
Preparative and Catalytic Properties of MoVI Mononuclear and Metallosupramolecular Coordination Assemblies Bearing Hydrazonato Ligands





Abstract
1. Introduction
2. Results and Discussion
2.1. Synthesis and Structural Characteriation
2.1.1. Hydrazones
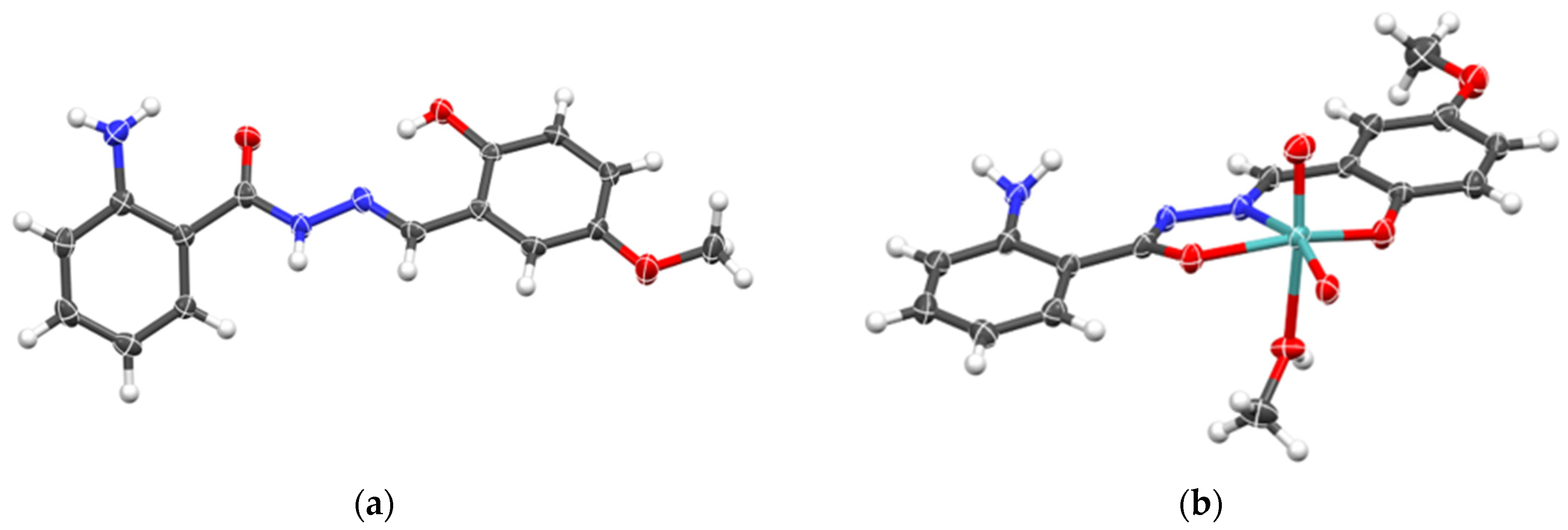
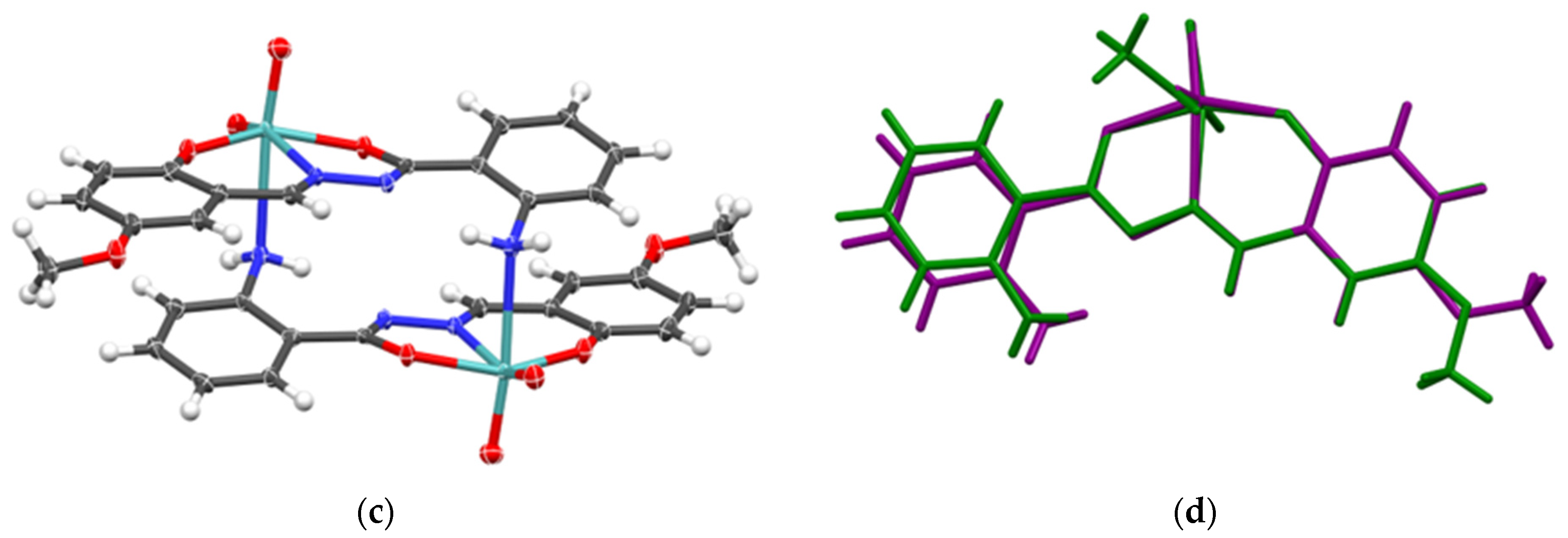
2.1.2. Metallosupramolecular Coordination Assemblies
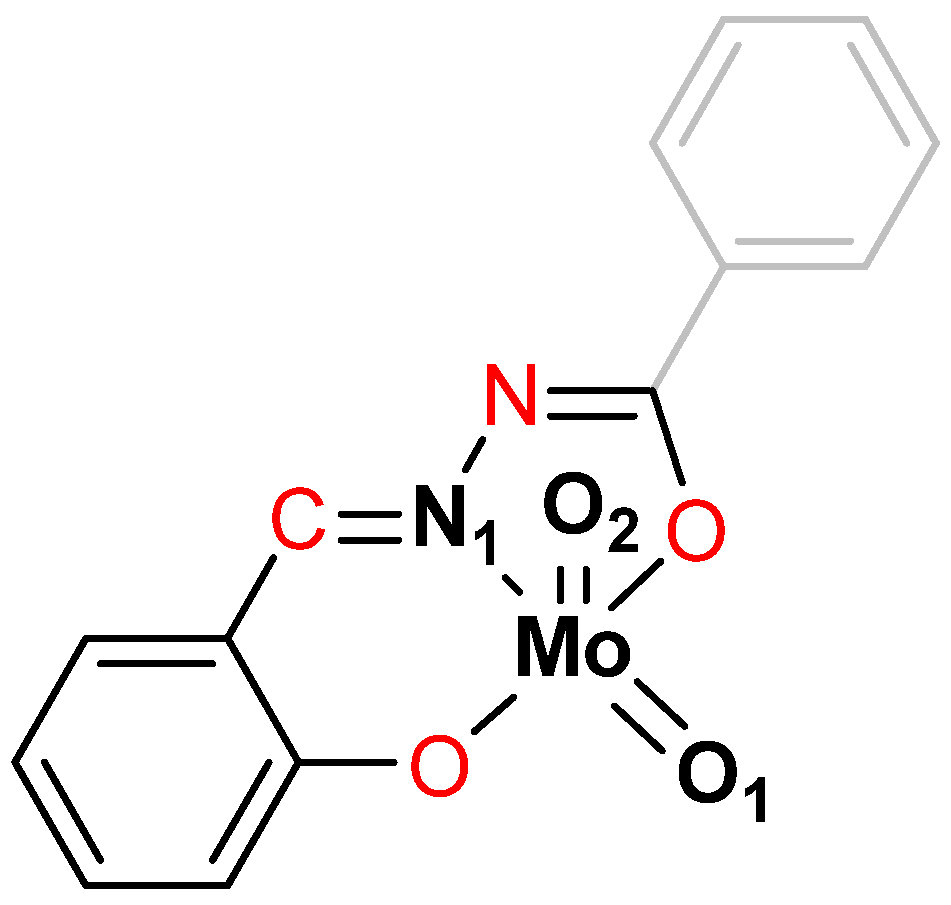
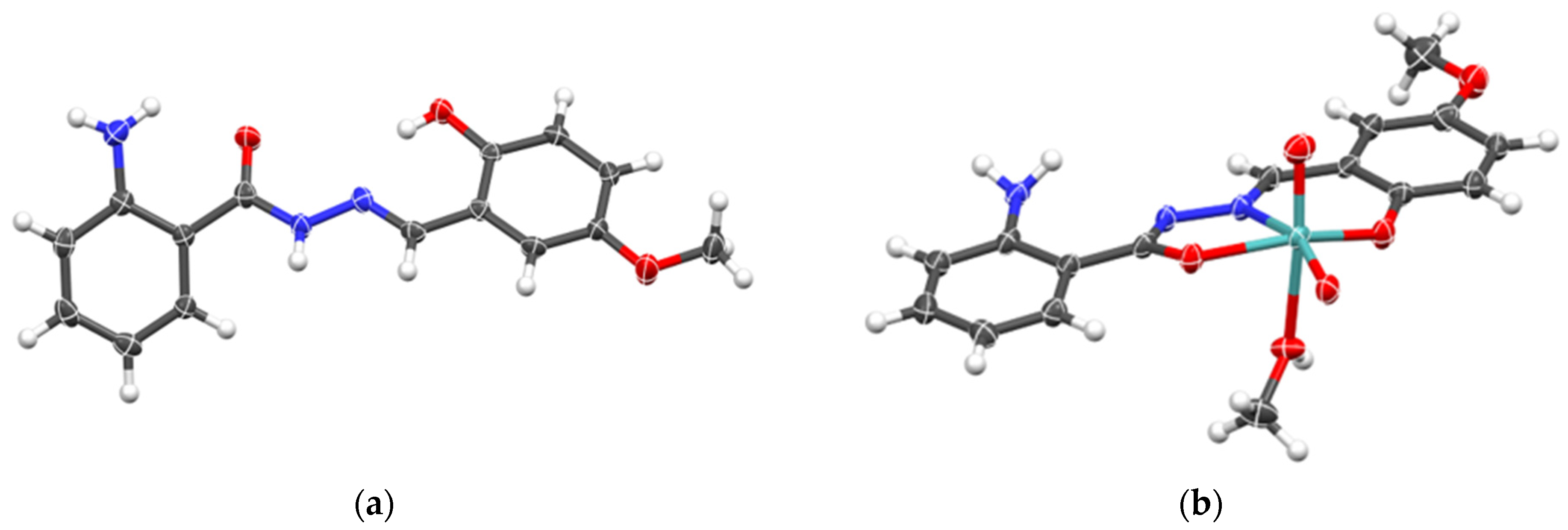
2.1.3. Mononuclear Coordination Assemblies
2.2. Spectroscopic Characterization
2.3. Thermal Analysis
2.4. Cyclooctene Epoxidation with TBHP
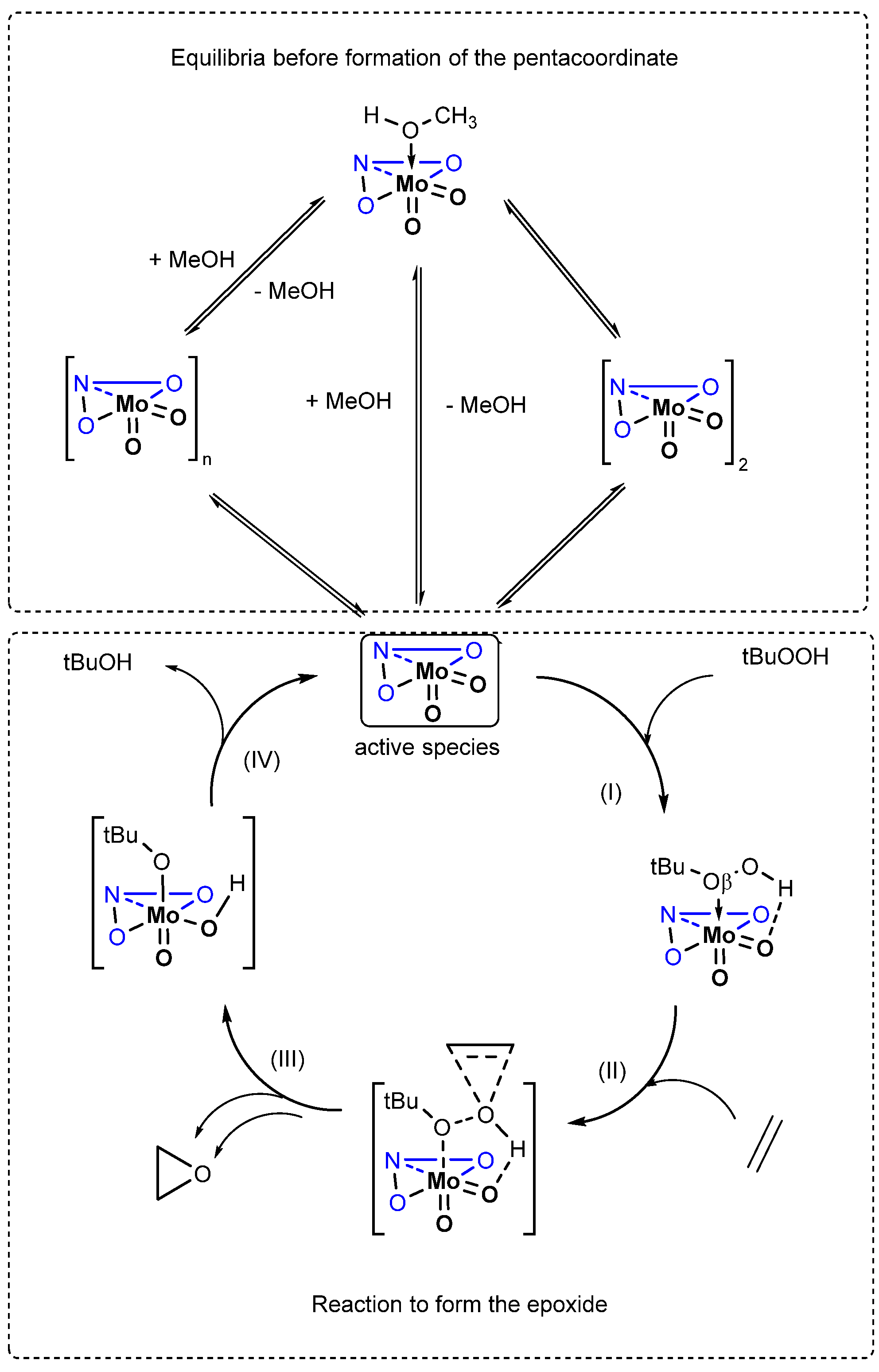
2.5. Density Functional Theory (DFT) Calculations
3. Materials and Methods
3.1. Preparative Part
3.2. Synthesis of H2L1 and H2L2
3.3. Synthesis of H2L3 and H2L4
3.4. Synthesis of 1–4
3.5. Synthesis of 1a–4a
3.6. Physical Methods
3.7. Theoretical Calculations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chen, L.; Chen, Q.H.; Wu, M.Y.; Jiang, F.L.; Hong, M.C. Controllable coordination-driven self-assembly: From discrete metallocages to infinite cage-based frameworks. Acc. Chem. Res. 2015, 48, 201–210. [Google Scholar] [CrossRef]
- Lescop, C. Coordination-Driven Syntheses of Compact Supramolecular Metallacycles toward Extended Metallo-organic Stacked Supramolecular Assemblies. Acc. Chem. Res. 2017, 50, 885–894. [Google Scholar] [CrossRef]
- Northrop, B.H.; Zheng, Y.R.; Chi, K.W.; Stang, P.J. Self-organization in coordination-driven self-assembly. Acc. Chem. Res. 2009, 42, 1554–1563. [Google Scholar] [CrossRef]
- Cook, T.R.; Stang, P.J. Recent Developments in the Preparation and Chemistry of Metallacycles and Metallacages via Coordination. Chem. Rev. 2015, 115, 7001–7045. [Google Scholar] [CrossRef]
- Cook, T.R.; Zheng, Y.R.; Stang, P.J. Metal–Organic Frameworks and Self-Assembled Supramolecular Coordination Complexes: Comparing and Contrasting the Design, Synthesis, and Functionality of Metal–Organic Materials. Chem. Rev. 2013, 113, 734–777. [Google Scholar] [CrossRef]
- Giuseppone, N.; Schmitt, J.L.; Lehn, J.M. Driven Evolution of a Constitutional Dynamic Library of Molecular Helices Toward the Selective Generation of [2 × 2] Gridlike Arrays under the Pressure of Metal Ion Coordination. J. Am. Chem. Soc. 2006, 128, 16748–16763. [Google Scholar] [CrossRef]
