, M.A. Systemic immunoglobulin light chain amyloidosis. Nat. Rev. Dis. Primers 2018, 4, 38. [Google Scholar] [CrossRef]
- Merlini, G.; Bellotti, V. Molecular mechanisms of amyloidosis. N. Engl. J. Med. 2003, 349, 583–596. [Google Scholar] [CrossRef] [PubMed]
- Gertz, M.A.; Landau, H.J.; Weiss, B.M. Organ response in patients with AL amyloidosis treated with NEOD001, an amyloid-directed monoclonal antibody. Am. J. Hematol. 2016, 91, E506–E508. [Google Scholar] [CrossRef] [PubMed]
- Hrncic, R.; Wall, J.; Wolfenbarger, D.A.; Murphy, C.L.; Schell, M.; Weiss, D.T.; Solomon, A. Antibody-mediated resolution of light chain-associated amyloid deposits. Am. J. Pathol. 2000, 157, 1239–1246. [Google Scholar] [CrossRef]
- O’Nuallain, B.; Allen, A.; Kennel, S.J.; Weiss, D.T.; Solomon, A.; Wall, J.S. Localization of a conformational epitope common to non-native and fibrillar immunoglobulin light chains. Biochemistry 2007, 46, 1240–1247. [Google Scholar] [CrossRef] [PubMed]
- Wall, J.S.; Kennel, S.J.; Stuckey, A.C.; Long, M.J.; Townsend, D.W.; Smith, G.T.; Wells, K.J.; Fu, Y.; Stabin, M.G.; Weiss, D.T.; et al. Radioimmunodetection of amyloid deposits in patients with AL amyloidosis. Blood 2010, 116, 2241–2244. [Google Scholar] [CrossRef]
- Edwards, C.V.; Rao, N.; Bhutani, D.; Mapara, M.; Radhakrishnan, J.; Shames, S.; Maurer, M.S.; Leng, S.; Solomon, A.; Lentzsch, S.; et al. Phase 1a/b study of monoclonal antibody CAEL-101 (11-1F4) in patients with AL amyloidosis. Blood 2021, 138, 2632–2641. [Google Scholar] [CrossRef]
- Brumshtein, B.; Esswein, S.R.; Landau, M.; Ryan, C.M.; Whitelegge, J.P.; Phillips, M.L.; Cascio, D.; Sawaya, M.R.; Eisenberg, D.S. Formation of amyloid fibers by monomeric light chain variable domains. J. Biol. Chem. 2014, 289, 27513–27525. [Google Scholar] [CrossRef]
- Zhao, J.; Zhang, B.; Zhu, J.; Nussinov, R.; Ma, B. Structure and energetic basis of overrepresented λ light chain in systemic light chain amyloidosis patients. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 2294–2303. [Google Scholar] [CrossRef] [PubMed]
- Piehl, D.W.; Blancas-Mejía, L.M.; Ramirez-Alvarado, M.; Rienstra, C.M. Solid-state NMR chemical shift assignments for AL-09 V(L) immunoglobulin light chain fibrils. Biomol. NMR Assign. 2017, 11, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Radamaker, L.; Lin, Y.H.; Annamalai, K.; Huhn, S.; Hegenbart, U.; Schönland, S.O.; Fritz, G.; Schmidt, M.; Fändrich, M. Cryo-EM structure of a light chain-derived amyloid fibril from a patient with systemic AL amyloidosis. Nat. Commun. 2019, 10, 1103. [Google Scholar] [CrossRef] [PubMed]
