
Searching for EGF Fragments Recreating the Outer Sphere of the Growth Factor Involved in Receptor Interactions

 ,
,
Abstract
1. Introduction
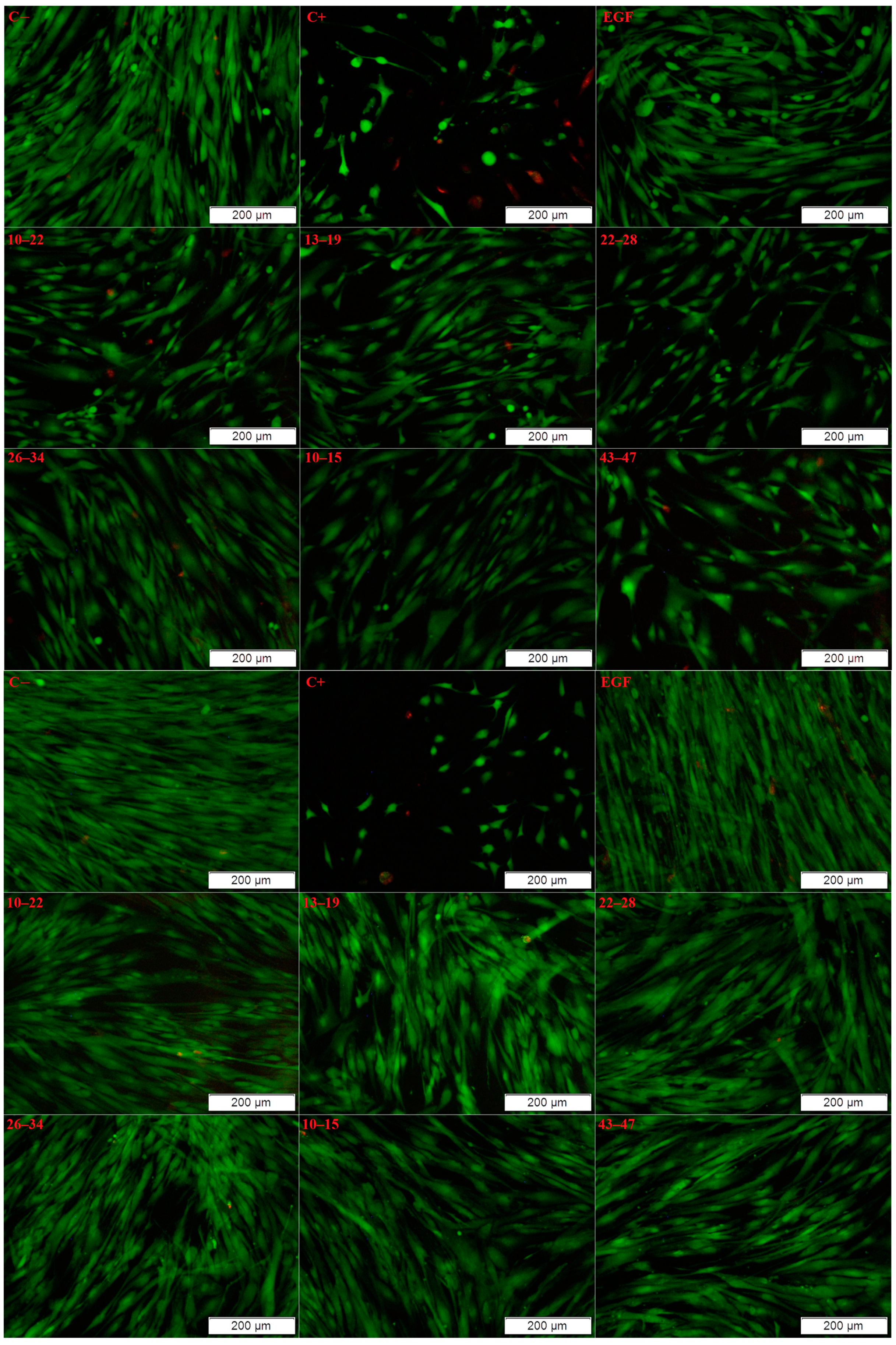
2. Results and Discussion
3. Materials and Methods
3.1. Synthesis of a Library of Overlapping EGF Fragments Immobilized on a Cellulose Matrix
3.1.1. Immobilization of 2,4-Dichloro-6-methoxy-1,3,5-triazine on Cellulose
3.1.2. Incorporation of Fmoc-Gly-OH on Cellulose as a Fragment of Triazine Linker
3.1.3. Synthesis of Decapeptides Using Modified SPOT Technique
3.1.4. Cleavage of Protecting Groups
3.2. Dot Blot Test
3.3. SPPS Synthesis of Selected EGF Fragments
3.3.1. Loading of First Amino Acid
3.3.2. Peptide Synthesis
3.3.3. Peptide Cleavage from Resin
- Synthesis of 10HDGYCLHDGVCMY22
- Synthesis of 13YCLHDGV19
- Synthesis of 22YIEALDK28
- Synthesis of 23IEAL26
- Synthesis of 34VVGYIGERCQYRDL47
- Synthesis of 38IGERCQ43
- Synthesis of 26LDKYACNCV34
- Synthesis of 10HDGYCL15
- Synthesis of 10HDGYCL15+29Y+41R
- Synthesis of 43QYRDL47
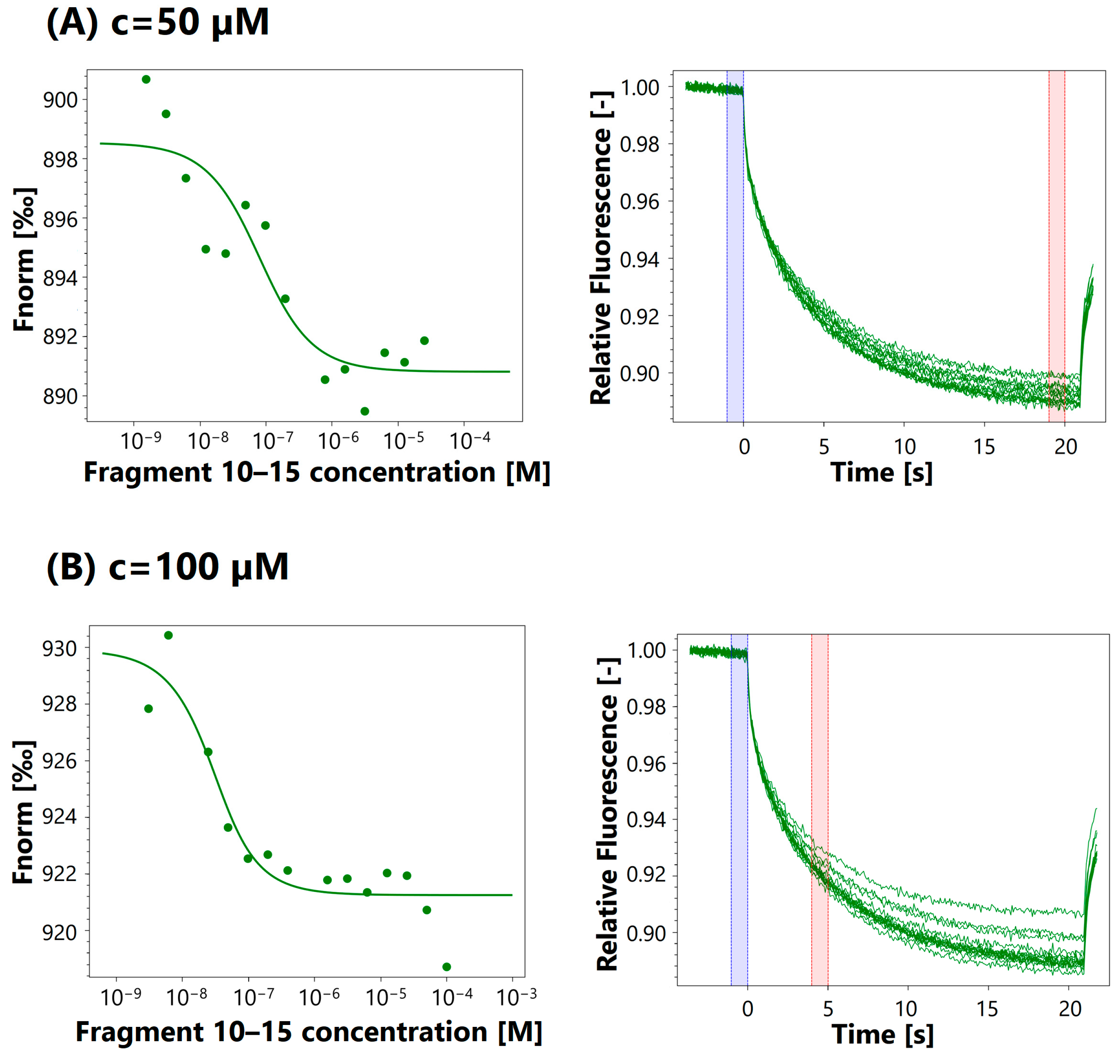
3.4. MST Analysis
3.4.1. Protein Labeling
3.4.2. Binding Assay
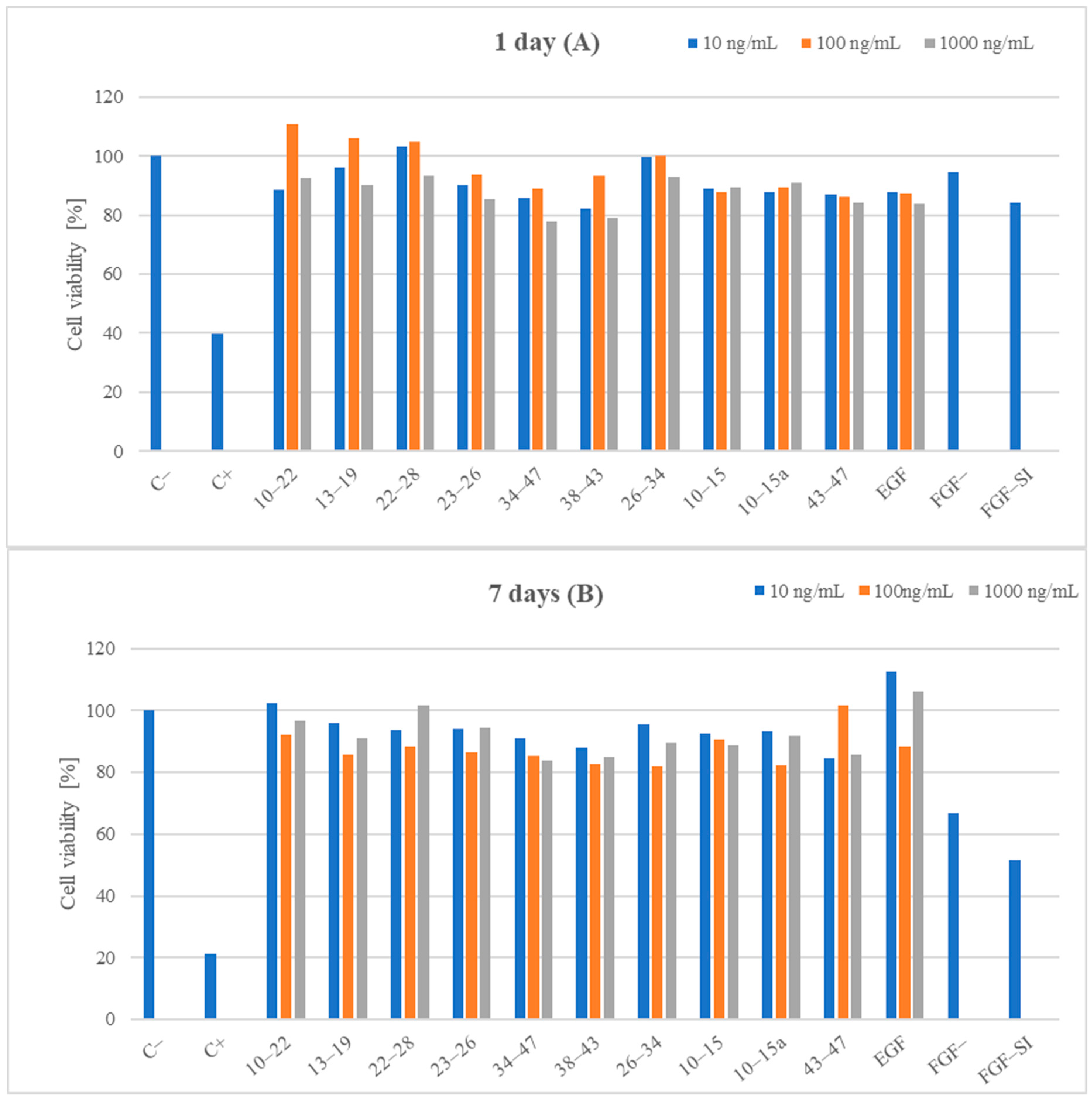
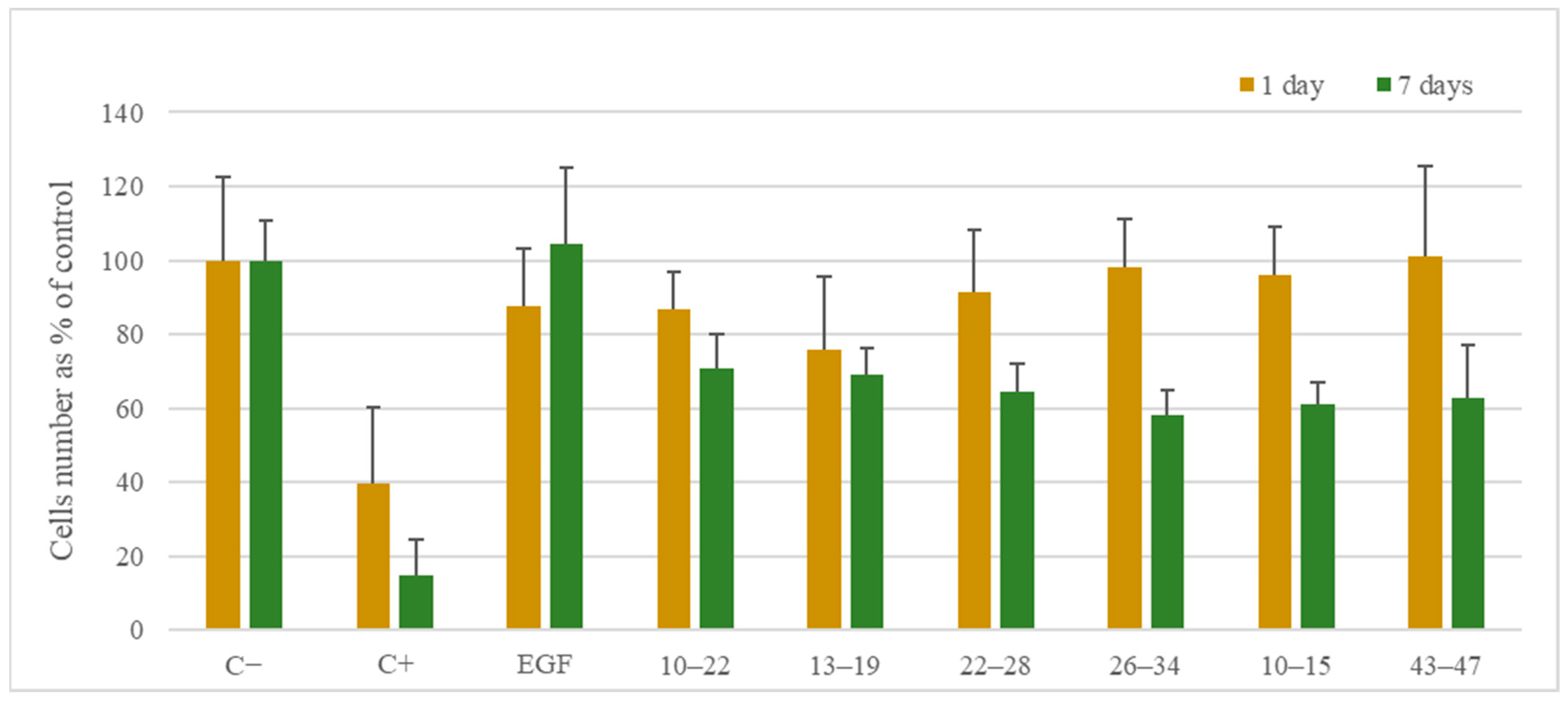
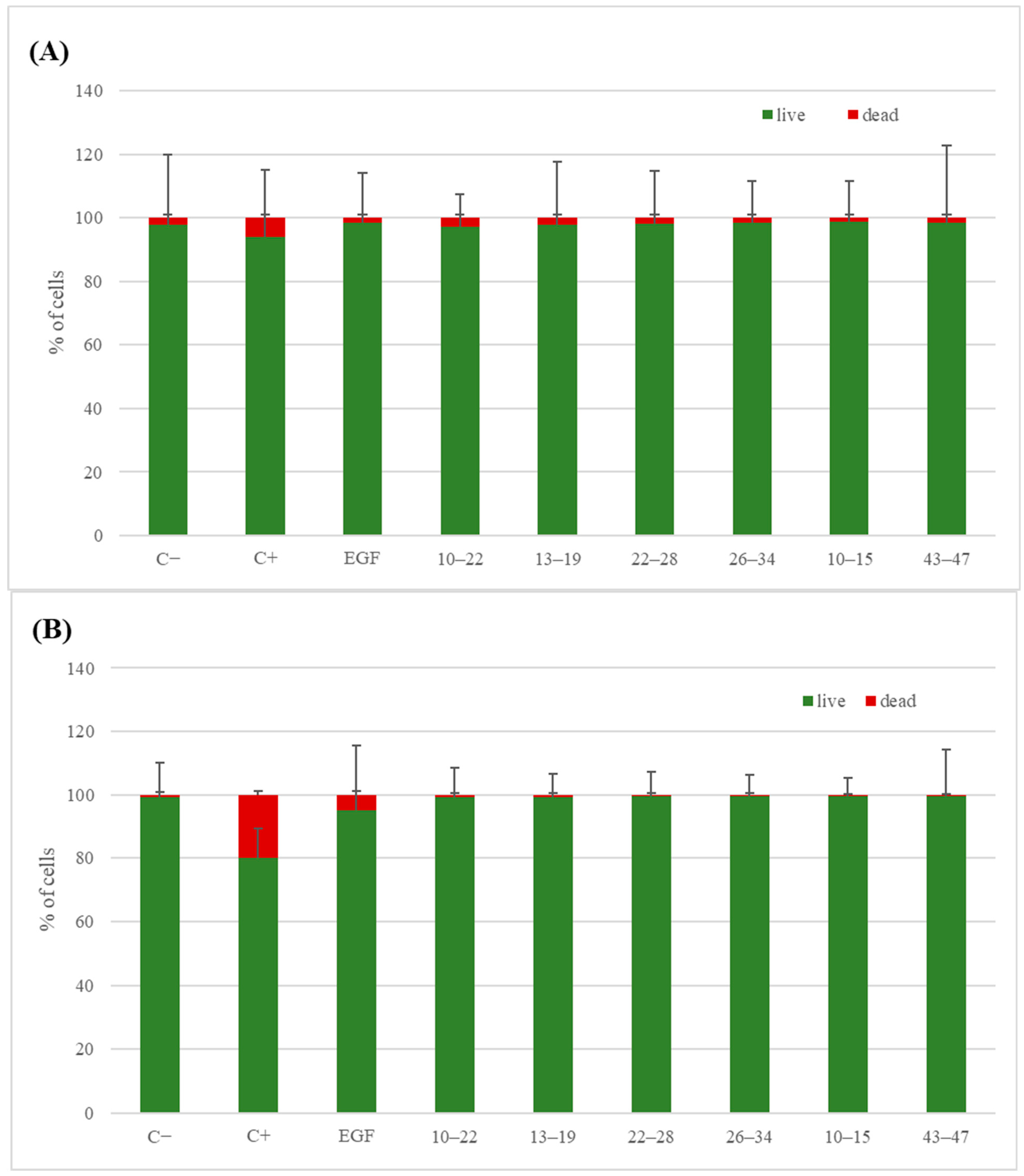
3.5. Biological Studies
3.5.1. Cell Viability
3.5.2. Live/Dead Test
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Giurgiu, M.; Reinhard, J.; Brauner, B.; Dunger-Kaltenbach, I.; Fobo, G.; Frishman, G.; Montrone, C.; Ruepp, A. CORUM: The comprehensive resource of mammalian protein complexes. Nucleic Acids Res. 2019, 47, D559–D563. [Google Scholar] [CrossRef]
- Ruepp, A.; Waegele, B.; Lechner, M.; Brauner, B.; Dunger-Kaltenbach, I.; Fobo, G.; Frishman, G.; Montrone, C.; Mewes, H.W. CORUM: The comprehensive resource of mammalian protein complexes. Nucleic Acids Res. 2010, 38, D497–D501. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Morris, J.H.; Cook, H.; Kuhn, M.; Wyder, S.; Simonovic, M.; Santos, A.; Doncheva, N.T.; Roth, A.; Bork, P.; et al. The STRING database in 2017: Quality-controlled proteinprotein association networks, made broadly accessible. Nucleic Acids Res. 2017, 45, D362–D368. [Google Scholar] [CrossRef]
- Martino, E.; Chiarugi, S.; Margheriti, F.; Garau, G. Mapping, Structure and Modulation of PPI. Front. Chem. 2021, 9, 718405. [Google Scholar] [CrossRef]
- Vidal, M.; Cusick, M.E.; Barabási, A.-L. Interactome Networks and Human Disease. Cell 2011, 144, 986–998. [Google Scholar] [CrossRef] [PubMed]
- Maniaci, C.; Ciulli, A. Bifunctional Chemical Probes Inducing Protein–Protein Interactions. Curr. Opin. Chem. Biol. 2019, 52, 145–156. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Zhou, Q.; He, J.; Jiang, Z.; Peng, C.; Tong, R.; Shi, J. Recent Advances in the Development of Protein–Protein Interactions Modulators: Mechanisms and Clinical Trials. Sig. Transduct. Target. Ther. 2020, 5, 213. [Google Scholar] [CrossRef] [PubMed]
- Huttlin, E.L.; Bruckner, R.J.; Navarrete-Perea, J.; Cannon, J.R.; Baltier, K.; Gebreab, F.; Gygi, M.P.; Thornock, A.; Zarraga, G.; Tam, S.; et al. Dual Proteome-Scale Networks Reveal Cell-Specific Remodeling of the Human Interactome. Cell 2021, 184, 3022–3040. [Google Scholar] [CrossRef] [PubMed]
- Ringe, D. What Makes a Binding Site a Binding Site. Curr. Opin. Struct. Biol. 1995, 5, 825–829. [Google Scholar] [CrossRef]
- Rajamani, D.; Thiel, S.; Vajda, S.; Camacho, C.J. Anchor Residues in Protein–Protein Interactions. Proc. Natl. Acad. Sci. USA 2004, 101, 11287–11292. [Google Scholar] [CrossRef]
- Ran, X.; Gestwicki, J.E. Inhibitors of Protein–Protein Interactions (PPIs): An Analysis of Scaffold Choices and Buried Surface Area. Curr. Opin. Chem. Biol. 2018, 44, 75–86. [Google Scholar] [CrossRef]
