
Implementation and Validation of an OpenMM Plugin for the Deep Potential Representation of Potential Energy


Abstract
1. Introduction
2. Results
2.1. Design and Implementation of the OpenMM Deepmd Plugin
2.1.1. General Architecture
2.1.2. Python Class DeepPotentialModel
2.2. Validation of the OpenMM Deepmd Plugin
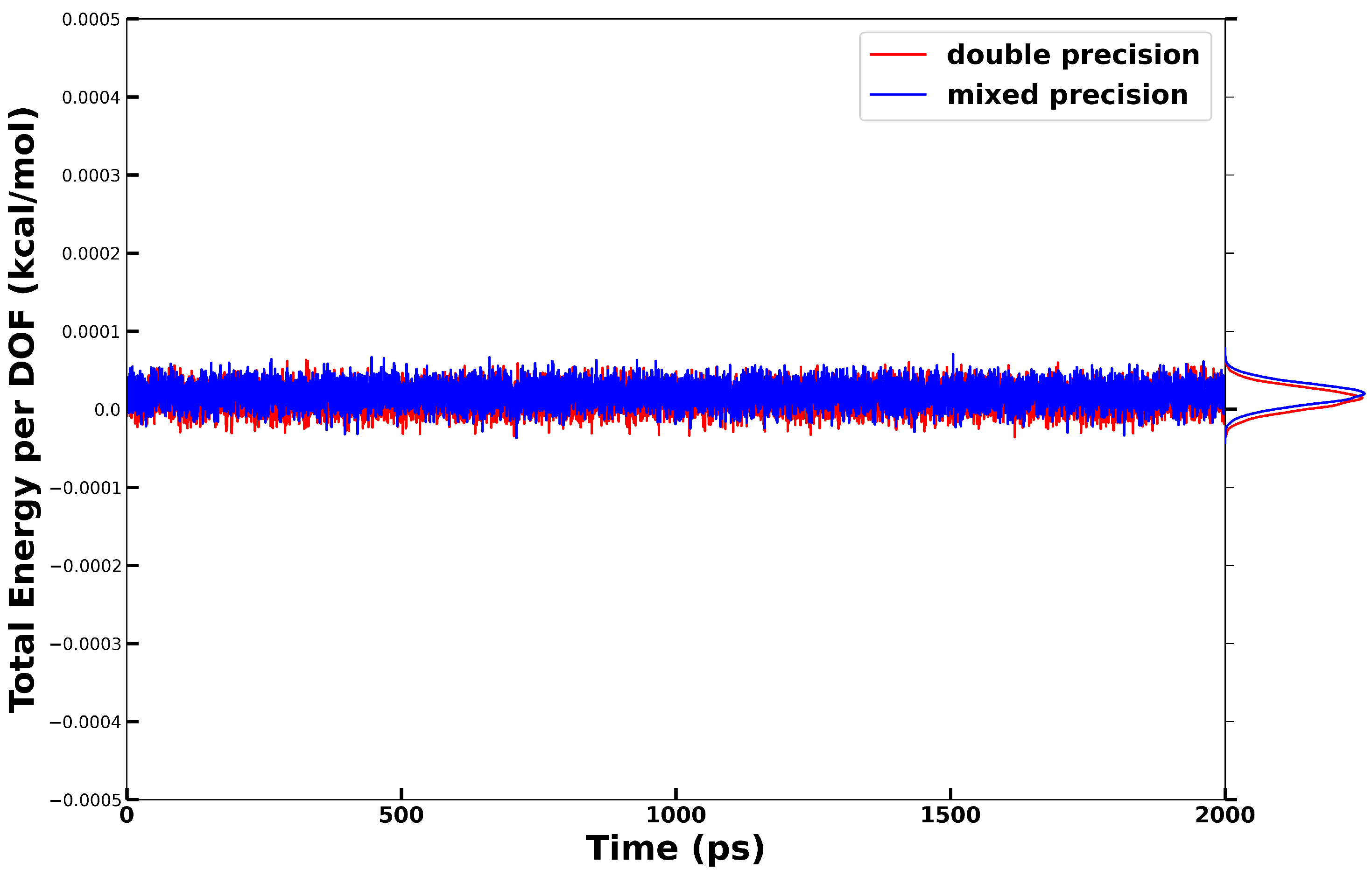
2.2.1. Energy Conservation in NVE Simulations
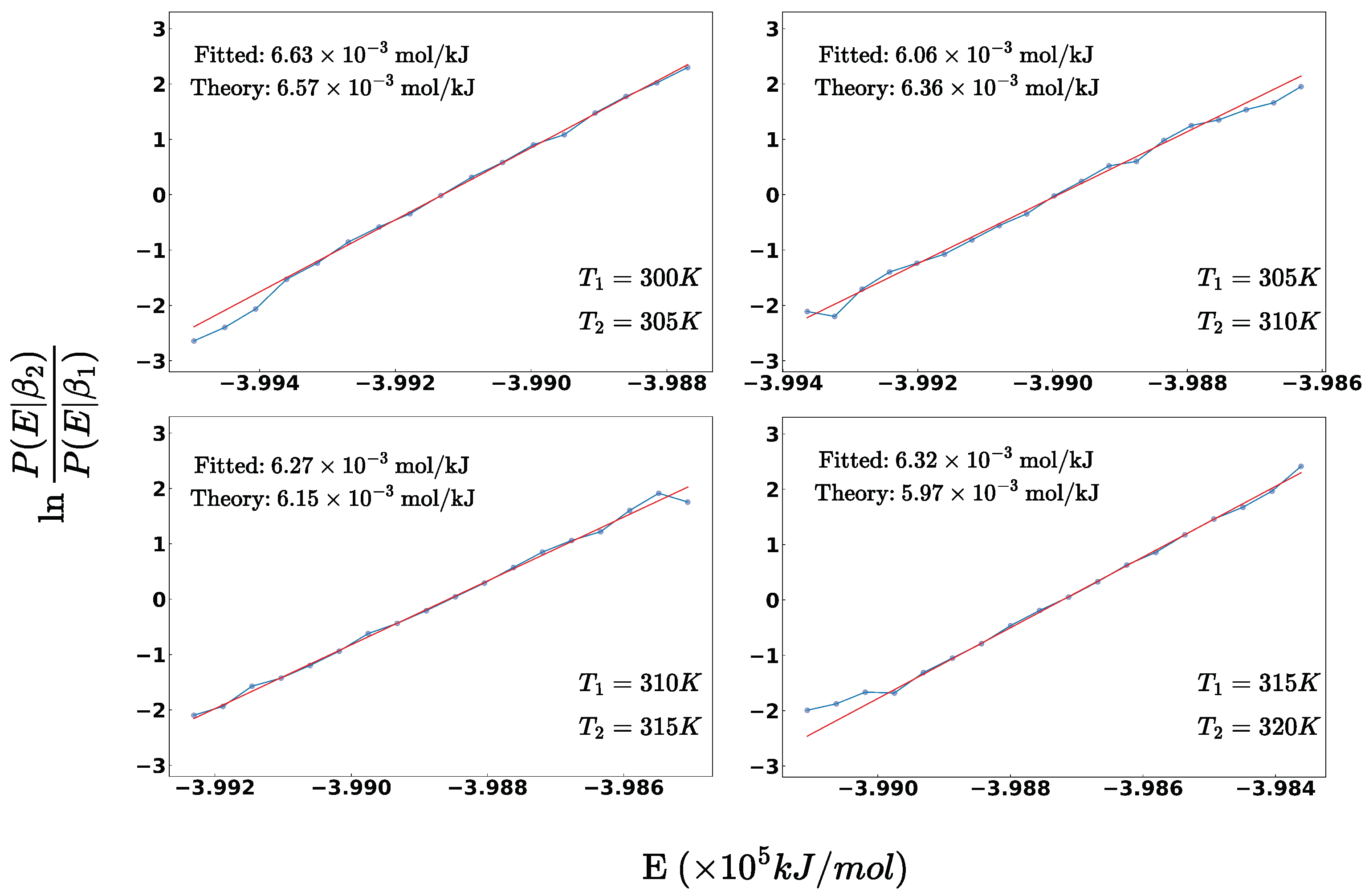
2.2.2. Thermodynamic Validation for Canonical Ensemble
2.2.3. Structural Property of Bulk Water
2.2.4. Diffusion Coefficients of Water
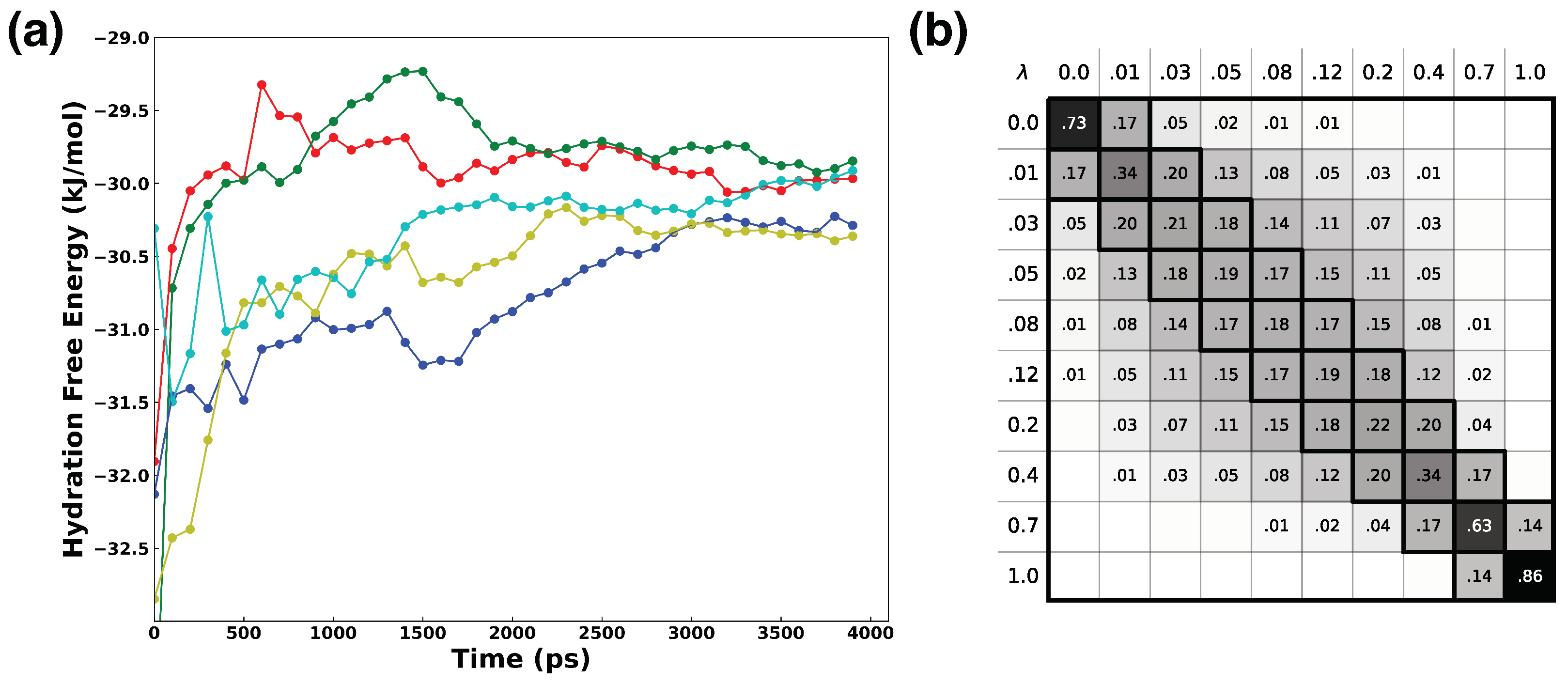
2.2.5. Hydration Free Energy of Water
2.2.6. Performance Profiling
3. Materials and Methods
3.1. Conventional MD Simulations with the DP Model
| Listing 1. Conventional MD simulations with the DP model. |
 |
3.2. Alchemical Simulations with the DP Model
| Listing 2. Alchemical simulations with the DP model. |
 |
3.3. Hybrid DP/MM Simulations with Fixed or Adaptive DP Regions
| Listing 3. DP/MM simulations with pre-selected particle. |
 |
| Listing 4. DP/MM simulations with adaptive selected particles. |
 |
4. Discussion
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bottaro, S.; Lindorff-Larsen, K. Biophysical experiments and biomolecular simulations: A perfect match? Science 2018, 361, 355–360. [Google Scholar] [CrossRef]
- Huang, J.; MacKerell, A.D. Force field development and simulations of intrinsically disordered proteins. Curr. Op. Struct. Biol. 2018, 48, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Elliott, J. Molecular dynamics simulations of the elastic properties of polymer/carbon nanotube composites. Comput. Mater. Sci. 2007, 39, 315–323. [Google Scholar] [CrossRef]
- Car, R.; Parrinello, M. Unified approach for molecular dynamics and density-functional theory. Phys. Rev. Lett. 1985, 55, 2471. [Google Scholar] [CrossRef]
- Yang, Y.I.; Shao, Q.; Zhang, J.; Yang, L.; Gao, Y.Q. Enhanced sampling in molecular dynamics. J. Chem. Phys. 2019, 151, 070902. [Google Scholar] [CrossRef] [PubMed]
- Case, D.A.; Cheatham, T.E., III; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M., Jr.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef] [PubMed]
