
What Is Hidden in Patients with Unknown Nephropathy? Genetic Screening Could Be the Missing Link in Kidney Transplantation Diagnosis and Management

 , ,
, ,  , , ,
, , ,  ,
,  ,
,  and
and
Abstract
1. Introduction
2. Results
2.1. Pathogenic Variants in Genes Related to Nephrotic Syndromes (NS) Solved 10% of the Cohort
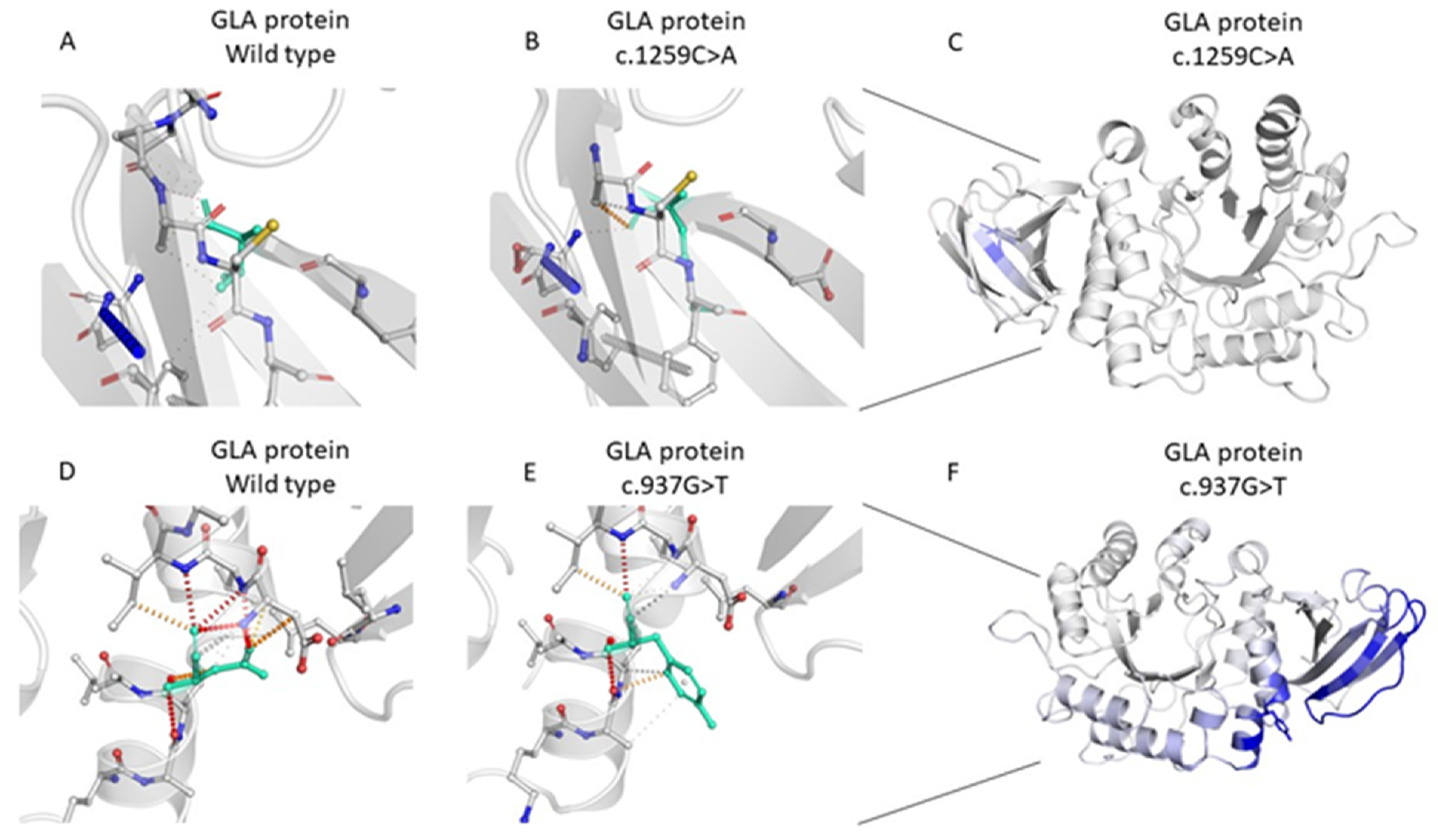
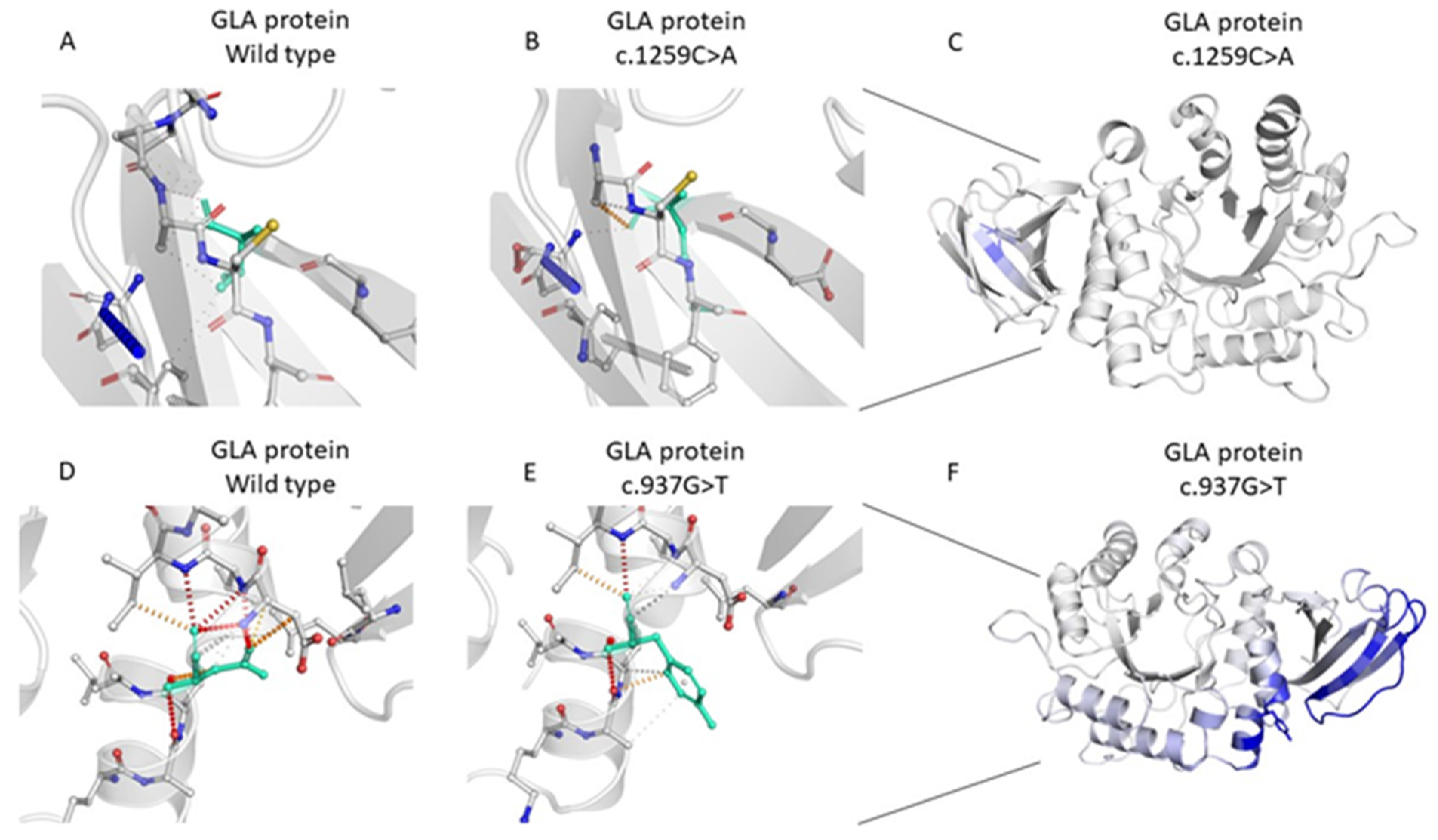
2.2. Genetic Analysis of the GLA Variants
3. Discussion
4. Materials and Methods
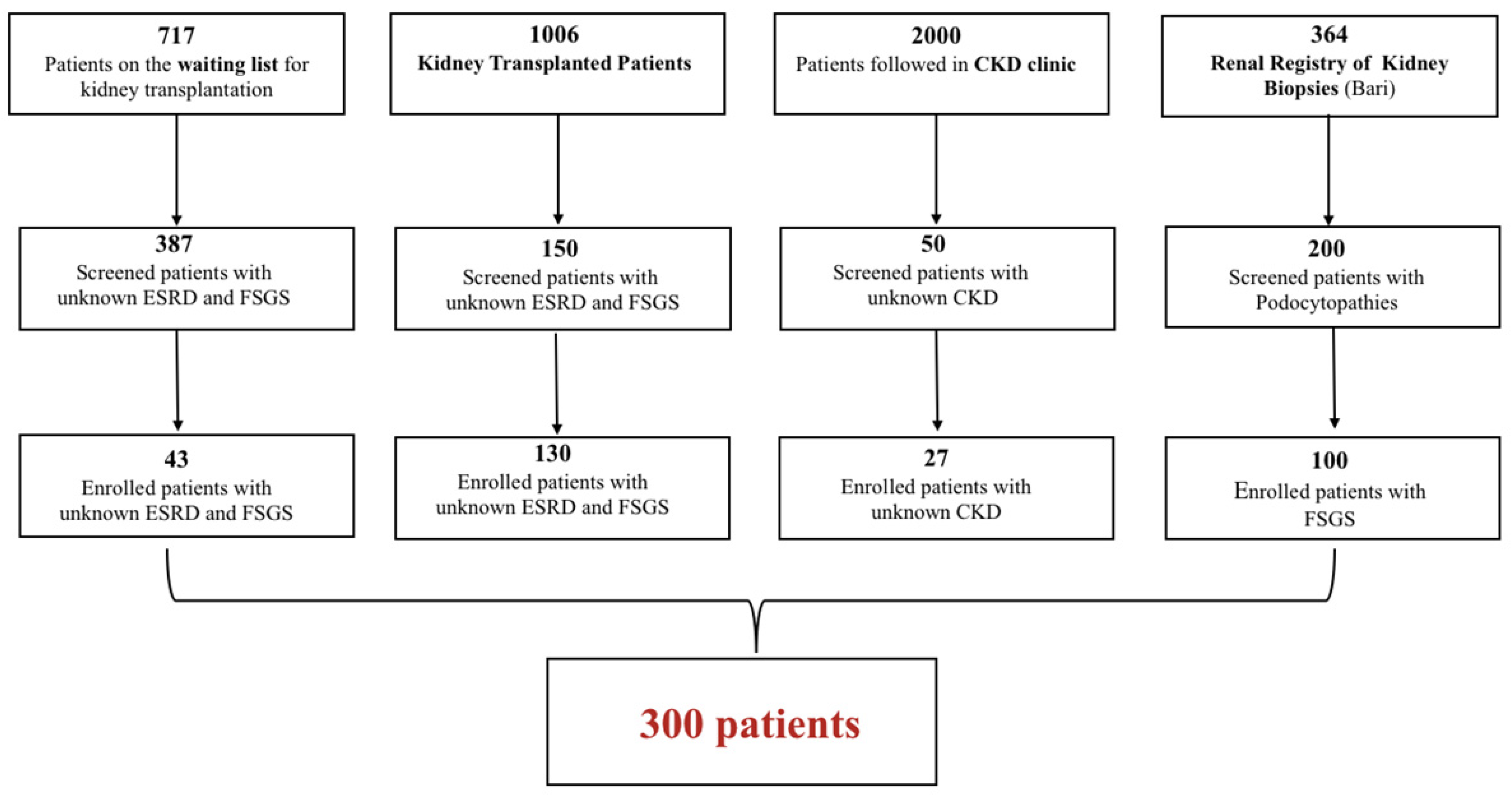
4.1. Patient Population
4.2. Gene Selection for the Custom-Panel Design
4.3. DNA Extraction and Next Generation Sequencing (NGS)
4.4. Annotation and Variants Analysis
4.5. Further Analysis in Patients with Suspected GLA Variants
4.6. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hill, N.R.; Fatoba, S.T.; Oke, J.L.; Hirst, J.A.; O’Callaghan, C.A.; Lasserson, D.S.; Hobbs, F.D.R. Global prevalence of chronic kidney disease—A systematic review and meta-analysis. PLoS ONE 2016, 11, e0158765. [Google Scholar] [CrossRef] [PubMed]
- Quaglia, M.; Musetti, C.; Ghiggeri, G.M.; Fogazzi, G.B.; Settanni, F.; Boldorini, R.L.; Lazzarich, E.; Airoldi, A.; Izzo, C.; Giordano, M.; et al. Unexpectedly high prevalence of rare genetic disorders in kidney transplant recipients with an unknown causal nephropathy. Clin. Transplant. 2014, 28, 995–1003. [Google Scholar] [CrossRef] [PubMed]
- Limou, S.; Vince, N.; Parsa, A. Lessons from CKD-related genetic association studies–moving forward. Clin. J. Am. Soc. Nephrol. 2018, 13, 140–152. [Google Scholar] [CrossRef] [PubMed]
- Kramer, A.; Pippias, M.; Noordzij, M.; Stel, V.S.; Afentakis, N.; Ambühl, P.M.; Andrusev, A.M.; Fuster, E.A.; Monzón, F.E.A.; Åsberg, A.; et al. The European Renal Association—European Dialysis and Transplant Association (ERA-EDTA) Registry Annual Report 2015: A summary. Clin. Kidney J. 2018, 11, 108–122. [Google Scholar] [CrossRef] [PubMed]