- Nooren, I.M.A.; Thornton, J.M. New Embo Member’s Review: Diversity of Protein–Protein Interactions. EMBO J. 2003, 22, 3486–3492. [Google Scholar] [CrossRef] [PubMed]
- Maurer, M.S.; Schwartz, J.H.; Gundapaneni, B.; Elliott, P.M.; Merlini, G.; Waddington-Cruz, M.; Kristen, A.V.; Grogan, M.; Witteles, R.; Damy, T.; et al. Tafamidis Treatment for Patients with Transthyretin Amyloid Cardiomyopathy. N. Engl. J. Med. 2018, 379, 1007–1016. [Google Scholar] [CrossRef] [PubMed]
- Speer, S.L.; Zheng, W.; Jiang, X.; Chu, I.-T.; Guseman, A.J.; Liu, M.; Pielak, G.J.; Li, C. The Intracellular Environment Affects Protein–Protein Interactions. Proc. Natl. Acad. Sci. USA 2021, 118, e2019918118. [Google Scholar] [CrossRef] [PubMed]
- Perkins, J.R.; Diboun, I.; Dessailly, B.H.; Lees, J.G.; Orengo, C. Transient Protein–Protein Interactions: Structural, Functional, and Network Properties. Structure 2010, 18, 1233–1243. [Google Scholar] [CrossRef] [PubMed]
- Marcotte, E.M.; Pellegrini, M.; Yeates, T.O.; Eisenberg, D. A census of protein repeats. J. Mol. Biol. 1999, 293, 151–160. [Google Scholar] [CrossRef]
- Headd, J.J.; Ban, Y.E.A.; Brown, P.; Edelsbrunner, H.; Vaidya, M.; Rudolph, J. Protein—Protein Interfaces: Properties, Preferences, and Projections research articles. J. Proteome Res. 2007, 6, 2576–2586. [Google Scholar] [CrossRef]
- Ngounou Wetie, A.G.; Sokolowska, I.; Woods, A.G.; Roy, U.; Loo, J.A.; Darie, C.C. Investigation of stable and transient protein–protein interactions: Past, present, and future. Proteomics 2013, 13, 538–557. [Google Scholar] [CrossRef]
- Wu, Y.; Li, Q.; Chen, X.Z. Detecting protein–protein interactions by far western blotting. Nat. Protoc. 2007, 2, 3278–3284. [Google Scholar] [CrossRef]
- Rao, V.S.; Srinivas, K.; Sujini, G.N.; Kumar, G.N.S. Protein–Protein Interaction Detection: Methods and Analysis. Int. J. Proteom. 2014, 2014, 1–12. [Google Scholar] [CrossRef]
- Lee, W.C.; Lee, K.H. Applications of affinity chromatography in proteomics. Anal. Biochem. 2004, 324, 1–10. [Google Scholar] [CrossRef]
- Uetz, P.; Giot, L.; Cagney, G.; Mansfield, T.A.; Judson, R.S.; Knight, J.R.; Lockshon, D.; Narayan, V.; Srinivasan, M.; Pochart, P.; et al. A comprehensive analysis of protein–protein interactions in Saccharomyces cerevisiae. Nature 2000, 403, 623–627. [Google Scholar] [CrossRef]
- Dong, M.; Yang, L.L.; Williams, K.; Fisher, S.J.; Hall, S.C.; Biggin, M.D.; Jin, J.; Witkowska, H.E. A “tagless” strategy for identification of stable protein complexes genome-wide by multidimensional orthogonal chromatographic separation and iTRAQ reagent tracking. J. Proteome Res. 2008, 7, 1836–1849. [Google Scholar] [CrossRef] [PubMed]
- Huttlin, E.L.; Ting, L.; Bruckner, R.J.; Gebreab, F.; Gygi, M.P.; Szpyt, J.; Tam, S.; Zarraga, G.; Colby, G.; Baltier, K.; et al. The BioPlex network: A systematic exploration of the human interactome. Cell 2015, 162, 425–440. [Google Scholar] [CrossRef] [PubMed]
- Zuiderweg, E.R.P. Mapping protein–protein interactions in solution by NMR spectroscopy. Biochemistry 2002, 41, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Vaynberg, J.; Qin, J. Weak protein–protein interactions as probed by NMR spectroscopy. Trends Biotechnol. 2006, 24, 22–27. [Google Scholar] [CrossRef] [PubMed]
- Hanlon, A.D.; Larkin, M.I.; Reddick, R.M. Free-solution, label-free protein–protein interactions characterized by dynamic light scattering. Biophys. J. 2010, 98, 297–304. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Piszczek, G. Measuring the affinity of protein–protein interactions on a single-molecule level by mass photometry. Anal. Biochem. 2020, 592, 113575. [Google Scholar] [CrossRef] [PubMed]