- Brooks, B.R.; Brooks, C.L., III; Mackerell, A.D., Jr.; Nilsson, L.; Petrella, R.J.; Roux, B.; Won, Y.; Archontis, G.; Bartels, C.; Boresch, S.; et al. CHARMM: The biomolecular simulation program. J. Comput. Chem. 2009, 30, 1545–1614. [Google Scholar] [CrossRef] [PubMed]
- Nelson, M.T.; Humphrey, W.; Gursoy, A.; Dalke, A.; Kalé, L.V.; Skeel, R.D.; Schulten, K. NAMD: A parallel, object-oriented molecular dynamics program. Int. J. Supercomput. Appl. High Perform. Comput. 1996, 10, 251–268. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef]
- Eastman, P.; Galvelis, R.; Peláez, R.P.; Abreu, C.R.; Farr, S.E.; Gallicchio, E.; Gorenko, A.; Henry, M.M.; Hu, F.; Huang, J.; et al. OpenMM 8: Molecular Dynamics Simulation with Machine Learning Potentials. arXiv 2023, arXiv:2310.03121. [Google Scholar] [CrossRef]
- Harvey, M.J.; Giupponi, G.; Fabritiis, G.D. ACEMD: Accelerating biomolecular dynamics in the microsecond time scale. J. Chem. Theory Comput. 2009, 5, 1632–1639. [Google Scholar] [CrossRef] [PubMed]
- Shaw, D.E.; Adams, P.J.; Azaria, A.; Bank, J.A.; Batson, B.; Bell, A.; Bergdorf, M.; Bhatt, J.; Butts, J.A.; Correia, T.; et al. Anton 3: Twenty microseconds of molecular dynamics simulation before lunch. In Proceedings of the International Conference for High Performance Computing, Networking, Storage and Analysis, St. Louis, MO, USA, 14–19 November 2021; pp. 1–11. [Google Scholar]
- Shaw, D.E.; Grossman, J.; Bank, J.A.; Batson, B.; Butts, J.A.; Chao, J.C.; Deneroff, M.M.; Dror, R.O.; Even, A.; Fenton, C.H.; et al. Anton 2: Raising the bar for performance and programmability in a special-purpose molecular dynamics supercomputer. In Proceedings of the SC’14: Proceedings of the International Conference for High Performance Computing, Networking, Storage and Analysis, New Orleans, LA, USA, 16–21 November 2014; IEEE: Piscataway, NJ, USA, 2014; pp. 41–53. [Google Scholar]
- Unke, O.T.; Chmiela, S.; Sauceda, H.E.; Gastegger, M.; Poltavsky, I.; Schuett, K.T.; Tkatchenko, A.; Mueller, K.R. Machine learning force fields. Chem. Rev. 2021, 121, 10142–10186. [Google Scholar] [CrossRef] [PubMed]
- Friederich, P.; Häse, F.; Proppe, J.; Aspuru-Guzik, A. Machine-learned potentials for next-generation matter simulations. Nat. Mater. 2021, 20, 750–761. [Google Scholar] [CrossRef]
- Chmiela, S.; Tkatchenko, A.; Sauceda, H.E.; Poltavsky, I.; Schütt, K.T.; Müller, K.R. Machine learning of accurate energy-conserving molecular force fields. Sci. Adv. 2017, 3, e1603015. [Google Scholar] [CrossRef] [PubMed]