- Swuec, P.; Lavatelli, F.; Tasaki, M.; Paissoni, C.; Rognoni, P.; Maritan, M.; Brambilla, F.; Milani, P.; Mauri, P.; Camilloni, C.; et al. Cryo-EM structure of cardiac amyloid fibrils from an immunoglobulin light chain AL amyloidosis patient. Nat. Commun. 2019, 10, 1269. [Google Scholar] [CrossRef] [PubMed]
- Radamaker, L.; Baur, J.; Huhn, S.; Haupt, C.; Hegenbart, U.; Schönland, S.; Bansal, A.; Schmidt, M.; Fändrich, M. Cryo-EM reveals structural breaks in a patient-derived amyloid fibril from systemic AL amyloidosis. Nat. Commun. 2021, 12, 875. [Google Scholar] [CrossRef] [PubMed]
- Radamaker, L.; Karimi-Farsijani, S.; Andreotti, G.; Baur, J.; Neumann, M.; Schreiner, S.; Berghaus, N.; Motika, R.; Haupt, C.; Walther, P.; et al. Role of mutations and post-translational modifications in systemic AL amyloidosis studied by cryo-EM. Nat. Commun. 2021, 12, 6434. [Google Scholar] [CrossRef]
- O’Nuallain, B.; Wetzel, R. Conformational Abs recognizing a generic amyloid fibril epitope. Proc. Natl. Acad. Sci. USA 2002, 99, 1485–1490. [Google Scholar] [CrossRef]
- Krishnan, R.; Hefti, F.; Tsubery, H.; Lulu, M.; Proschitsky, M.; Fisher, R. Conformation as the Therapeutic Target for Neurodegenerative Diseases. Curr. Alzheimer Res. 2017, 14, 393–402. [Google Scholar] [CrossRef]
- Stravalaci, M.; Tapella, L.; Beeg, M.; Rossi, A.; Joshi, P.; Pizzi, E.; Mazzanti, M.; Balducci, C.; Forloni, G.; Biasini, E.; et al. The Anti-Prion Antibody 15B3 Detects Toxic Amyloid-β Oligomers. J. Alzheimers Dis. 2016, 53, 1485–1497. [Google Scholar] [CrossRef]
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, J.; McNamee, C.; et al. Drug repurposing: Progress, challenges and recommendations. Nat. Rev. Drug Discov. 2019, 18, 41–58. [Google Scholar] [CrossRef] [PubMed]
- Ma, B.; Zhao, J.; Nussinov, R. Conformational selection in amyloid-based immunotherapy: Survey of crystal structures of antibody-amyloid complexes. Biochim. Biophys. Acta 2016, 1860, 2672–2681. [Google Scholar] [CrossRef] [PubMed]
- Wall, J.; Schell, M.; Murphy, C.; Hrncic, R.; Stevens, F.J.; Solomon, A. Thermodynamic instability of human lambda 6 light chains: Correlation with fibrillogenicity. Biochemistry 1999, 38, 14101–14108. [Google Scholar] [CrossRef] [PubMed]