- Kopec, J.; Schneider, G. Comparison of fluorescence and light scattering based methods to assess formation and stability of protein–protein complexes. J. Struct. Biol. 2011, 175, 216–223. [Google Scholar] [CrossRef]
- Karlssonz, R.; Fält, A. Experimental design for kinetic analysis of protein–protein interactions with surface plasmon resonance biosensors. J. Immunol. Methods 1997, 200, 121–133. [Google Scholar] [CrossRef]
- Lopez, A.J.; Quoika, P.K.; Linke, M.; Hummer, G. Quantifying protein–protein interactions in molecular simulations. Chemrxiv 2019, 2019, 11421201. [Google Scholar]
- Jerabek-Willemsen, M.; André, T.; Wanner, R.; Roth, H.M.; Duhr, S.; Baaske, P.; Breitsprecher, D. MicroScale Thermophoresis: Interaction analysis and beyond. J. Mol. Struct. 2014, 1077, 101–113. [Google Scholar] [CrossRef]
- Dunham, W.H.; Mullin, M.; Gingras, A.C. Affinity-purification coupled to mass spectrometry: Basic principles and strategies. Proteomics 2012, 12, 1576–1590. [Google Scholar] [CrossRef] [PubMed]
- Roux, K.J.; Kim, D.I.; Raida, M.; Burke, B. A promiscuous biotin ligase fusion protein identifies proximal and interacting proteins in mammalian cells. J. Cell Biol. 2012, 196, 801–810. [Google Scholar] [CrossRef] [PubMed]
- Branon, T.C.; Bosch, J.A.; Sanchez, A.D.; Udeshi, N.D.; Svinkina, T.; Carr, S.A.; Feldman, J.L.; Perrimon, N.; Ting, A.Y. Efficient proximity labeling in living cells and organisms with TurboID. Nat. Biotechnol. 2018, 36, 880–887. [Google Scholar] [CrossRef] [PubMed]
- Miura, K. An Overview of Current Methods to Confirm Protein- Protein Interactions. Protein Pept. Lett. 2018, 25, 728–733. [Google Scholar] [CrossRef] [PubMed]
- Figeys, D.; McBroom, L.D.; Moran, M.F. Mass spectrometry for the study of protein–protein interactions. Methods 2001, 24, 230–239. [Google Scholar] [CrossRef]
- Ivanov, A.S.; Zgoda, V.G.; Archakov, A.I. Technologies of Protein Interactomics: A Review. Russ. J. Bioorgan. Chem. 2011, 37, 4–16. [Google Scholar] [CrossRef]
- Holguin-Cruz, J.A.; Foster, L.J.; Gsponer, J. Where protein structure and cell diversity meet. Trends Cell Biol. 2022, 32, 996–1007. [Google Scholar] [CrossRef]
- Dittmar, G.; Hernandez, D.P.; Kowenz-Leutz, E.; Kirchner, M.; Kahlert, G.; Wesolowski, R.; Baum, K.; Knoblich, M.; Hofstätter, M.; Muller, A.; et al. PRISMA: Protein interaction screen on peptide matrix reveals interaction footprints and modifications- dependent interactome of intrinsically disordered C/EBPβ. iScience 2019, 13, 351–370. [Google Scholar] [CrossRef]
- Meyer, K.; Kirchner, M.; Uyar, B.; Cheng, J.-Y.; Russo, G.; Hernandez-Miranda, L.R.; Szymborska, A.; Zauber, H.; Rudolph, I.-M.; Willnow, T.E.; et al. Mutations in disordered regions can cause disease by creating dileucine motifs. Cell 2018, 175, 239–253. [Google Scholar] [CrossRef]
- Vermeulen, M. Identifying chromatin readers using a SILAC-based histone peptide pull-down approach. Methods Enzymol. 2012, 512, 137–160. [Google Scholar]
- Eberl, H.C.; Spruijt, C.G.; Kelstrup, C.D.; Vermeulen, M.; Mann, M. A map of general and specialized chromatin readers in mouse tissues generated by label-free interaction proteomics. Mol. Cell 2013, 49, 368–378. [Google Scholar] [CrossRef]
- Vermeulen, M.; Mulder, K.W.; Denissov, S.; Pijnappel, W.W.M.P.; van Schaik, F.M.A.; Varier, R.A.; Baltissen, M.P.A.; Stunnenberg, H.G.; Mann, M.; Timmers, H.T.M. Selective anchoring of TFIID to nucleosomes by trimethylation of histone H3 lysine 4. Cell 2007, 131, 58–69. [Google Scholar] [CrossRef]
- Rivas, J.D.L.; De Las, R.J.; Fontanillo, C. Protein–protein interactions essentials: Key concepts to building and analyzing interactome networks. PLoS Comput. Biol. 2010, 6, e1000807. [Google Scholar]
- Mann, M.; Jensen, O.N. Proteomic analysis of post-translational modifications. Nat. Biotechnol. 2003, 21, 255–261. [Google Scholar] [CrossRef] [PubMed]