- Bassanese, G.; Wlodkowski, T.; Servais, A.; Heidet, L.; Roccatello, D.; Emma, F.; Levtchenko, E.; Ariceta, G.; Bacchetta, J.; Capasso, G.; et al. The European Rare Kidney Disease Registry (ERKReg): Objectives, design and initial results. Orphanet J. Rare Dis. 2021, 16, 251. [Google Scholar] [CrossRef] [PubMed]
- Devuyst, O.; Knoers, N.V.A.M.; Remuzzi, G.; Schaefer, F. Rare inherited kidney diseases: Challenges, opportunities, and perspectives. Lancet 2014, 383, 1844–1859. [Google Scholar] [CrossRef]
- Vivante, A.; Hildebrandt, F. Exome Sequencing Frequently Reveals the Cause of Early-Onset Chronic Kidney Disease. Nat. Rev. Nephrol. 2016, 12, 133–146. [Google Scholar] [CrossRef]
- Groopman, E.E.; Marasa, M.; Cameron-Christie, S.; Petrovski, S.; Aggarwal, V.S.; Milo-Rasouly, H.; Li, Y.; Zhang, J.; Nestor, J.; Krithivasan, P.; et al. Diagnostic Utility of Exome Sequencing for Kidney Disease. N. Engl. J. Med. 2019, 380, 142–151. [Google Scholar] [CrossRef]
- Connaughton, D.M.; Kennedy, C.; Shril, S.; Mann, N.; Murray, S.L.; Williams, P.A.; Conlon, E.; Nakayama, M.; van der Ven, A.T.; Ityel, H.; et al. Monogenic causes of chronic kidney disease in adults. Kidney Int. 2019, 95, 914–928. [Google Scholar] [CrossRef]
- Lata, S.; Marasa, M.; Li, Y.; Fasel, D.A.; Groopman, E.; Jobanputra, V.; Rasouly, H.; Mitrotti, A.; Westland, R.; Verbitsky, M.; et al. Whole-Exome Sequencing in Adults with Chronic Kidney Disease. Ann. Intern. Med. 2018, 168, 100. [Google Scholar] [CrossRef]
- Knoers, N.; Antignac, C.; Bergmann, C.; Dahan, K.; Giglio, S.; Heidet, L.; Lipska-Ziętkiewicz, B.S.; Noris, M.; Remuzzi, G.; Vargas-Poussou, R.; et al. Genetic testing in the diagnosis of chronic kidney disease: Recommendations for clinical practice. Nephrol. Dial. Transplant. 2022, 37, 239–254. [Google Scholar] [CrossRef] [PubMed]