- Schwede, T.; Kopp, J.; Guex, N.; Peitsch, M.C. SWISS-MODEL: An automated protein homology-modeling server. Nucleic Acids Res. 2003, 31, 3381–3385. [Google Scholar] [CrossRef]
- Wang, Z.; Huang, W.; Liu, M.; Kennel, S.J.; Wall, J.S.; Cheng, X. Computational investigation of the binding of a designed peptide to λ light chain amyloid fibril. Phys. Chem. Chem. Phys. 2021, 23, 20634–20644. [Google Scholar] [CrossRef] [PubMed]
- de Vries, S.J.; van Dijk, M.; Bonvin, A.M. The HADDOCK web server for data-driven biomolecular docking. Nat. Protoc. 2010, 5, 883–897. [Google Scholar] [CrossRef] [PubMed]
- Pietal, M.J.; Tuszynska, I.; Bujnicki, J.M. PROTMAP2D: Visualization, comparison and analysis of 2D maps of protein structure. Bioinformatics 2007, 23, 1429–1430. [Google Scholar] [CrossRef]
- Wang, M.; Zhu, D.; Zhu, J.; Nussinov, R.; Ma, B. Local and global anatomy of antibody-protein antigen recognition. J. Mol. Recognit. 2018, 31, e2693. [Google Scholar] [CrossRef]
- Ultsch, M.; Li, B.; Maurer, T.; Mathieu, M.; Adolfsson, O.; Muhs, A.; Pfeifer, A.; Pihlgren, M.; Bainbridge, T.W.; Reichelt, M.; et al. Structure of Crenezumab Complex with Aβ Shows Loss of β-Hairpin. Sci. Rep. 2016, 6, 39374. [Google Scholar] [CrossRef]
- Bowers, P.M.; Boyle, W.J.; Damoiseaux, R. The Use of Somatic Hypermutation for the Affinity Maturation of Therapeutic Antibodies. Methods Mol. Biol. 2018, 1827, 479–489. [Google Scholar] [CrossRef]
- Rabia, L.A.; Desai, A.A.; Jhajj, H.S.; Tessier, P.M. Understanding and overcoming trade-offs between antibody affinity, specificity, stability and solubility. Biochem. Eng. J. 2018, 137, 365–374. [Google Scholar] [CrossRef]
- Saito, R.; Saito, Y.; Nakazawa, H.; Hattori, T.; Kumagai, I.; Umetsu, M.; Makabe, K. Impact in stability during sequential CDR grafting to construct camelid VHH antibodies against zinc oxide and gold. J. Biochem. 2018, 164, 21–25. [Google Scholar] [CrossRef] [PubMed]
- Dudgeon, K.; Rouet, R.; Kokmeijer, I.; Schofield, P.; Stolp, J.; Langley, D.; Stock, D.; Christ, D. General strategy for the generation of human antibody variable domains with increased aggregation resistance. Proc. Natl. Acad. Sci. USA 2012, 109, 10879–10884. [Google Scholar] [CrossRef]
- Perchiacca, J.M.; Lee, C.C.; Tessier, P.M. Optimal charged mutations in the complementarity-determining regions that prevent domain antibody aggregation are dependent on the antibody scaffold. Protein Eng. Des. Sel. 2014, 27, 29–39. [Google Scholar] [CrossRef]