- Seo, J.; Lee, K.-J. Post-translational modifications and their biological functions: Proteomic analysis and systematic approaches. J. Biochem. Mol. Biol. 2004, 37, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Yakubu, R.R.; Nieves, E.; Weiss, L.M. The methods employed in mass spectrometric analysis of posttranslational modifications (PTMs) and protein–protein interactions (PPIs). Adv. Exp. Med. Biol. 2019, 1140, 169–198. [Google Scholar] [PubMed]
- Deshaies, R.J.; Ferrell, J.E., Jr. Multisite phosphorylation and the countdown to S phase. Cell 2001, 107, 819–822. [Google Scholar] [CrossRef] [PubMed]
- Lemmon, M.A.; Ladbury, J.E.; Mandiyan, V.; Zhou, M.; Schlessinger, J. Independent binding of peptide ligands to the SH2 and SH3 domains of Grb2. J. Biol. Chem. 1994, 269, 31653–31658. [Google Scholar] [CrossRef]
- Ward, C.W.; Gough, K.H.; Rashke, M.; Wan, S.S.; Tribbick, G.; Wang, J.-X. Systematic mapping of potential binding sites for Shc and Grb2 SH2 domains on insulin receptor substrate-1 and the receptors for insulin, epidermal growth factor, platelet-derived growth factor, and fibroblast growth factor. J. Biol. Chem. 1996, 271, 5603–5609. [Google Scholar] [CrossRef] [PubMed]
- Babu, M.M.; Kriwacki, R.W.; Pappu, R.V. Structural biology. Versatility from protein disorder. Science 2012, 337, 1460–1461. [Google Scholar] [CrossRef] [PubMed]
- Forman-Kay, J.D.; Mittag, T. From sequence and forces to structure, function, and evolution of intrinsically disordered proteins. Structure 2013, 21, 1492–1499. [Google Scholar] [CrossRef] [PubMed]
- Dunker, A.K.; Babu, M.M.; Barbar, E.; Blackledge, M.; Bondos, S.E.; Dosztányi, Z.; Dyson, H.J.; Forman-Kay, J.; Fuxreiter, M.; Gsponer, J.; et al. What’s in a name? Why these proteins are intrinsically disordered: Why these proteins are intrinsically disordered. Intrinsically Disord. Proteins 2013, 1, e24157. [Google Scholar] [CrossRef]
- Dyson, H.J.; Wright, P.E. Intrinsically unstructured proteins and their functions. Nat. Rev. Mol. Cell. Biol. 2005, 6, 197–208. [Google Scholar] [CrossRef]
- Tompa, P. Intrinsically disordered proteins: A 10-year recap. Trends Biochem. Sci. 2012, 37, 509–516. [Google Scholar] [CrossRef]
- Uversky, V.N.; Dunker, A.K. Understanding protein non-folding. Biochim. Biophys. Acta 2010, 1804, 1231–1264. [Google Scholar] [CrossRef]
- van der Lee, R.; Buljan, M.; Lang, B.; Weatheritt, R.J.; Daughdrill, G.W.; Dunker, A.K.; Fuxreiter, M.; Gough, J.; Gsponer, J.; Jones, D.T.; et al. Classification of intrinsically disordered regions and proteins. Chem. Rev. 2014, 114, 6589–6631. [Google Scholar] [CrossRef]
- Zhou, J.; Oldfield, C.J.; Yan, W.; Shen, B.; Dunker, A.K. Intrinsically disordered domains: Sequence ➔ disorder ➔ function relationships. Protein Sci. 2019, 28, 1652–1663. [Google Scholar] [CrossRef] [PubMed]
- Davey, N.E.; Cowan, J.L.; Shields, D.C.; Gibson, T.J.; Coldwell, M.J.; Edwards, R.J. SLiMPrints: Conservation-based discovery of functional motif fingerprints in intrinsically disordered protein regions. Nucleic Acids Res. 2012, 40, 10628–10641. [Google Scholar] [CrossRef] [PubMed]
- Kriwacki, R.W.; Hengst, L.; Tennant, L.; Reed, S.I.; Wright, P.E. Structural studies of p21Waf1/Cip1/Sdi1 in the free and Cdk2-bound state: Conformational disorder mediates binding diversity. Proc. Natl. Acad. Sci. USA 1996, 93, 11504–11509. [Google Scholar] [CrossRef]
- Wright, P.E.; Dyson, H.J. Intrinsically unstructured proteins : Reassessing the protein structure -function paradigm. J. Mol. Biol. 1999, 293, 321–331. [Google Scholar] [CrossRef]
- Uversky, V.N.; Gillespie, J.R.; Fink, A.L. Why are “natively unfolded” proteins unstructured under physiologic conditions? Proteins: Struct. Funct. Bioinf. 2000, 41, 415–427. [Google Scholar] [CrossRef]