- Dietrich-Buchecker, C.; Colasson, B.; Fujita, M.; Hori, A.; Geum, N.; Sakamoto, S.; Yamaguchi, K.; Sauvage, J.P. Quantitative Formation of [2]Catenanes Using Copper(I) and Palladium(II) as Templating and Assembling Centers: The Entwining Route and the Threading Approach. J. Am. Chem. Soc. 2003, 125, 5717–5725. [Google Scholar] [CrossRef]
- Berl, V.; Huc, I.; Khoury, R.G.; Krische, M.J.; Lehn, J.M. Interconversion of single and double helices formed from synthetic molecular strands. Nature 2000, 407, 720–723. [Google Scholar] [CrossRef]
- Levchenko, A.A.; Yee, C.K.; Parikh, A.N.; Navrotsky, A. Energetics of Self-Assembly and Chain Confinement in Silver Alkanethiolates: Enthalpy–Entropy Interplay. Chem. Mater. 2005, 17, 5428–5438. [Google Scholar] [CrossRef]
- Kunz, V.; Lindner, J.O.; Schulze, M.; Röhr, M.I.S.; Schmidt, D.; Mitrić, R.; Würthner, F. Cooperative water oxidation catalysis in a series of trinuclear metallosupramolecular ruthenium macrocycles. Energy Environ. Sci. 2017, 10, 2137–2153. [Google Scholar] [CrossRef]
- Kim, S.; Cho, K.-B.; Lee, Y.-M.; Chen, J.; Fukuzumi, S.; Nam, W. Factors Controlling the Chemoselectivity in the Oxidation of Olefins by Nonheme Manganese(IV)-Oxo Complexes. J. Am. Chem. Soc. 2016, 138, 10654–10663. [Google Scholar] [CrossRef]
- Liu, Y.; Xuan, W.; Zhang, H.; Cui, Y. Chirality- and Threefold-Symmetry-Directed Assembly of Homochiral Octupolar Metal–Organoboron Frameworks. Inorg. Chem. 2009, 48, 10018–10023. [Google Scholar] [CrossRef]
- Liang, L.-L.; Ren, S.-B.; Zhang, J.; Li, Y.-Z.; Du, H.-B.; You, X.-Z. Two Thermostable Three-Dimensional Homochiral Metal–Organic Polymers with Quartz Topology. Cryst. Growth Des. 2010, 10, 1307–1311. [Google Scholar] [CrossRef]
- Bark, T.; von Zelewsky, A.; Rappoport, D.; Neuburger, M.; Schaffner, S.; Lacour, J.; Jodry, J. Synthesis and Stereochemical Properties of Chiral Square Complexes of Iron(II). Chem. Eur. J. 2004, 10, 4839–4845. [Google Scholar] [CrossRef]
- Demadis, K.D.; Papadaki, M.; Aranda, M.A.G.; Cabeza, A.; Olivera-Pastor, P.; Sanakis, Y. Stepwise Topotactic Transformations (1D to 3D) in Copper Carboxyphosphonate Materials: Structural Correlations. Cryst. Growth Des. 2010, 10, 357–364. [Google Scholar] [CrossRef]