- Lambert, M.P.; Barlow, A.K.; Chromy, B.A.; Edwards, C.; Freed, R.; Liosatos, M.; Morgan, T.E.; Rozovsky, I.; Trommer, B.; Viola, K.L.; et al. Diffusible, nonfibrillar ligands derived from Abeta1-42 are potent central nervous system neurotoxins. Proc. Natl. Acad. Sci. USA 1998, 95, 6448–6453. [Google Scholar] [CrossRef]
- McLean, C.A.; Cherny, R.A.; Fraser, F.W.; Fuller, S.J.; Smith, M.J.; Beyreuther, K.; Bush, A.I.; Masters, C.L. Soluble pool of Abeta amyloid as a determinant of severity of neurodegeneration in Alzheimer’s disease. Ann. Neurol. 1999, 46, 860–866. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Dickson, D.W.; Trojanowski, J.Q.; Lee, V.M. The levels of soluble versus insoluble brain Abeta distinguish Alzheimer’s disease from normal and pathologic aging. Exp. Neurol. 1999, 158, 328–337. [Google Scholar] [CrossRef]
- Wolwertz, M.L.; Nguyen, P.T.; Quittot, N.; Bourgault, S. Probing the role of λ6 immunoglobulin light chain dimerization in amyloid formation. Biochim. Biophys. Acta 2016, 1864, 409–418. [Google Scholar] [CrossRef] [PubMed]
- McWilliams-Koeppen, H.P.; Foster, J.S.; Hackenbrack, N.; Ramirez-Alvarado, M.; Donohoe, D.; Williams, A.; Macy, S.; Wooliver, C.; Wortham, D.; Morrell-Falvey, J.; et al. Light Chain Amyloid Fibrils Cause Metabolic Dysfunction in Human Cardiomyocytes. PLoS ONE 2015, 10, e0137716. [Google Scholar] [CrossRef]
- Shi, J.; Guan, J.; Jiang, B.; Brenner, D.A.; Monte, F.D.; Ward, J.E.; Connors, L.H.; Sawyer, D.B.; Semigran, M.J.; Macgillivray, T.E.; et al. Amyloidogenic light chains induce cardiomyocyte contractile dysfunction and apoptosis via a non-canonical p38α MAPK pathway. Proc. Natl. Acad. Sci. USA 2010, 107, 4188–4193. [Google Scholar] [CrossRef]
- Çalık, A.N.; Özcan, K.S.; Yüksel, G.; Güngör, B.; Aruğarslan, E.; Varlibas, F.; Ekmekci, A.; Osmonov, D.; Tatlısu, M.A.; Karaca, M.; et al. Altered diastolic function and aortic stiffness in Alzheimer’s disease. Clin. Interv. Aging 2014, 9, 1115–1121. [Google Scholar] [CrossRef]
- Glabe, C.G. Conformation-dependent antibodies target diseases of protein misfolding. Trends Biochem. Sci. 2004, 29, 542–547. [Google Scholar] [CrossRef]