- Tompa, P. Intrinsically unstructured proteins. Trends Biochem. Sci. 2002, 27, 527–533. [Google Scholar] [CrossRef]
- Dunker, A.K.; Oldfield, C.J.; Meng, J.; Romero, P.; Yang, J.Y.; Walton Chen, J.; Vacic, V.; Obradovic, Z.; Uversky, V.N. The unfoldomics decade: An update on intrinsically disordered proteins. BMC Genom. 2008, 9, S1. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. Intrinsically disordered proteins and their “mysterious” (meta)physics. Front. Phys. 2019, 7, 10. [Google Scholar] [CrossRef]
- Ramberger, E.; Sapozhnikova, V.; Kowenz-Leutz, E.; Zimmermann, K.; Nicot, N.; Nazarov, P.V.; Perez-Hernandez, D.; Reimer, U.; Mertins, P.; Dittmar, G. PRISMA and BioID disclose a motifs-based interactome of the intrinsically disordered transcription factor C/EBPα. iScience 2021, 24, 102686. [Google Scholar] [CrossRef] [PubMed]
- Owen, I.; Shewmaker, F. The role of post-translational modifications in the phase transitions of intrinsically disordered proteins. Int. J. Mol. Sci. 2019, 20, 5501. [Google Scholar] [CrossRef]
- Bah, A.; Forman-Kay, J.D. Modulation of intrinsically disordered protein function by post-translational modifications. J. Biol. Chem. 2016, 291, 6696–6705. [Google Scholar] [CrossRef]
- Olsen, J.V.; Blagoev, B.; Gnad, F.; Macek, B.; Kumar, C.; Mortensen, P.; Mann, M. Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell 2006, 127, 635–648. [Google Scholar] [CrossRef]
- Kramer, A.; Schneider-Mergener, J. Synthesis and screening of peptide libraries on continuous cellulose membrane supports. Methods Mol. Biol. 1998, 87, 25–39. [Google Scholar]
- Hernandezm, D.P.; Dittmar, G. Peptide array–based interactomics. Anal. Bioanal. Chem. 2021, 413, 5561–5566. [Google Scholar] [CrossRef] [PubMed]
- Wenschuh, H.; Volkmer-Engert, R.; Schmidt, M.; Schulz, M.; Schneider-Mergener, J.; Reineke, U. Coherent membrane supports for parallel microsynthesis and screening of bioactive peptides. Biopolymers 2000, 55, 188–206. [Google Scholar] [CrossRef] [PubMed]
- Starr, T.N.; Picton, L.K.; Thornton, J.W. Alternative evolutionary histories in the sequence space of an ancient protein. Nature 2017, 549, 409–413. [Google Scholar] [CrossRef]
- Ha, J.-H.; Presti, M.F.; Loh, S.N. A single protein disruption site results in efficient reassembly by multiple engineering methods. Biophys. J. 2019, 117, 56–65. [Google Scholar] [CrossRef]
- Dwyer, M.A.; Hellinga, H.W. Periplasmic binding proteins: A versatile superfamily for protein engineering. Curr. Opin. Struct. Biol. 2004, 14, 495–504. [Google Scholar] [CrossRef]
- Weiergräber, O.; Schneider-Mergener, J.; Grötzinger, J.; Wollmer, A.; Küster, A.; Exner, M.; Heinrich, P.C. Use of immobilized synthetic peptides for the identification of contact sites between human interleukin-6 and its receptor. FEBS Lett. 1996, 379, 122–126. [Google Scholar] [CrossRef] [PubMed]
- Reineke, U.; Sabat, R.; Volk, H.-D.; Schneider-Mergener, J. Mapping of the interleukin-10/interleukin-10 receptor combining site. Protein Sci. 2008, 7, 951–960. [Google Scholar] [CrossRef]
- Lin, J.-S.; Lai, E.-M. Protein–Protein Interactions: Co-Immunoprecipitation. Methods Mol. Biol. 2017, 1615, 211–219. [Google Scholar]
- Carpenter, G.; Cohen, S. Epidermal growth factor. J. Biol. Chem. 1990, 265, 7709–7712. [Google Scholar] [CrossRef]
- Kajikawa, K.; Yasui, W.; Sumiyoshi, H.; Yoshida, K.; Nakayama, H.; Ayhan, A.; Yokozaki, H.; Ito, H.; Tahara, E. Expression of epidermal growth factor in human tissues. Immunohistochemical and biochemical analysis. Virchows Archiv. Eur. J. Pathol. 1991, 418, 27–32. [Google Scholar] [CrossRef]
- Venturi, S.; Venturi, M. Iodine in evolution of salivary glands and in oral health. Nutr. Health 2009, 20, 119–134. [Google Scholar] [CrossRef]