- Li, Z.-Y.; Dai, J.-W.; Damjanović, M.; Shiga, T.; Wang, J.-H.; Zhao, J.; Oshio, H.; Yamashita, M.; Bu, X.-H. Structure Switching and Modulation of the Magnetic Properties in Diarylethene-Bridged Metallosupramolecular Compounds by Controlled Coordination-Driven Self-Assembly. Angew. Chem. Int. Ed. 2019, 58, 4339–4344. [Google Scholar] [CrossRef]
- Estrader, M.; Uber, J.S.; Barrios, L.A.; Garcia, J.; Lloyd-Williams, P.; Roubeau, O.; Teat, S.J.; Aromi, G. A Magneto-optical Molecular Device: Interplay of Spin Crossover, Luminescence, Photomagnetism, and Photochromism. Angew. Chem. Int. Ed. 2017, 56, 15622–15627. [Google Scholar] [CrossRef]
- Duan, J.; Jin, W.; Krishna, R. Natural Gas Purification Using a Porous Coordination Polymer with Water and Chemical Stability. Inorg. Chem. 2015, 54, 4279–4284. [Google Scholar] [CrossRef]
- Gupta, N.K.; Kim, E.J.; Bae, J.; Kim, K.S. Probing the origin and stability of bivalency in copper based porous coordination network and its application for H2S gas capture. Sci. Rep. 2022, 12, 15388. [Google Scholar] [CrossRef]
- Saha, M.L.; Yan, X.Z.; Stang, P.J. Photophysical Properties of Organoplatinum(II) Compounds and Derived Self-Assembled Metallacycles and Metallacages: Fluorescence and its Applications. Acc. Chem. Res. 2016, 49, 2527–2539. [Google Scholar] [CrossRef]
- Rösner, B.; Milek, M.; Witt, A.; Gobaut, B.; Torelli, P.; Fink, R.H.; Khusniyarov, M.M. Reversible Photoswitching of a Spin-Crossover Molecular Complex in the Solid State at Room Temperature. Angew. Chem. Int. Ed. 2015, 54, 12976–12980. [Google Scholar] [CrossRef] [PubMed]
- Pisk, J.; Agustin, D.; Vrdoljak, V.; Poli, R. Epoxidation Processes by Pyridoxal Dioxomolybdenum(VI) (Pre)Catalysts Without Organic Solvent. Adv. Synth. Catal. 2011, 353, 2910–2914. [Google Scholar] [CrossRef]
- Judmaier, M.E.; Holzer, C.; Volpe, M.; Mösch-Zanetti, N.C. Molybdenum(VI) dioxo complexes employing Schiff base ligands with an intramolecular donor for highly selective olefin epoxidation. Inorg. Chem. 2012, 51, 9956–9966. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.X.; Li, W.H. Synthesis, Crystal Structures, and Catalytic Property of Dioxomolybdenum(VI) Complexes with Hydrazones. Russ. J. Coord. Chem. 2012, 38, 92–98. [Google Scholar] [CrossRef]