- Mitrotti, A.; Giliberti, M.; Di Leo, V.; di Bari, I.; Pontrelli, P.; Gesualdo, L. Hidden genetics behind glomerular scars: An opportunity to understand the heterogeneity of focal segmental glomerulosclerosis? Pediatr. Nephrol. 2023, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Jahan, S.; Sarathchandran, S.; Akhter, S.; Goldblatt, J.; Stark, S.; Crawford, D.; Mallett, A.; Thomas, M. Prevalence of Fabry disease in dialysis patients: Western Australia Fabry disease screening study—The FoRWARD study. Orphanet J. Rare Dis. 2020, 15, 10. [Google Scholar] [CrossRef] [PubMed]
- Svarstad, E.; Bostad, L.; Kaarbøe, O.; Houge, G.; Tøndel, C.; Lyngdal, P.; Iversen, B. Focal and segmental glomerular sclerosis (FSGS) in a man and a woman with Fabry’s disease. Clin. Nephrol. 2005, 63, 394–401. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, R.; Oak, N.; Plon, S.E. Evaluation of in silico algorithms for use with ACMG/AMP clinical variant interpretation guidelines. Genome Biol. 2017, 18, 225. [Google Scholar] [CrossRef] [PubMed]
- Ioannidis, N.M.; Rothstein, J.H.; Pejaver, V.; Middha, S.; McDonnell, S.K.; Baheti, S.; Musolf, A.; Li, Q.; Holzinger, E.; Karyadi, D.; et al. REVEL: An Ensemble Method for Predicting the Pathogenicity of Rare Missense Variants. Am. J. Hum. Genet. 2016, 99, 877–885. [Google Scholar] [CrossRef] [PubMed]
- Dong, C.; Wei, P.; Jian, X.; Gibbs, R.; Boerwinkle, E.; Wang, K.; Liu, X. Comparison and integration of deleteriousness prediction methods for nonsynonymous SNVs in whole exome sequencing studies. Hum. Mol. Genet. 2015, 24, 2125–2137. [Google Scholar] [CrossRef]
- Cooper, G.M.; Stone, E.A.; Asimenos, G.; Green, E.D.; Batzoglou, S.; Sidow, A. Distribution and intensity of constraint in mammalian genomic sequence. Genome Res. 2005, 15, 901–913. [Google Scholar] [CrossRef]