- Guglietta, A.; Sullivan, P.B. Clinical applications of epidermal growth factor. Eur. J. Gastroenterol. Hepatol. 1995, 7, 945–950. [Google Scholar] [CrossRef]
- Real, F.X.; Rettig, W.J.; Chesa, P.G.; Melamed, M.R.; Old, L.J.; Mendelsohn, J. Expression of Epidermal Growth Factor Receptor in Human Cultured Cells and Tissues: Relationship to Cell Lineage and Stage of Differentiation. Cancer Res. 1986, 46, 4726–4731. [Google Scholar]
- Berlanga-Acosta, J.; Gavilondo-Cowley, J.; del Barco-Herrera, D.G.; Martín-Machado, J.; Guillen-Nieto, G. Epidermal Growth Factor (EGF) and Platelet-Derived Growth Factor (PDGF) as Tissue Healing Agents: Clarifying Concerns about their Possible Role in Malignant Transformation and Tumor Progression. J. Carcinog. Mutagen. 2011, 2, 100–115. [Google Scholar] [CrossRef]
- Iwakura, Y.; Piao, Y.-S.; Mizuno, M.; Takei, N. Influences of dopaminergic lesion on epidermal growth factor-ErbB signals in Parkinson’s disease and its model: Neurotrophic implication in nigrostriatal neurons. J. Neurochem. 2005, 93, 974–983. [Google Scholar] [CrossRef] [PubMed]
- Lim, N.S.; Swanson, C.R.; Cherng, H.-R.; Unger, T.L.; Xie, S.X.; Weintraub, D.; Marek, K.; Stern, M.B.; Siderowf, A.; Trojanowski, J.Q.; et al. Plasma EGF and cognitive decline in Parkinson’s disease and Alzheimer’s disease. Ann. Clin. Transl. Neurol. 2016, 3, 346–355. [Google Scholar] [CrossRef] [PubMed]
- Kwon, Y.B.; Kim, H.W.; Roh, D.H.; Yoon, S.Y.; Baek, R.M.; Kim, J.Y.; Kweon, H.Y.; Lee, K.G.; Park, Y.H.; Lee, J.H. Topical application of epidermal growth factor accelerates wound healing by myofibroblast proliferation and collagen synthesis in rat. J. Vet. Sci. 2006, 7, 105–109. [Google Scholar] [CrossRef] [PubMed]
- Berlanga-Acosta, J.; Fernández-Montequín, J.I.; López-Mola, E.; López-Saura, P.A.; del Río-Martín, A.; Valenzuela-Silva, C.; Valdivia-Arencibia, J.; Muzio, V.; Raíces-Pérez, M.; Silva-Rodríguez, R.; et al. Heberprot-P: A novel product for treating advanced diabetic foot ulcer. MEDICC Rev. 2013, 15, 11–15. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Geng, Z.; Ma, K.; Sun, X.; Fu, X. Efficacy of Topical Recombinant Human Epidermal Growth Factor for Treatment of Diabetic Foot Ulcer: A Systematic Review and Meta-Analysis. Int. J. Low. Extrem. Wounds 2016, 15, 120–125. [Google Scholar] [CrossRef] [PubMed]
- Ogiso, H.; Ishitani, R.; Nureki, O.; Fukai, S.; Yamanaka, M.; Kim, J.-H.; Saito, K.; Sakamoto, A.; Inoue, M.; Shirouzu, M.; et al. Crystal structure of the complex of human epidermal growth factor and receptor extracellular domains. Cell 2002, 110, 775–787. [Google Scholar] [CrossRef]
- Frank, R. Spot-synthesis: An easy technique for the positionally addressable, parallel chemical synthesis on a membrane support. Tetrahedron 1992, 48, 9217–9232. [Google Scholar] [CrossRef]
- Fraczyk, J.; Walczak, M.; Kaminski, Z.J. New methodology for automated SPOT synthesis of peptides on cellulose using 1,3,5-triazine derivatives as linkers and as coupling reagents. J. Pept. Sci. 2018, 24, e3063. [Google Scholar] [CrossRef] [PubMed]
- Kolesinska, B.; Rozniakowski, K.K.; Fraczyk, J.; Relich, I.; Papini, A.M.; Kaminski, Z.J. The Effect of Counterion and Tertiary Amine on the Efficiency of N -Triazinylammonium Sulfonates in Solution and Solid-Phase Peptide Synthesis. Eur. J. Org. Chem. 2015, 2015, 401–408. [Google Scholar] [CrossRef]
- Boisguerin, P.; Leben, R.; Ay, B.; Radziwill, G.; Moelling, K.; Dong, L.; Volkmer-Engert, R. An Improved Method for the Synthesis of Cellulose Membrane-Bound Peptides with Free C Termini Is Useful for PDZ Domain Binding Studies. Chem. Biol. 2004, 11, 449–459. [Google Scholar] [CrossRef] [PubMed]