- Hatami, A.; Monjazeb, S.; Glabe, C. The Anti-Amyloid-β Monoclonal Antibody 4G8 Recognizes a Generic Sequence-Independent Epitope Associated with α-Synuclein and Islet Amyloid Polypeptide Amyloid Fibrils. J. Alzheimers Dis. 2016, 50, 517–525. [Google Scholar] [CrossRef] [PubMed]
- de Matos, A.M.; de Macedo, M.P.; Rauter, A.P. Bridging Type 2 Diabetes and Alzheimer’s Disease: Assembling the Puzzle Pieces in the Quest for the Molecules with Therapeutic and Preventive Potential. Med. Res. Rev. 2018, 38, 261–324. [Google Scholar] [CrossRef]
- Zhao, J.; Nussinov, R.; Ma, B. Mechanisms of recognition of amyloid-β (Aβ) monomer, oligomer, and fibril by homologous antibodies. J. Biol. Chem. 2017, 292, 18325–18343. [Google Scholar] [CrossRef]
- Chaudhury, S.; Berrondo, M.; Weitzner, B.D.; Muthu, P.; Bergman, H.; Gray, J.J. Benchmarking and analysis of protein docking performance in Rosetta v3.2. PLoS ONE 2011, 6, e22477. [Google Scholar] [CrossRef] [PubMed]
- Kalé, L.; Skeel, R.; Bhandarkar, M.; Brunner, R.; Gursoy, A.; Krawetz, N.; Phillips, J.; Shinozaki, A.; Varadarajan, K.; Schulten, K.; et al. NAMD2: Greater scalability for parallel molecular dynamics. J. Comput. Phys. 1999, 151, 283–312. [Google Scholar] [CrossRef]
- MacKerell, A.D.; Bashford, D.; Bellott, M.; Dunbrack, R.L.; Evanseck, J.D.; Field, M.J.; Fischer, S.; Gao, J.; Guo, H.; Ha, S.; et al. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B 1998, 102, 3586–3616. [Google Scholar] [CrossRef]
- Lee, M.S.; Feig, M.; Salsbury, F.R., Jr.; Brooks, C.L., 3rd. New analytic approximation to the standard molecular volume definition and its application to generalized Born calculations. J. Comput. Chem. 2003, 24, 1348–1356. [Google Scholar] [CrossRef]
- Marín-Argany, M.; Rivera-Hernández, G.; Martí, J.; Villegas, S. An anti-Aβ (amyloid β) single-chain variable fragment prevents amyloid fibril formation and cytotoxicity by withdrawing Aβ oligomers from the amyloid pathway. Biochem. J. 2011, 437, 25–34. [Google Scholar] [CrossRef]
- Raffen, R.; Dieckman, L.J.; Szpunar, M.; Wunschl, C.; Pokkuluri, P.R.; Dave, P.; Stevens, P.W.; Cai, X.; Schiffer, M.; Stevens, F.J.; et al. Physicochemical consequences of amino acid variations that contribute to fibril formation by immunoglobulin light chains. Protein Sci. 1999, 8, 509–517. [Google Scholar] [CrossRef] [PubMed]

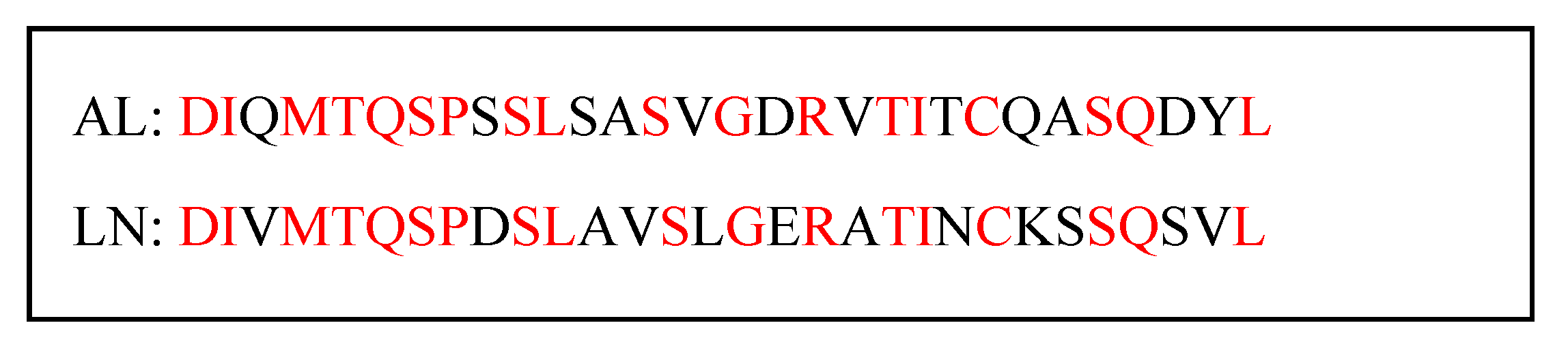

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
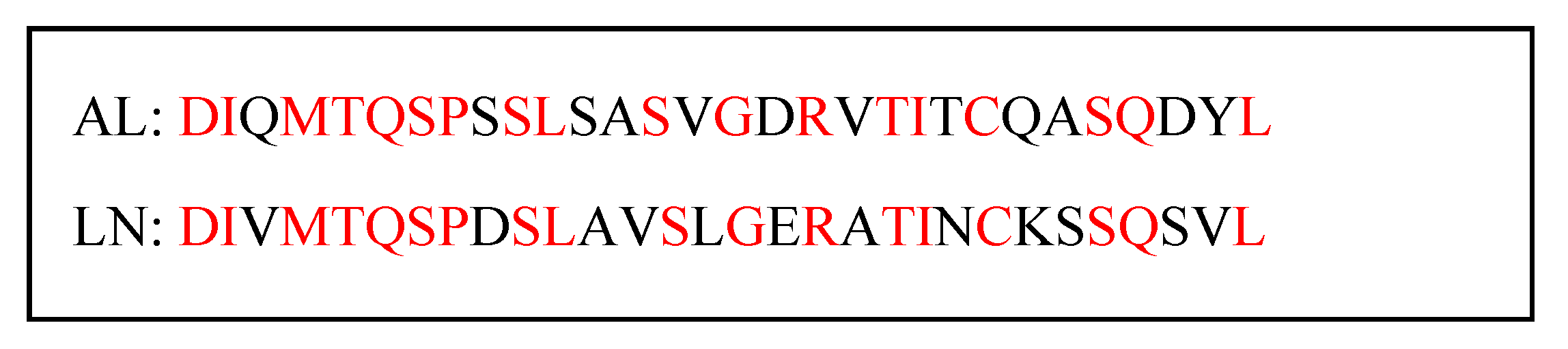{kind=link}