- Chmiela, S.; Sauceda, H.E.; Müller, K.R.; Tkatchenko, A. Towards exact molecular dynamics simulations with machine-learned force fields. Nat. Commun. 2018, 9, 3887. [Google Scholar] [CrossRef] [PubMed]
- Rupp, M.; Tkatchenko, A.; Müller, K.R.; Von Lilienfeld, O.A. Fast and accurate modeling of molecular atomization energies with machine learning. Phys. Rev. Lett. 2012, 108, 058301. [Google Scholar] [CrossRef]
- Bartók, A.P.; Payne, M.C.; Kondor, R.; Csányi, G. Gaussian approximation potentials: The accuracy of quantum mechanics, without the electrons. Phys. Rev. Lett. 2010, 104, 136403. [Google Scholar] [CrossRef]
- Bartók, A.P.; Kondor, R.; Csányi, G. On representing chemical environments. Phys. Rev. B 2013, 87, 184115. [Google Scholar] [CrossRef]
- Faber, F.A.; Christensen, A.S.; Huang, B.; Von Lilienfeld, O.A. Alchemical and structural distribution based representation for universal quantum machine learning. J. Chem. Phys. 2018, 148, 241717. [Google Scholar] [CrossRef]
- Blank, T.B.; Brown, S.D.; Calhoun, A.W.; Doren, D.J. Neural network models of potential energy surfaces. J. Chem. Phys. 1995, 103, 4129–4137. [Google Scholar] [CrossRef]
- Brown, D.F.; Gibbs, M.N.; Clary, D.C. Combining ab initio computations, neural networks, and diffusion Monte Carlo: An efficient method to treat weakly bound molecules. J. Chem. Phys. 1996, 105, 7597–7604. [Google Scholar] [CrossRef]
- Tafeit, E.; Estelberger, W.; Horejsi, R.; Moeller, R.; Oettl, K.; Vrecko, K.; Reibnegger, G. Neural networks as a tool for compact representation of ab initio molecular potential energy surfaces. J. Mol. Graph. 1996, 14, 12–18. [Google Scholar] [CrossRef]
- Behler, J.; Parrinello, M. Generalized neural-network representation of high-dimensional potential-energy surfaces. Phys. Rev. Lett. 2007, 98, 146401. [Google Scholar] [CrossRef]
- Behler, J. Constructing high-dimensional neural network potentials: A tutorial review. Int. J. Quantum Chem. 2015, 115, 1032–1050. [Google Scholar] [CrossRef]
- Jose, K.J.; Artrith, N.; Behler, J. Construction of high-dimensional neural network potentials using environment-dependent atom pairs. J. Chem. Phys. 2012, 136, 194111. [Google Scholar] [CrossRef]