- Walczak, M.; Frączyk, J.; Kamiński, Z.; Kolesińska, B. Functionalized cellulose as a matrix for the synthesis of library of molecular receptors useful for screening of compounds with anti-histamine activity. Eng. Biomater. 2017, 20, 2–6. [Google Scholar]
- Hilpert, K.; Volkmer-Engert, R.; Walter, T.; Hancock, R.E.W. High-throughput generation of small antibacterial peptides with improved activity. Nat. Biotechnol. 2005, 23, 1008–1012. [Google Scholar] [CrossRef]
- Volkmer, R. Synthesis and application of peptide arrays: Quo vadis SPOT technology. ChemBioChem 2009, 10, 1431–1442. [Google Scholar] [CrossRef]
- Frank, R.; Overwin, H. SPOT synthesis. Ep. Mapp. Protoc. Humana Press. 1996, 66, 149–169. [Google Scholar]
- Hoffmann, S.; Frank, R. A new safety-catch peptide-resin linkage for the direct release of peptides into aqueous buffers. Tetrahedron Lett. 1994, 35, 7763–7766. [Google Scholar] [CrossRef]
- Kamiński, Z.J.; Relich, I.; Konieczna, I.; Kaca, W.; Kolesińska, B. Cross-Reactivity of Polyclonal Antibodies against Canavalia Ensiformis (Jack Bean) Urease and H. Pylori Urease Subunit A Fragments. Chem. Biodiv. 2017, 14, e1700444. [Google Scholar] [CrossRef] [PubMed]
- Björkelund, H.; Gedda, L.; Andersson, K. Comparing the Epidermal Growth Factor Interaction with Four Different Cell Lines: Intriguing Effects Imply Strong Dependency of Cellular Context. PLoS ONE 2011, 6, e16536. [Google Scholar] [CrossRef] [PubMed]
- Khavrutskii, L.; Yeh, J.; Timofeeva, O.; Tarasov, S.G.; Pritt, S.; Stefanisko, K.; Tarasova, N. Protein purification-free method of binding affinity determination by microscale thermophoresis. J. Vis. Exp. 2013, 78, e50541. [Google Scholar]
- Singh, M.; McKenzie, K.; Ma, X. Effect of dimethyl sulfoxide on in vitro proliferation of skin fibroblast cells. J. Biotech Res. 2017, 8, 78–82. [Google Scholar]
- Pieklarz, K.; Galita, G.; Tylman, M.; Maniukiewicz, W.; Kucharska, E.; Majsterek, I.; Modrzejewska, Z. Physico-Chemical Properties and Biocompatibility of Thermosensitive Chitosan Lactate and Chitosan Chloride Hydrogels Developed for Tissue Engineering Application. J. Funct. Biomater. 2021, 12, 37. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | Intensity (Staining Value) | Sequence | Position | Intensity (Staining Value) | Sequence |
|---|---|---|---|---|---|
| A1 | − (14.8) | H-1NSDSECPLSH10-matrix | B3 | +/− (63.8) | H-23IEALDKYACN32-matrix |
| A2 | − (22.3) | H-2SDSECPLSHD11-matrix | B4 | − (30.5) | H-24EALDKYACNC33-matrix |
| A3 | +/− (71.7) | H-3DSECPLSHDG12-matrix | B5 | − (29.6) | H-25ALDKYACNCV34-matrix |
| A4 | +/− (55.2) | H-4SECPLSHDGY13-matrix | B6 | +/− (46.6) | H-26LDKYACNCVV35-matrix |
| A5 | +/− (64.5) | H-5ECPLSHDGYC14-matrix | B7 | +/− (55.3) | H-27DKYACNCVVG36-matrix |
| A6 | +/− (58.4) | H-6CPLSHDGYCL15-matrix | B8 | +/− (63.5) | H-28KYACNCVVGY37-matrix |
| A7 | +/− (62.2) | H-7PLSHDGYCLH16-matrix | B9 | + (91.1) | H-29YACNCVVGYI38-matrix |
| A8 | +/− (59.3) | H-8LSHDGYCLHD17-matrix | B10 | +/− (58.5) | H-30ACNCVVGYIG39-matrix |
| A9 | +/− (63.8) | H-9SHDGYCLHDG18-matrix | B11 | +/− (60.3) | H-31CNCVVGYIGE40-matrix |
| A10 | + (91.3) | H-10HDGYCLHDGV19-matrix | B12 | +/− (56.3) | H-32NCVVGYIGER41-matrix |
| A11 | + (90.5) | H-11DGYCLHDGVC20-matrix | B13 | +/− (65.7) | H-33CVVGYIGERC42-matrix |
| A12 | + (92.4) | H-12GYCLHDGVCM21-matrix | B14 | +/− (69.4) | H-34VVGYIGERCQ43-matrix |
| A13 | + (93.0) | H-13YCLHDGVCMY22-matrix | B15 | + (91.0) | H-35VGYIGERCQY44-matrix |
| A14 | − (31.6) | H-14CLHDGVCMYI23-matrix | B16 | + (98.0) | H-36GYIGERCQYR45-matrix |
| A15 | − (41.6) | H-15LHDGVCMYIE24-matrix | B17 | + (106.2) | H-37YIGERCQYRD46-matrix |
| A16 | − (44.8) | H-16HDGVCMYIEA25-matrix | B18 | +/− (76.1) | H-38IGERCQYRDL47-matrix |