- Rodrigues, C.H.; Pires, D.E.; Ascher, D.B. DynaMut: Predicting the impact of mutations on protein conformation, flexibility and stability. Nucleic Acids Res. 2018, 46, W350–W355. [Google Scholar] [CrossRef]
- Pires, D.E.; Ascher, D.B. mCSM–NA: Predicting the effects of mutations on protein–nucleic acids interactions. Nucleic Acids Res. 2017, 45, W241–W246. [Google Scholar] [CrossRef]
- Pires, D.E.V.; Ascher, D.B.; Blundell, T.L. DUET: A server for predicting effects of mutations on protein stability using an integrated computational approach. Nucleic Acids Res. 2014, 42, W314–W319. [Google Scholar] [CrossRef] [PubMed]
- Baydakova, G.; Ilyushkina, A.; Moiseev, S.; Bychkov, I.; Nikitina, N.; Buruleva, T.A.; Zakharova, E. α-Galactosidase A/lysoGb3 ratio as a potential marker for Fabry disease in females. Clin. Chim. Acta 2020, 501, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Smid, B.E.; van der Tol, L.; Biegstraaten, M.; Linthorst, G.E.; Hollak, C.E.M.; Poorthuis, B.J.H.M. Plasma globotriaosylsphingosine in relation to phenotypes of Fabry disease. J. Med. Genet. 2015, 52, 262–268. [Google Scholar] [CrossRef]
- Effraimidis, G.; Rasmussen, Å.K.; Bundgaard, H.; Sørensen, S.S.; Feldt-Rasmussen, U. Is the alpha-galactosidase A variant p.Asp313Tyr (p.D313Y) pathogenic for Fabry disease? A systematic review. J. Inherit. Metab. Dis. 2020, 43, 922–933. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Bikbov, B.; Purcell, C.A.; Levey, A.S.; Smith, M.; Abdoli, A.; Abebe, M.; Adebayo, O.M.; Afarideh, M.; Agarwal, S.K.; Agudelo-Botero, M.; et al. Global, regional, and national burden of chronic kidney disease, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2020, 395, 709–733. [Google Scholar] [CrossRef]