- Zhang, L.; Han, J.; Wang, H.; Saidi, W.; Car, R.; Weinan, E. End-to-end symmetry preserving inter-atomic potential energy model for finite and extended systems. In Proceedings of the Advances in Neural Information Processing Systems, Montreal, QC, Canada, 3–8 December 2018; pp. 4436–4446. [Google Scholar]
- Zhang, Y.; Hu, C.; Jiang, B. Embedded atom neural network potentials: Efficient and accurate machine learning with a physically inspired representation. J. Phys. Chem. Lett. 2019, 10, 4962–4967. [Google Scholar] [CrossRef]
- Schütt, K.T.; Arbabzadah, F.; Chmiela, S.; Müller, K.R.; Tkatchenko, A. Quantum-chemical insights from deep tensor neural networks. Nat. Commun. 2017, 8, 13890. [Google Scholar] [CrossRef] [PubMed]
- Schütt, K.; Kindermans, P.J.; Sauceda Felix, H.E.; Chmiela, S.; Tkatchenko, A.; Müller, K.R. Schnet: A continuous-filter convolutional neural network for modeling quantum interactions. Adv. Neural Inf. Process. Syst. 2017, 30, 992–1002. [Google Scholar]
- Unke, O.T.; Meuwly, M. PhysNet: A neural network for predicting energies, forces, dipole moments, and partial charges. J. Chem. Theory Comput. 2019, 15, 3678–3693. [Google Scholar] [CrossRef] [PubMed]
- Lubbers, N.; Smith, J.S.; Barros, K. Hierarchical modeling of molecular energies using a deep neural network. J. Chem. Phys. 2018, 148, 241715. [Google Scholar] [CrossRef] [PubMed]
- Haghighatlari, M.; Li, J.; Guan, X.; Zhang, O.; Das, A.; Stein, C.J.; Heidar-Zadeh, F.; Liu, M.; Head-Gordon, M.; Bertels, L.; et al. NewtonNet: A Newtonian message passing network for deep learning of interatomic potentials and forces. arXiv 2021, arXiv:2108.02913. [Google Scholar] [CrossRef] [PubMed]
- Yao, K.; Herr, J.E.; Toth, D.W.; Mckintyre, R.; Parkhill, J. The TensorMol-0.1 model chemistry: A neural network augmented with long-range physics. Chem. Sci. 2018, 9, 2261–2269. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Wang, Z.; Zou, N.; Ye, M.; Duan, W.; Xu, Y. Deep neural network representation of density functional theory Hamiltonian. arXiv 2021, arXiv:2104.03786. [Google Scholar]
- Wang, X.; Xu, Y.; Zheng, H.; Yu, K. A Scalable Graph Neural Network Method for Developing an Accurate Force Field of Large Flexible Organic Molecules. J. Phys. Chem. Lett. 2021, 12, 7982–7987. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.S.; Isayev, O.; Roitberg, A.E. ANI-1: An extensible neural network potential with DFT accuracy at force field computational cost. Chem. Sci. 2017, 8, 3192–3203. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.S.; Nebgen, B.T.; Zubatyuk, R.; Lubbers, N.; Devereux, C.; Barros, K.; Tretiak, S.; Isayev, O.; Roitberg, A.E. Approaching coupled cluster accuracy with a general-purpose neural network potential through transfer learning. Nat. Commun. 2019, 10, 2903. [Google Scholar] [CrossRef] [PubMed]
- Gilmer, J.; Schoenholz, S.S.; Riley, P.F.; Vinyals, O.; Dahl, G.E. Neural message passing for quantum chemistry. In Proceedings of the International Conference on Machine Learning, PMLR, Sydney, Australia, 6–11 August 2017; pp. 1263–1272. [Google Scholar]
- Ding, Y.; Yu, K.; Huang, J. Data science techniques in biomolecular force field development. Curr. Opin. Struct. Biol. 2023, 78, 102502. [Google Scholar] [CrossRef]
- Andrade, M.F.C.; Ko, H.Y.; Zhang, L.; Car, R.; Selloni, A. Free energy of proton transfer at the water–TiO2 interface from ab initio deep potential molecular dynamics. Chem. Sci. 2020, 11, 2335–2341. [Google Scholar] [CrossRef]
- Yang, M.; Karmakar, T.; Parrinello, M. Liquid-Liquid Critical Point in Phosphorus. arXiv 2021, arXiv:2104.14688. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, H.; Car, R.; Weinan, E. Phase Diagram of a Deep Potential Water Model. Phys. Rev. Lett. 2021, 126, 236001. [Google Scholar] [CrossRef]
- Lu, D.; Wang, H.; Chen, M.; Lin, L.; Car, R.; Weinan, E.; Jia, W.; Zhang, L. 86 PFLOPS Deep Potential Molecular Dynamics simulation of 100 million atoms with ab initio accuracy. Comput. Phys. Commun. 2021, 259, 107624. [Google Scholar] [CrossRef]
- Yue, S.; Muniz, M.C.; Calegari Andrade, M.F.; Zhang, L.; Car, R.; Panagiotopoulos, A.Z. When do short-range atomistic machine-learning models fall short? J. Chem. Phys. 2021, 154, 034111. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, H.; Muniz, M.C.; Panagiotopoulos, A.Z.; Car, R. A deep potential model with long-range electrostatic interactions. J. Chem. Phys. 2022, 156, 124107. [Google Scholar] [CrossRef]
- Pan, X.; Yang, J.; Van, R.; Epifanovsky, E.; Ho, J.; Huang, J.; Pu, J.; Mei, Y.; Nam, K.; Shao, Y. Machine-Learning-Assisted Free Energy Simulation of Solution-Phase and Enzyme Reactions. J. Chem. Theory Comput. 2021, 17, 5745–5758. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhang, L.; Han, J.; Weinan, E. DeePMD-kit: A deep learning package for many-body potential energy representation and molecular dynamics. Comput. Phys. Commun. 2018, 228, 178–184. [Google Scholar] [CrossRef]