- Bagherzadeh, M.; Zare, M.; Amani, V.; Ellern, A.; Woo, L.K. Dioxo and oxo-peroxo molybdenum (VI) complexes bearing salicylidene 2-picoloyl hydrazone: Structures and catalytic performances. Polyhedron 2013, 53, 223–229. [Google Scholar] [CrossRef]
- Vrdoljak, V.; Prugovečki, B.; Matković-Čalogović, D.; Pisk, J.; Dreos, R.; Siega, P. Supramolecular Hexagon and Chain Coordination Polymer Containing the MoO22+ Core: Structural Transformation in the Solid State. Cryst. Growth Des. 2011, 11, 1244–1252. [Google Scholar] [CrossRef]
- Vrdoljak, V.; Prugovečki, B.; Matković-Čalogović, D.; Hrenar, T.; Dreos, R.; Siega, P. Three Polymorphic Forms of a Monomeric Mo(VI) Complex: Building Blocks for Two Metal–Organic Supramolecular Isomers. Intermolecular Interactions and Ligand Substituent Effects. Cryst. Growth Des. 2013, 13, 3773–3784. [Google Scholar] [CrossRef]
- Sinha, S.; Chakraborty, M.; Pramanik, N.R.; Raychaudhuri, T.K.; Mondal, T.K.; Sarkar, D.; Drew, M.G.B.; Ghosh, S.; Mandal, S.S. Dimer Formation by Symbiotic Donor–Acceptor Interaction Between Two Molecules of a Specially Designed Dioxomolybdenum(VI) Complex Containing Both Donor and Acceptor Centers—A Structural, Spectroscopic and DFT Study. Polyhedron 2013, 55, 192–200. [Google Scholar] [CrossRef]
- McCormick, L.J.; Abrahams, B.F.; Boughton, B.A.; Grannas, M.J.; Hudson, T.A.; Robson, R. Synthesis, Structure and Cation-Binding Properties of Some [4 + 4] Metallocyclic MO22+ (M = Mo or W) Derivatives of 9-Phenyl-2,3,7-trihydroxyfluor-6-one. Inorg. Chem. 2014, 53, 1721–1728. [Google Scholar] [CrossRef]
- Bikas, R.; Lippolis, V.; Noshiranzadeh, N.; Farzaneh-Bonab, H.; Blake, A.J.; Siczek, M.; Hosseini-Monfared, H.; Lis, T. Electronic Effects of Aromatic Rings on the Catalytic Activity of Dioxidomolybdenum(VI)–Hydrazone Complexes. Eur. J. Inorg. Chem. 2017, 2017, 999–1006. [Google Scholar] [CrossRef]
- Maurya, M.R.; Rana, L.; Avecilla, F. Catalytic Oxidation of Internal and Terminal Alkenes by Oxidoperoxidomolybdenum(VI) and Dioxidomolybdenum(VI) Complexes. Inorg. Chim. Acta 2015, 429, 138–147. [Google Scholar] [CrossRef]