- de Haan, A.; Eijgelsheim, M.; Vogt, L.; Knoers, N.V.A.M.; de Borst, M.H. Diagnostic Yield of Next-Generation Sequencing in Patients with Chronic Kidney Disease of Unknown Etiology. Front. Genet. 2019, 10, 1264. [Google Scholar] [CrossRef]
- El Ters, M.; e Vairo, F.P.; Prochnow, C.B.; Schinstock, C.; Dean, P.; Kemppainen, J.; Lazaridis, K.; Cosio, F.; Fervenza, F.C.; Cornell, L.; et al. Incorporation of Genetic Studies in the Kidney Transplant Evaluation Clinic: The Value of a Multidisciplinary Approach. Transplantation 2023, 107, 952–960. [Google Scholar] [CrossRef]
- Gibson, J.T.; Huang, M.; Dabrera, M.S.C.; Shukla, K.; Rothe, H.; Hilbert, P.; Deltas, C.; Storey, H.; Lipska-Ziętkiewicz, B.S.; Chan, M.M.Y.; et al. Genotype–phenotype correlations for COL4A3–COL4A5 variants resulting in Gly substitutions in Alport syndrome. Sci. Rep. 2022, 12, 2722. [Google Scholar] [CrossRef]
- Stokman, M.F.; Renkema, K.Y.; Giles, R.H.; Schaefer, F.; Knoers, N.V.; van Eerde, A.M. The expanding phenotypic spectra of kidney diseases: Insights from genetic studies. Nat. Rev. Nephrol. 2016, 12, 472–483. [Google Scholar] [CrossRef]
- Bullich, G.; Domingo-Gallego, A.; Vargas, I.; Ruiz, P.; Lorente-Grandoso, L.; Furlano, M.; Fraga, G.; Madrid, Á.; Ariceta, G.; Borregán, M.; et al. A kidney-disease gene panel allows a comprehensive genetic diagnosis of cystic and glomerular inherited kidney diseases. Kidney Int. 2018, 94, 363–371. [Google Scholar] [CrossRef] [PubMed]
- Howie, A.J. Genetic studies of focal segmental glomerulosclerosis: A waste of scientific time? Pediatr. Nephrol. 2020, 35, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Li, M.M.; Datto, M.; Duncavage, E.J.; Kulkarni, S.; Lindeman, N.I.; Roy, S.; Tsimberidou, A.M.; Vnencak-Jones, C.L.; Wolff, D.J.; Younes, A.; et al. Standards and Guidelines for the Interpretation and Reporting of Sequence Variants in Cancer: A Joint Consensus Recommendation of the Association for Molecular Pathology, American Society of Clinical Oncology, and College of American Pathologists. J. Mol. Diagn. 