| A17 | +/− (88.9) | H-17DGVCMYIEAL26-matrix | B19 | − (28.9) | H-39GERCQYRDLK48-matrix |
| A18 | + (116.0) | H-18GVCMYIEALD27-matrix | B20 | +/− (87.3) | H-40ERCQYRDLKW49-matrix |
| A19 | ++ (128.0) | H-19VCMYIEALDK28-matrix | C1 | + (91.5) | H-41RCQYRDLKWW50-matrix |
| A20 | ++ (132.8) | H-20CMYIEALDKY29-matrix | C2 | + (91.4) | H-42CQYRDLKWWE51-matrix |
| B1 | ++ (130.3) | H-21MYIEALDKYA30-matrix | C3 | + (99.4) | H-43QYRDLKWWEL52-matrix |
| B2 | ++ (122.4) | H-22YIEALDKYAC31-matrix | C4 | + (104.2) | H-44YRDLKWWELR53-matrix |
| From the Literature | Found | |
|---|---|---|
| fragment 1 | 18GVCMYIEALDKYACNC33 | 17DGVCMYIEALDKYACN32 |
| 27DKYACNCVVGYIG39 | ||
| fragment 2 | 10HDGYCL15 + 29Y + 41R | 1NSDSECPLSHDGYCLHDGVCMY22 |
| fragment 3 | 43QYRDL47 | 33CVVGYIGERCQYRDL47 |
| 40ERCQYRDLKWWELR53 |
| Fragment | Number of Amino Acids | Purity (%) Based on HPLC | Molar Weight (g/mol) | Measured m/z |
|---|---|---|---|---|
| 10HDGYCLHDGVCMY22 | 13 | 91.7 | 1512.72 | 510.2 (3+) |
| 764.8 (2+) | ||||
| 1529.6 (+) (product with oxidized Met) | ||||
| 13YCLHDGV19 | 7 | 89.5 | 805.91 | 403.7 (2+) |
| 806.4 (+) | ||||
| 22YIEALDK28 | 7 | 83.0 | 850.97 | 426.3 (2+) |
| 851.5 (+) | ||||
| 23IEAL26 | 4 | 96.8 dimer | 444.53 | 445.3 (+) |
| 34VVGYIGERCQYRDL47 | 14 | 87.7 | 1670.92 | 557.6 (3+) |
| 835.9 (2+) | ||||
| 38IGERCQ43 | 6 | 90 | 704.81 | 353.2 (2+) |
| 705.4 (+) | ||||
| 26LDKYACNCV34 | 9 | 79.2 | 1028.22 | 514.7 (2+) |
| 1028.5 (+) | ||||
| 10HDGYCL15 | 6 | 84.5 | 706.27 | 707.3 (+) |
| 10HDGYCL15+29Y+41R | 8 | 81.5 | 1025.44 | 513.8 (2+) |
| 1026.5 (+) | ||||
| 43QYRDL47 | 5 | 75.1 | 693.35 | 347.7 (2+) |
| 694.4 (+) |
| Fragment | Assay Concentration | Kd |
|---|---|---|
| 10–22 | 50 μM | 70.5 nM |
| 100 μM | 8.2 μM | |
| 463 μM | 21.2 μM | |
| 1 mM | 35.9 μM | |
| 3 mM | 416 μM | |
| 10–15 | 50 μM | 66.4 nM |
| 100 μM | 17.4 nM | |
| 250 μM | 48.4 μM | |
| 2.5 mM | 431 μM | |
| 5 mM | 663 μM | |
| 6.02 mM | 715 μM | |
| 10 mM | 533 μM | |
| 22–28 | 10 μM | - |
| 250 μM | 4.02 nM | |
| 500 μM | - | |
| 3 mM | 1.96 mM | |
| 5 mM | 1.31 mM | |
| 10 mM | 1.65 mM | |
| 23–26 | 1.25 μM | - |
| 2.5 μM | - | |
| 50 μM | - | |
| 100 μM | - | |
| 5 mM | - | |
| 10 mM | 5.09 mM | |
| 34–47 | 50 μM | 5.92 μM |
| 141 μM | 4.62 μM | |
| 350 μM | 1.21 μM | |
| 705 μM | 2.17 μM | |
| 1 mM | 530 nM | |
| 38–43 | 10 μM | 709 nM |
| 500 μM | 154 nM | |
| 2.5 mM | 108 nM | |
| 5 mM | 157 nM | |
| 43–47 | 500 μM | 4.93 μM |
| 10 mM | 125 μM | |
| EGF | 500 nM | 10.6 nM |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Czerczak-Kwiatkowska, K.; Kaminska, M.; Fraczyk, J.; Majsterek, I.; Kolesinska, B. Searching for EGF Fragments Recreating the Outer Sphere of the Growth Factor Involved in Receptor Interactions. Int. J. Mol. Sci. 2024, 25, 1470. https://doi.org/10.3390/ijms25031470
Czerczak-Kwiatkowska K, Kaminska M, Fraczyk J, Majsterek I, Kolesinska B. Searching for EGF Fragments Recreating the Outer Sphere of the Growth Factor Involved in Receptor Interactions. International Journal of Molecular Sciences. 2024; 25(3):1470. https://doi.org/10.3390/ijms25031470
Chicago/Turabian StyleCzerczak-Kwiatkowska, Katarzyna, Marta Kaminska, Justyna Fraczyk, Ireneusz Majsterek, and Beata Kolesinska. 2024. "Searching for EGF Fragments Recreating the Outer Sphere of the Growth Factor Involved in Receptor Interactions" International Journal of Molecular Sciences 25, no. 3: 1470. https://doi.org/10.3390/ijms25031470
APA StyleCzerczak-Kwiatkowska, K., Kaminska, M., Fraczyk, J., Majsterek, I., & Kolesinska, B. (2024). Searching for EGF Fragments Recreating the Outer Sphere of the Growth Factor Involved in Receptor Interactions. International Journal of Molecular Sciences, 25(3), 1470. https://doi.org/10.3390/ijms25031470