- Biswal, D.; Pramanik, N.R.; Chakrabati, S.; Drew, M.G.B.; Sarkar, B.; Maurya, M.R.; Mukherjee, S.K.; Chowdhury, P. New Polymeric, Dimeric and Mononuclear Dioxidomolybdenum(VI) Complexes with an ONO Donor Ligand: Crystal Structures, DFT Calculations, Catalytic Performance and Protein Binding study of the Ligand. New J. Chem. 2017, 41, 4116–4137. [Google Scholar] [CrossRef]
- Pisk, J.; Rubčić, M.; Kuzman, D.; Cindrić, M.; Agustin, D.; Vrdoljak, V. Molybdenum(VI) complexes of hemilabile aroylhydrazone ligands as efficient catalysts for greener cyclooctene epoxidation: An experimental and theoretical approach. New J. Chem. 2019, 43, 5531–5542. [Google Scholar] [CrossRef]
- Cvijanović, D.; Pisk, J.; Pavlović, G.; Šišak-Jung, D.; Matković-Čalogović, D.; Cindrić, M.; Agustin, D.; Vrdoljak, V. Discrete Mononuclear and Dinuclear Compounds Containing a MoO22+ Core and 4-Aminobenzhydrazone Ligands: Synthesis, Structure and Organic-Solvent-Free Epoxidation Activity. New J. Chem. 2019, 43, 1791–1802. [Google Scholar] [CrossRef]
- Pisk, J.; Agustin, D.; Vrdoljak, V. Tetranuclear molybdenum(VI) hydrazonato epoxidation (pre)catalysts: Is water always the best choice? Catal. Commun. 2020, 142, 106027. [Google Scholar] [CrossRef]
- Morlot, J.; Uyttebroeck, N.; Agustin, D.; Poli, R. Solvent-Free Epoxidation of Olefins Catalyzed by “[MoO2(SAP)]”: A New Mode of tert-Butylhydroperoxide Activation. ChemCatChem 2013, 5, 601–611. [Google Scholar] [CrossRef]
- Prasanna, M.K.; Sithambaresan, M.; Pradeepkumar, K.; Kurup, M.R.P. N′-[(E)-2-Hydroxy-5-methoxybenzyl idene]pyridine-4-carbohydrazide monohydrate. Acta Cryst. 2013, E69, o881. [Google Scholar] [CrossRef]
- Kargar, H.; Kia, R.; Akkurt, M.; Buyukgungor, O. 5-Chloro-2-hydroxy benzaldehyde thiosemicarbazone. Acta Cryst. 2010, E66, o2982. [Google Scholar] [CrossRef]
- Krämer, R.; Lehn, J.M.; Marquis-Rigault, A. Self-recognition in helicate self-assembly: Spontaneous formation of helical metal complexes from mixtures of ligands and metal ions. Proc. Natl. Acad. Sci. USA 1993, 90, 5394–5398. [Google Scholar] [CrossRef]