- Plimpton, S. Fast parallel algorithms for short-range molecular dynamics. J. Comput. Phys. 1995, 117, 1–19. [Google Scholar] [CrossRef]
- Dral, P.O. MLatom: A program package for quantum chemical research assisted by machine learning. J. Comput. Chem. 2019, 40, 2339–2347. [Google Scholar] [CrossRef] [PubMed]
- Dral, P.O.; Ge, F.; Xue, B.X.; Hou, Y.F.; Pinheiro, M.; Huang, J.; Barbatti, M. MLatom 2: An integrative platform for atomistic machine learning. Top. Curr. Chem. 2021, 379, 27. [Google Scholar] [CrossRef] [PubMed]
- Eastman, P.; Pande, V. OpenMM: A hardware-independent framework for molecular simulations. Comput. Sci. Eng. 2010, 12, 34–39. [Google Scholar] [CrossRef] [PubMed]
- Eastman, P.; Pande, V.S. Efficient nonbonded interactions for molecular dynamics on a graphics processing unit. J. Comput. Chem. 2010, 31, 1268–1272. [Google Scholar] [CrossRef] [PubMed]
- Kondratyuk, N.; Nikolskiy, V.; Pavlov, D.; Stegailov, V. GPU-accelerated molecular dynamics: State-of-art software performance and porting from Nvidia CUDA to AMD HIP. Int. J. High Perform. Comput. Appl. 2021, 35, 312–324. [Google Scholar] [CrossRef]
- Harger, M.; Li, D.; Wang, Z.; Dalby, K.; Lagardère, L.; Piquemal, J.P.; Ponder, J.; Ren, P. Tinker-OpenMM: Absolute and relative alchemical free energies using AMOEBA on GPUs. J. Comput. Chem. 2017, 38, 2047–2055. [Google Scholar] [CrossRef]
- Huang, J.; Lemkul, J.A.; Eastman, P.K.; MacKerell, A.D., Jr. Molecular dynamics simulations using the drude polarizable force field on GPUs with OpenMM: Implementation, validation, and benchmarks. J. Comput. Chem. 2018, 39, 1682–1689. [Google Scholar] [CrossRef]
- Qiu, Y.; Smith, D.G.; Boothroyd, S.; Jang, H.; Hahn, D.F.; Wagner, J.; Bannan, C.C.; Gokey, T.; Lim, V.T.; Stern, C.D.; et al. Development and Benchmarking of Open Force Field v1. 0.0—the Parsley Small-Molecule Force Field. J. Chem. Theory Comput. 2021, 17, 6262–6280. [Google Scholar] [CrossRef]
- OpenMM Tensorflow Plugin. 2021. Available online: https://github.com/openmm/openmm-tensorflow (accessed on 10 January 2023).
- OpenMM Torch Plugin. 2021. Available online: https://github.com/openmm/openmm-torch (accessed on 10 January 2023).