2017, 19, 4–23. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, H.; Taguchi, A.; Mikame, M.; Lu, H.; Tada, N.; Ishijima, M.; Kaneko, H.; Kawai, M.; Goto, S.; Saito, A.; et al. Low bone mineral density due to secondary hyperparathyroidism in the GlatmTg(CAG-A4GALT) mouse model of Fabry disease. FASEB BioAdvances 2020, 2, 365–381. [Google Scholar] [CrossRef] [PubMed]
- Creixell, P.; Schoof, E.M.; Tan, C.S.H.; Linding, R. Mutational properties of amino acid residues: Implications for evolvability of phosphorylatable residues. Philos. Trans. R. Soc. B Biol. Sci. 2012, 367, 2584–2593. [Google Scholar] [CrossRef] [PubMed]
- Colon, C.; Ortolano, S.; Melcon-Crespo, C.; Alvarez, J.V.; Lopez-Suarez, O.E.; Couce, M.L.; Fernández-Lorenzo, J.R. Newborn screening for Fabry disease in the north-west of Spain. Eur. J. Pediatr. 2017, 176, 1075–1081. [Google Scholar] [CrossRef] [PubMed]
- Froissart, R. Fabry disease: D313Y is an α-galactosidase A sequence variant that causes pseudodeficient activity in plasma. Mol. Genet. Metab. 2003, 80, 307–314. [Google Scholar] [CrossRef]
- Schiffmann, R.; Fuller, M.; Clarke, L.A.; Aerts, J.M. Is it Fabry disease? Genet. Med. 2016, 18, 1181–1185. [Google Scholar] [CrossRef]
- Oliveira, J.P.; Ferreira, S. Multiple phenotypic domains of Fabry disease and their relevance for establishing genotype–phenotype correlations. Appl. Clin. Genet. 2019, 12, 35–50. [Google Scholar] [CrossRef]
- Ries, M.; Gal, A. Genotype–Phenotype Correlation in Fabry Disease. 2006. Available online: http://www.ncbi.nlm.nih.gov/pubmed/15776423 (accessed on 21 December 2023).