- Xu, W.-X.; Yuan, Y.-M.; Li, W.-H. Syntheses, crystal structures, and catalysis by polymeric dioxomolybdenum(VI) complexes with similar (iso)nicotinohydrazones. J. Coord. Chem. 2013, 66, 2726–2735. [Google Scholar] [CrossRef]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P. The Cambridge Structural Database. Acta Crystallogr. Sect. B Struct. Sci. 2016, 72, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Benkhäuser, C.; Lützen, A. Self-assembly of heteroleptic dinuclear metallosupramolecular kites from multivalent ligands via social self-sorting. Beilstein J. Org. Chem. 2015, 11, 693–700. [Google Scholar] [CrossRef]
- Pisk, J.; Hrenar, T.; Rubčić, M.; Pavlović, G.; Damjanović, V.; Lovrić, J.; Cindrić, M.; Vrdoljak, V. Comparative studies on conventional and solvent free synthesis toward hydrazones: Application of PXRD and chemometric data analysis in mechanochemical reaction monitoring. CrystEngComm 2018, 20, 1804–1817. [Google Scholar] [CrossRef]
- Heydaria, N.; Bikas, R.; Shateriana, M.; Lis, T. Green solvent free epoxidation of olefins by a heterogenised hydrazone-dioxidotungsten(VI) coordination compound. RSC Adv. 2022, 12, 4813–4827. [Google Scholar] [CrossRef]
- Heydaria, N.; Bikas, R.; Shateriana, M.; Lis, T. Selective oxidation of benzyl alcohols by silica-supported heterogeneous catalyst containing dioxidotungsten(VI) core. Appl. Organomet. Chem. 2023, 37, e6939. [Google Scholar] [CrossRef]
- Vrdoljak, V.; Mandarić, M.; Hrenar, T.; Đilović, I.; Pisk, J.; Pavlović, G.; Cindrić, M.; Agustin, D. Geometrically constrained molybdenum(VI) metallosupramolecular architectures: Conventional Synthesis versus vapor and thermally induced solid-state structural transformations. Cryst. Growth. Des. 2019, 19, 3000–3011. [Google Scholar] [CrossRef]
- Bafti, A.; Razum, M.; Topić, E.; Agustin, D.; Pisk, J.; Vrdoljak, V. Implication of oxidant activation on olefin epoxidation catalysed by Molybdenum catalysts with aroylhydrazonato ligands: Experimental and theoretical studies. Mol. Catal. 2021, 512, 111764. [Google Scholar] [CrossRef]
- Chen, J.-J.; McDonald, J.W.; Newton, W.E. Synthesis of molybdenum(IV) and molybdenum(V) complexes using oxo abstraction by phosphines. Mechanistic implications. Inorg. Chem. 1976, 15, 2612–2615. [Google Scholar] [CrossRef]
- Degen, T.; Sadki, M.; Bron, E.; König, U.; Nénert, G. The High Score suite. Powder Diffr. 2014, 29, S13–S18. [Google Scholar] [CrossRef]
- CrysAlisPro. Software System, Version 1.171.43.90; Rigaku Oxford Diffraction: Abingdon, UK, 2023.