| PosA | SetA | PosB | SetB | Energy (kcal/mol) | Dist (Å) |
|---|---|---|---|---|---|
| 15 | Leu128 | 32 | Tyr32 | −2.75 | 3.83 |
| 12 | Ala125 | 55 | Lys55 | −2.71 | 3.84 |
| 3 | Val3 | 37 | Tyr37 | −.24 | 4.17 |
| 1 | Asp1 | 37 | Tyr37 | −1.04 | 3.69 |
| 109 | Lys222 | 33 | Asn33 | −1.01 | 3.99 |
| 17 | Glu130 | 32 | Tyr32 | −0.95 | 3.99 |
| 19 | Ala19 | 54 | Tyr54 | −0.86 | 3.93 |
| 20 | Thr20 | 58 | Asn58 | −0.86 | 3.9 |
| 78 | Thr78 | 58 | Asn58 | −0.57 | 4.17 |
| 3 | Val3 | 36 | Thr36 | −0.52 | 4.36 |
| 18 | Arg18 | 53 | Ile53 | −0.46 | 4.24 |
| 4 | Met4 | 35 | Asp35 | −0.4 | 3.47 |
| 15 | Leu128 | 52 | Asn52 | −0.3 | 4.24 |
| 15 | Leu128 | 53 | Ser53 | −0.25 | 3.6 |
| 112 | Ile225 | 31 | Tyr31 | −0.21 | 3.95 |
| 17 | Glu17 | 32 | Tyr32 | −0.2 | 4.38 |
| 113 | Lys226 | 99 | Val99 | −0.2 | 4.09 |
| 11 | Leu124 | 32 | Ser32 | −0.19 | 3.93 |
| 16 | Gly16 | 2 | Val2 | −0.13 | 4.15 |
| 81 | Ile81 | 54 | Tyr54 | −0.11 | 3.74 |
| 10 | Ser123 | 31 | Tyr31 | −0.1 | 3.88 |
| 15 | Leu128 | 51 | Ile51 | −0.1 | 4.37 |
| 78 | Thr78 | 57 | Ser57 | −0.06 | 4.26 |
| 17 | Glu130 | 31 | Ser31 | −0.02 | 3.71 |
| 16 | Gly16 | 101 | Tyr101 | −0.01 | 4.11 |
| 13 | Val126 | 31 | Tyr31 | 0 | 4.47 |
| 113 | Lys113 | 31 | Ser31 | 0 | 4.44 |
| 5 | Thr5 | 33 | Asn33 | 0 | 3.78 |
| 111 | Glu224 | 37 | Tyr37 | 0 | 4.49 |
| 84 | Leu197 | 53 | Ser53 | 0 | 4.46 |
| 85 | Gln198 | 57 | Ser57 | 0 | 4.41 |
| 60 | Arg60 | 65 | Asp65 | 0 | 4.33 |
| 113 | Lys226 | 102 | Thr102 | 0 | 4.49 |
| Chain | Contact | ∆∆E (kcal/mol) | |
|---|---|---|---|
| ASP35 | L | 2 | 50.7 |
| LYS55 | L | 4 | 41.7 |
| ARG59 | L | 3 | 31.4 |
| ASN33 | L | 5 | 27.3 |
| TYR31 | L | 4 | 23.6 |
| GLU1 | H | 2 | 23.2 |
| ASN58 | L | 3 | 17.2 |
| SER53 | H | 2 | 15.2 |
| TYR54 | L | 4 | 14.2 |
| SER31 | H | 2 | 13.6 |
| TYR37 | L | 3 | 12.3 |
| ASN52 | H | 2 | 11.6 |
| SER32 | L | 3 | 3.3 |
| ASN54 | H | 2 | 2.1 |
| WT | 0 | ||
| TYR32 | H | 4 | −7.1 |
| SER61 | L | 3 | −9.4 |
| TYR101 | H | 2 | −25 |
| Antibody | Aβ42 Oligomers | AL Fibrils | ||||
|---|---|---|---|---|---|---|
| ka (1/Ms) | kd (1/s) | KD (nM) | ka (1/Ms) | kd (1/s) | KD (nM) | |
| M5 | 5.341 × 104 | 5.248 × 10−4 | 9.826 | 2.625 × 104 | 2.370 × 10−4 | 9.030 |
| M10 | 3.694 × 104 | 1.141 × 10−3 | 30.9 | 3.542 × 104 | 2.519 × 10−5 | 0.7112 |
| M2 | 5.257 × 104 | 6.600 × 10−4 | 12.55 | / | / | / |
| M11 | / | / | / | 3.181 × 104 | 6.255 × 10−5 | 1.996 |