- Galvelis, R.; Varela-Rial, A.; Doerr, S.; Fino, R.; Eastman, P.; Markland, T.E.; Chodera, J.D.; De Fabritiis, G. NNP/MM: Accelerating Molecular Dynamics Simulations with Machine Learning Potentials and Molecular Mechanics. J. Chem. Inf. Model. 2023, 23, 5701–5708. [Google Scholar] [CrossRef]
- Ding, Y.; Huang, J. DP/MM: A Hybrid Model for Zinc-Protein Interactions in Molecular Dynamics. J. Phys. Chem. Lett. 2024, 15, 616–627. [Google Scholar] [CrossRef]
- Zeng, J.; Giese, T.J.; Ekesan, S.; York, D.M. Development of range-corrected deep learning potentials for fast, accurate quantum mechanical/molecular mechanical simulations of chemical reactions in solution. J. Chem. Theory Comput. 2021, 17, 6993–7009. [Google Scholar] [CrossRef] [PubMed]
- Zeng, J.; Tao, Y.; Giese, T.J.; York, D.M. QDπ: A quantum deep potential interaction model for drug discovery. J. Chem. Theory Comput. 2023, 19, 1261–1275. [Google Scholar] [CrossRef]
- Rufa, D.A.; Bruce Macdonald, H.E.; Fass, J.; Wieder, M.; Grinaway, P.B.; Roitberg, A.E.; Isayev, O.; Chodera, J.D. Towards chemical accuracy for alchemical free energy calculations with hybrid physics-based machine learning/molecular mechanics potentials. bioRxiv 2020. [Google Scholar] [CrossRef]
- Shirts, M.R. Simple quantitative tests to validate sampling from thermodynamic ensembles. J. Chem. Theory Comput. 2013, 9, 909–926. [Google Scholar] [CrossRef] [PubMed]
- Basconi, J.E.; Shirts, M.R. Effects of temperature control algorithms on transport properties and kinetics in molecular dynamics simulations. J. Chem. Theory Comput. 2013, 9, 2887–2899. [Google Scholar] [CrossRef]
- Merz, P.T.; Shirts, M.R. Testing for physical validity in molecular simulations. PLoS ONE 2018, 13, e0202764. [Google Scholar] [CrossRef]
- Gray, C.G.; Gubbins, K.E.; Joslin, C.G. Theory of Molecular Fluids: Volume 2: Applications; Oxford University Press: Oxford, UK, 2011; Volume 10. [Google Scholar]
- Mcquarrie, D. Statistical Mechanics; Harper & Row: New York, NY, USA, 1965. [Google Scholar]
- Levine, B.G.; Stone, J.E.; Kohlmeyer, A. Fast analysis of molecular dynamics trajectories with graphics processing units—Radial distribution function histogramming. J. Comput. Phys. 2011, 230, 3556–3569. [Google Scholar] [CrossRef] [PubMed]
- Wade, A.D.; Wang, L.P.; Huggins, D.J. Assimilating radial distribution functions to build water models with improved structural properties. J. Chem. Inf. Model. 2018, 58, 1766–1778. [Google Scholar] [CrossRef] [PubMed]
- Chiba, S.; Okuno, Y.; Honma, T.; Ikeguchi, M. Force-field parametrization based on radial and energy distribution functions. J. Comput. Chem. 2019, 40, 2577–2585. [Google Scholar] [CrossRef] [PubMed]
- Lyubartsev, A.P.; Laaksonen, A. Calculation of effective interaction potentials from radial distribution functions: A reverse Monte Carlo approach. Phys. Rev. E 1995, 52, 3730. [Google Scholar] [CrossRef] [PubMed]
- Michaud-Agrawal, N.; Denning, E.J.; Woolf, T.B.; Beckstein, O. MDAnalysis: A toolkit for the analysis of molecular dynamics simulations. J. Comput. Chem. 2011, 32, 2319–2327. [Google Scholar] [CrossRef] [PubMed]
- Soper, A. The radial distribution functions of water and ice from 220 to 673 K and at pressures up to 400 MPa. Chem. Phys. 2000, 258, 121–137. [Google Scholar] [CrossRef]
- Kulschewski, T.; Pleiss, J. A molecular dynamics study of liquid aliphatic alcohols: Simulation of density and self-diffusion coefficient using a modified OPLS force field. Mol. Simul. 2013, 39, 754–767. [Google Scholar] [CrossRef]
- Wang, J.; Hou, T. Application of molecular dynamics simulations in molecular property prediction II: Diffusion coefficient. J. Comput. Chem. 2011, 32, 3505–3519. [Google Scholar] [CrossRef]
- Borodin, O. Polarizable force field development and molecular dynamics simulations of ionic liquids. J. Phys. Chem. B 2009, 113, 11463–11478. [Google Scholar] [CrossRef] [PubMed]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef] [PubMed]
- Yeh, I.C.; Hummer, G. System-size dependence of diffusion coefficients and viscosities from molecular dynamics simulations with periodic boundary conditions. J. Phys. Chem. B 2004, 108, 15873–15879. [Google Scholar] [CrossRef]
- Mills, R. Self-diffusion in normal and heavy water in the range 1–45∘. J. Phys. Chem. 1973, 77, 685–688. [Google Scholar] [CrossRef]