- Sheldrick, G.M. SHELXT–Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal Structure Refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A Complete Structure Solution, Refinement and Analysis Program. Journal of Applied Crystallography. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 16, Revision B.01; Gaussian, Inc.: Wallingford, CT, USA, 2013. [Google Scholar]
- Becke, D. Density–functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.T.; Yang, W.T.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B Condens. Matter Mater. Phys. 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Miehlich, B.; Savin, A.; Stoll, H.; Preuss, H. Results obtained with the correlation energy density functionals of becke and Lee, Yang and Parr. Chem. Phys. Lett. 1989, 157, 200–206. [Google Scholar] [CrossRef]
- Ditchfield, R.; Hehre, W.J.; Pople, J.A. Self–Consistent Molecular–Orbital Methods. IX. An Extended Gaussian-Type Basis for Molecular-Orbital Studies of Organic Molecules. J. Chem. Phys. 1971, 54, 724–728. [Google Scholar] [CrossRef]
- Hehre, W.; Ditchfield, R.; Pople, J. Self—Consistent Molecular Orbital Methods. XII. Further Extensions of Gaussian—Type Basis Sets for Use in Molecular Orbital Studies of Organic Molecules. J. Chem. Phys. 1972, 56, 2257–2261. [Google Scholar] [CrossRef]
- Hariharan, P.C.; Pople, J. The influence of polarization functions on molecular orbital hydrogenation energies. Theor. Chim. Acta 1973, 28, 213–222. [Google Scholar] [CrossRef]
- Hariharan, P.C.; Pople, J.A. Accuracy of AHn equilibrium geometries by single determinant molecular orbital theory. Mol. Phys. 1974, 27, 209–214. [Google Scholar] [CrossRef]
- Stevens, W.J.; Basc, H.; Krauss, M. Compact effective potentials and efficient shared–exponent basis sets for the first- and second-row atoms. J. Chem. Phys. 1984, 81, 6026–6033. [Google Scholar] [CrossRef]
- Stevens, W.J.; Krauss, M.; Basch, H.; Jasien, P.G. Relativistic compact effective potentials and efficient, shared-exponent basis sets for the third-, fourth-, and fifth-row atoms. Can. J. Chem. 1992, 70, 612–630. [Google Scholar] [CrossRef]

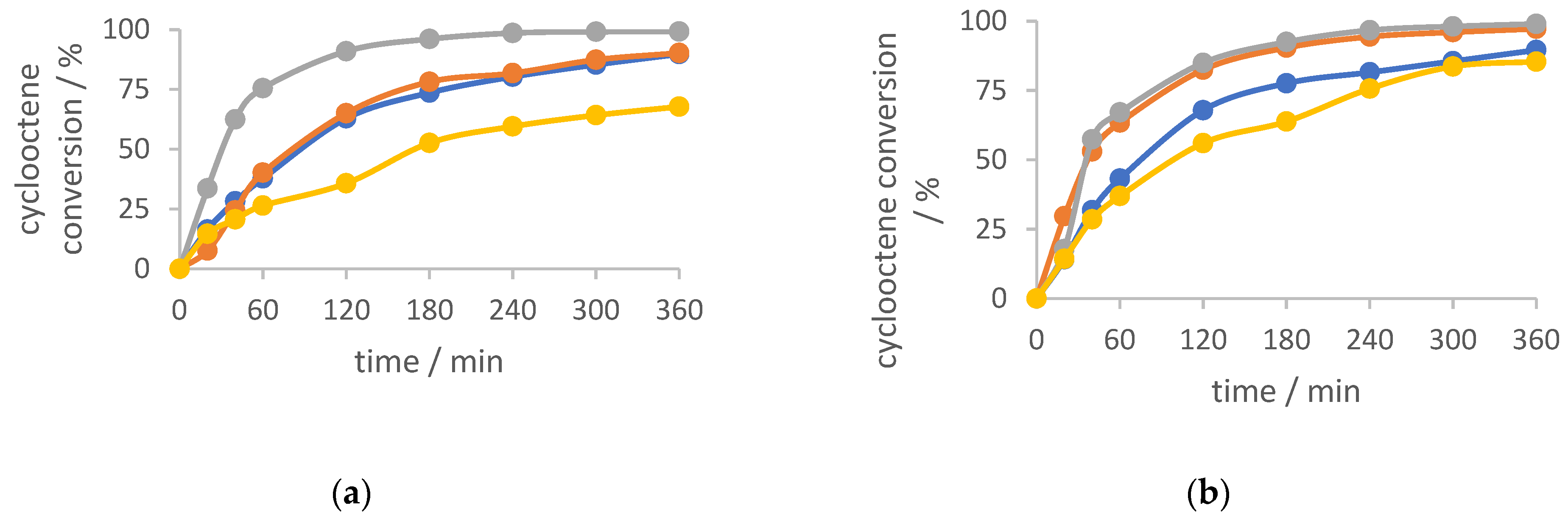

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | Con. a/% | Sel. b/% | TON c | TOF20min d |
|---|---|---|---|---|
| 1 | 89 | 78 | 348 | 191 |
| 2 | 90 | 84 | 360 | 91 |
| 3 | 99 | 85 | 399 | 415 |
| 4 | 67 | 90 | 180 | 279 |
| 1a | 89 | 89 | 359 | 350 |
| 2a | 97 | 85 | 389 | 356 |
| 3a | 99 | 92 | 395 | 214 |
| 4a | 85 | 98 | 341 | 191 |
| Catalyst | Con./% | Sel./% | TON | TOF20min | Ref. |
|---|---|---|---|---|---|
| [MoO2(L2′)]n | 27 | 56 | 113 | 72 | [46] |
| [MoO2(L2″)]n | 49 | 67 | 192 | 119 | [46] |
| ][MoO2(L3′)]2 | 90 | 92 | 372 | 343 | [33] |
| [MoO2(L3″)(MeOH)] | 85 | 90 | 349 | 383 | [33] |
| [MoO2(L3″)]2 | 87 | 91 | 330 | 345 | [33] |
| [MoO2(L4′)(MeOH)] | 56 | 86 | 229 | 75 | [34] |