| M15 | / | / | / | 3131 | 3.901 × 10−6 | 1.246 |
| M18 | / | / | / | 1.501 × 104 | 6.268 × 10−5 | 4.177 |
| M29 | 3.497 × 104 | 1.257 × 10−3 | 35.93 | 1.189 × 104 | 1.493 × 10−4 | 12.56 |
| M30 | 6.029 × 104 | 1.1818 × 10−3 | 30.15 | / | / | / |
| wild-type | 7.611 × 105 | 1.164 × 10−3 | 1.53 | NB | NB | NB |
| Protein | Observed Mass (Da) | Expected Mass (Da) | Mass Error (mDa) | Mass Error (ppm) |
|---|---|---|---|---|
| M5 | 143,201.8825 | 143,192.63260 | 9249.9 | 64.6 |
| M10 | 143,226.5483 | 143,216.65400 | 9894.3 | 69.1 |
| M29 | 143,505.5634 | 143,503.01770 | 2545.7 | 17.7 |
| M5-LC | 24,041.0548 | 24,040.52080 | 534.0 | 22.2 |
| M5-HC | 47,574.5082 | 47,571.92250 | 2585.7 | 54.3 |
| M10-LC | 24,040.521 | 24,041.2514 | 730.4 | 30.4 |
| M10-HC | 47,583.933 | 47,585.6194 | 1686.4 | 35.4 |
| M29-LC | 24,041.7617 | 24,040.52080 | 1240.9 | 51.6 |
| M29-HC | 47,729.4542 | 47,727.11510 | 2339.1 | 49.0 |
| Capillary | Tm Onset | Tm | Tagg Onset | Tagg Scattering Signal |
|---|---|---|---|---|
| M5 | 62.6 °C | 68.9 °C | 67.4 °C | 233 |
| M10 | 62.3 °C | 68.5 °C | 67.5 °C | 267 |
| M29 | 62.4 °C | 67.8 °C | 67.1 °C | 149 |
| Wild-type | 63.2 °C | 71.3 °C | 70.0 °C | 235 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bai, J.; Li, X.; Zhao, J.; Zong, H.; Yuan, Y.; Wang, L.; Zhang, X.; Ke, Y.; Han, L.; Xu, J.; et al. Re-Engineering Therapeutic Anti-Aβ Monoclonal Antibody to Target Amyloid Light Chain. Int. J. Mol. Sci. 2024, 25, 1593. https://doi.org/10.3390/ijms25031593
Bai J, Li X, Zhao J, Zong H, Yuan Y, Wang L, Zhang X, Ke Y, Han L, Xu J, et al. Re-Engineering Therapeutic Anti-Aβ Monoclonal Antibody to Target Amyloid Light Chain. International Journal of Molecular Sciences. 2024; 25(3):1593. https://doi.org/10.3390/ijms25031593
Chicago/Turabian StyleBai, Jingyi, Xi Li, Jun Zhao, Huifang Zong, Yuan Yuan, Lei Wang, Xiaoshuai Zhang, Yong Ke, Lei Han, Jianrong Xu, and et al. 2024. "Re-Engineering Therapeutic Anti-Aβ Monoclonal Antibody to Target Amyloid Light Chain" International Journal of Molecular Sciences 25, no. 3: 1593. https://doi.org/10.3390/ijms25031593
APA StyleBai, J., Li, X., Zhao, J., Zong, H., Yuan, Y., Wang, L., Zhang, X., Ke, Y., Han, L., Xu, J., Ma, B., Zhang, B., & Zhu, J. (2024). Re-Engineering Therapeutic Anti-Aβ Monoclonal Antibody to Target Amyloid Light Chain. International Journal of Molecular Sciences, 25(3), 1593. https://doi.org/10.3390/ijms25031593