- Raabe, G.; Sadus, R.J. Molecular dynamics simulation of the effect of bond flexibility on the transport properties of water. J. Chem. Phys. 2012, 137, 104512. [Google Scholar] [CrossRef]
- Shirts, M.R.; Chodera, J.D. Statistically optimal analysis of samples from multiple equilibrium states. J. Chem. Phys. 2008, 129, 124105. [Google Scholar] [CrossRef] [PubMed]
- Milne, A.W.; Jorge, M. Polarization corrections and the hydration free energy of water. J. Chem. Theory Comput. 2018, 15, 1065–1078. [Google Scholar] [CrossRef]
- Wu, J.Z.; Azimi, S.; Khuttan, S.; Deng, N.; Gallicchio, E. Alchemical transfer approach to absolute binding free energy estimation. J. Chem. Theory Comput. 2021, 17, 3309–3319. [Google Scholar] [CrossRef]
- Jindal, S.; Chiriki, S.; Bulusu, S.S. Spherical harmonics based descriptor for neural network potentials: Structure and dynamics of Au147 nanocluster. J. Chem. Phys. 2017, 146 20, 204301. [Google Scholar] [CrossRef]
- Huang, J.; Zhang, L.; Wang, H.; Zhao, J.; Cheng, J.; Weinan, E. Deep potential generation scheme and simulation protocol for the Li10GeP2S12-type superionic conductors. J. Chem. Phys. 2021, 154, 094703. [Google Scholar] [CrossRef]
- Wu, J.; Yang, J.; Ma, L.Y.; Zhang, L.; Liu, S. Modular development of deep potential for complex solid solutions. Phys. Rev. B 2023, 107, 144102. [Google Scholar] [CrossRef]
- Wen, T.; Zhang, L.; Wang, H.; Weinan, E.; Srolovitz, D.J. Deep Potentials for Materials Science. Mater. Futur. 2022, 1, 022601. [Google Scholar] [CrossRef]
- Lahey, S.L.J.; Rowley, C.N. Simulating protein–ligand binding with neural network potentials. Chem. Sci. 2020, 11, 2362–2368. [Google Scholar] [CrossRef] [PubMed]
- Vant, J.W.; Lahey, S.L.J.; Jana, K.; Shekhar, M.; Sarkar, D.; Munk, B.H.; Kleinekathöfer, U.; Mittal, S.; Rowley, C.; Singharoy, A. Flexible fitting of small molecules into electron microscopy maps using molecular dynamics simulations with neural network potentials. J. Chem. Inf. Model. 2020, 60, 2591–2604. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Zhu, T.; Zhang, J.Z. Automatically constructed neural network potentials for molecular dynamics simulation of zinc proteins. Front. Chem. 2021, 9, 692200. [Google Scholar] [CrossRef]
- Lier, B.; Poliak, P.; Marquetand, P.; Westermayr, J.; Oostenbrink, C. BuRNN: Buffer Region Neural Network Approach for Polarizable-Embedding Neural Network/Molecular Mechanics Simulations. J. Phys. Chem. Lett. 2022, 13, 3812–3818. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Hardware | Num. of Particles | OpenMM (ns/day) | DP Evaluation Cost (μs) | Communication Cost (μs) | LAMMPS (ns/day) |
|---|---|---|---|---|---|
| 4 CPU cores | 192 | 1.84 | 7682.650 | 113.673 | 1.895 |
| + 1060 Ti | 768 | 0.554 | 31,201.579 | 121.686 | 0.569 |
| 3072 | 0.139 | 123,432.237 | 149.248 | 0.149 | |
| 6144 | 0.0687 | 250,992.324 | 171.257 | 0.076 | |
| 4 CPU cores | 192 | 2.73 | 6884.679 | 360.378 | 2.924 |
| + A40 | 768 | 1.51 | 11,500.541 | 361.795 | 1.376 |
| 3072 | 0.482 | 35,651.468 | 291.818 | 0.420 | |
| 6144 | 0.243 | 69,671.011 | 328.957 | 0.228 | |
| 9216 | 0.159 | 105,681.057 | 345.710 | 0.159 | |
| 12,288 | 0.121 | 143,443.777 | 370.841 | 0.123 | |
| 15,360 | 0.093 | 180,575.597 | 400.271 | 0.100 | |
| 18,432 | 0.074 | 228,041.861 | 426.523 | 0.084 | |
| 21,504 | 0.060 | 286,185.872 | 453.118 | 0.073 | |
| 24,576 | 0.056 | 312,219.517 | 488.567 | 0.063 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ding, Y.; Huang, J. Implementation and Validation of an OpenMM Plugin for the Deep Potential Representation of Potential Energy. Int. J. Mol. Sci. 2024, 25, 1448. https://doi.org/10.3390/ijms25031448
Ding Y, Huang J. Implementation and Validation of an OpenMM Plugin for the Deep Potential Representation of Potential Energy. International Journal of Molecular Sciences. 2024; 25(3):1448. https://doi.org/10.3390/ijms25031448
Chicago/Turabian StyleDing, Ye, and Jing Huang. 2024. "Implementation and Validation of an OpenMM Plugin for the Deep Potential Representation of Potential Energy" International Journal of Molecular Sciences 25, no. 3: 1448. https://doi.org/10.3390/ijms25031448
APA StyleDing, Y., & Huang, J. (2024). Implementation and Validation of an OpenMM Plugin for the Deep Potential Representation of Potential Energy. International Journal of Molecular Sciences, 25(3), 1448. https://doi.org/10.3390/ijms25031448