| [MoO2(L4″)(MeOH)] | 63 | 85 | 257 | 97 | [34] |
| [MoO2(L4′)]2 | 84 | 90 | 380 | 295 | [34] |
| [MoO2(L4″)]2 | 89 | 86 | 386 | 298 | [34] |
| Absolute Enthalpy (Relative) Values | |||||
|---|---|---|---|---|---|
| Steps | [MoO2(L1)] | [MoO2(L2)] | [MoO2(L3)] | [MoO2(L4)] | |
| (0) | Dimer decoordination | n.c. | n.c. | 5.9 | 5.5 |
| (0) | MeOH decoordination | 10.6 | 11.1 | 10.8 | 10.3 |
| (I) | TBHP coordination | −8.7 | −8.7 | −8.4 | −8.9 |
| (II) | TS barrier | 23.5 | 23.2 | 23.8 | 24.6 |
| (III) | Release of epoxide | −51.3 | −51.1 | −51.4 | −51.7 |
| (IV) | Regeneration of catalyst | −4.9 | −4.9 | −5.4 | −5.4 |
| [MoO2L(D)] | |||||
|---|---|---|---|---|---|
| D | MeOH | no | TBHP | TS | |
| Interatomic distances (Å) | |||||
| [Mo–Oβ] | L1 | 2.552 | - | 2.990 | 2.394 |
| L2 | 2.499 | - | 2.993 | 2.397 | |
| L3 | 2.504 | - | 3.018 | 2.412 | |
| L4 | 2.509 | - | 3.041 | 2.428 | |
| N1–Mo | L1 | 2.283 | 2.274 | 2.268 | 2.278 |
| L2 | 2.296 | 2.274 | 2.269 | 2.278 | |
| L3 | 2.293 | 2.273 | 2.268 | 2.275 | |
| L4 | 2.294 | 2.275 | 2.265 | 2.273 | |
| Mo=O1 (±plane) | L1 | 1.709 | 1.697 | 1.714 | 1.733 |
| L2 | 1.705 | 1.697 | 1.714 | 1.733 | |
| L3 | 1.706 | 1.698 | 1.715 | 1.734 | |
| L4 | 1.708 | 1.699 | 1.716 | 1.735 | |
| Mo–O2 (⊥plane) | L1 | 1.694 | 1.696 | 1.691 | 1.693 |
| L2 | 1.695 | 1.696 | 1.691 | 1.693 | |
| L3 | 1.696 | 1.697 | 1.692 | 1.693 | |
| L4 | 1.696 | 1.697 | 1.693 | 1.695 | |
| Interatomic angle (absolute values in °) | |||||
| OCNO Dihedral angle | L1 | 9.99 | 6.16 | 11.05 | 15.69 |
| L2 | 10.36 | 6.28 | 11.18 | 17.10 | |
| L3 | 10.45 | 6.05 | 11.99 | 16.39 | |
| L4 | 10.63 | 6.10 | 12.19 | 15.71 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mandarić, M.; Topić, E.; Agustin, D.; Pisk, J.; Vrdoljak, V. Preparative and Catalytic Properties of MoVI Mononuclear and Metallosupramolecular Coordination Assemblies Bearing Hydrazonato Ligands. Int. J. Mol. Sci. 2024, 25, 1503. https://doi.org/10.3390/ijms25031503
Mandarić M, Topić E, Agustin D, Pisk J, Vrdoljak V. Preparative and Catalytic Properties of MoVI Mononuclear and Metallosupramolecular Coordination Assemblies Bearing Hydrazonato Ligands. International Journal of Molecular Sciences. 2024; 25(3):1503. https://doi.org/10.3390/ijms25031503
Chicago/Turabian StyleMandarić, Mirna, Edi Topić, Dominique Agustin, Jana Pisk, and Višnja Vrdoljak. 2024. "Preparative and Catalytic Properties of MoVI Mononuclear and Metallosupramolecular Coordination Assemblies Bearing Hydrazonato Ligands" International Journal of Molecular Sciences 25, no. 3: 1503. https://doi.org/10.3390/ijms25031503
APA StyleMandarić, M., Topić, E., Agustin, D., Pisk, J., & Vrdoljak, V. (2024). Preparative and Catalytic Properties of MoVI Mononuclear and Metallosupramolecular Coordination Assemblies Bearing Hydrazonato Ligands. International Journal of Molecular Sciences, 25(3), 1503. https://doi.org/10.3390/ijms25031503