- Varela, P.; Kirsztajn, G.M.; Motta, F.L.; Martin, R.P.; Turaça, L.T.; Ferrer, H.L.F.; Gomes, C.P.; Nicolicht, P.; Marins, M.M.; Pessoa, J.G.; et al. Correlation between GLA variants and alpha-Galactosidase A profile in dried blood spot: An observational study in Brazilian patients. Orphanet J. Rare Dis. 2020, 15, 30. [Google Scholar] [CrossRef]
- Pisani, A.; Imbriaco, M.; Zizzo, C.; Albeggiani, G.; Colomba, P.; Alessandro, R.; Iemolo, F.; Duro, G. A classical phenotype of Anderson-Fabry disease in a female patient with intronic mutations of the GLA gene: A case report. BMC Cardiovasc. Disord. 2012, 12, 2–5. [Google Scholar] [CrossRef] [PubMed]
- Miller, E.M.; Patterson, N.E.; Zechmeister, J.M.; Bejerano-Sagie, M.; Delio, M.; Patel, K.; Ravi, N.; Quispe-Tintaya, W.; Maslov, A.; Simmons, N.; et al. Development and validation of a targeted next generation DNA sequencing panel outperforming whole exome sequencing for the identification of clinically relevant genetic variants. Oncotarget 2017, 8, 102033–102045. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wu, C.; Li, C.; Boerwinkle, E. dbNSFP v3.0: A One-Stop Database of Functional Predictions and Annotations for Human Nonsynonymous and Splice-Site SNVs. Hum. Mutat. 2016, 37, 235–241. [Google Scholar] [CrossRef] [PubMed]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef]
- Kircher, M.; Witten, D.M.; Jain, P.; O‘Roak, B.J.; Cooper, G.M.; Shendure, J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 2014, 46, 310–315. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| N. 300 | |
|---|---|
| Sex | 118 F 182 M |
| Phenotype | 162 Podocytopathies 138 Unknown CKD/ESRD * |
| Age of Onset of Kidney Disease | 35.65 ± 16 Whole cohort 37.17 ± 15.5 CKD/ESRD * 32.23 ± 16.9 FSGS * |
| Transplant | 130 Yes 170 No |
| Ethnicity | 298 Not Hispanic or Latino 2 Hispanic or Latino |
| Race | 298 White 2 Hispanic |
| Family History | 23 Yes 277 No/unknown |
| Sample | Candidate Gene Name | Chromosome | Genomic Variation | cDNA Variation | dbSNP | Allele Frequency | Genotype | Inheritance | Functional Effect | ACMG Interpretation | Phenotype * |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 35 | FN1 | 2 | g.216241356C>T | c.5752G>A | - | 0.0 | Het | AD | Missense | VUS | Unknown ESRD |
| 46 | COL4A3 | 2 | g.228173662T>C | c.4510T>C | rs201671013 | 0.0002350 | Het | AD/AR | Missense | VUS/Likely Pathogenic | Unknown ESRD |
| 51 | TTC21B | 2 | g.166786784C>T | c.985G>A | rs768266139 | 0.000008814 | Hom | AR | Missense | VUS/Likely Pathogenic | Unknown ESRD |
| 56 | INF2 | 14 | g.105167927_105167928insAGC | c.226_227insAGC | - | 0.0 | Het | AD | In-frame insertion | Pathogenic | Unknown ESRD |
| 57 | COL4A4 | 2 | g.227973547A>G | c.693+2T>C | - | 0.0 | Het | AD | Splice Donor Loss | Pathogenic | Unknown ESRD |
| 62 | NPHS2 | 1 | g.179521754_179521755del | c.855_856del | rs749740335 | 0.0001558 | Hom | AR | Frameshift | Pathogenic | Unknown ESRD |
| 63 | COL4A5 | X | g.107827760_107827761insTT | c.1032+16_1032+17dup | - | 0.0 | Het | AD/AR | Splicing Variant | VUS/Likely Pathogenic | Unknown ESRD |
| 67 | COL4A4 | 2 | g.227922281C>A | c.2419G>T | - | 0.0 | Het | AD/AR | Missense | VUS/Likely Pathogenic | FSGS |
| MYO1E | 15 | g.59430471A>G | c.4316-2A>G | - | 0.0 | Het | AR | Missense | VUS | ||
| 77 | COL4A5 | X | g.107930710A>G | c.4316-2A>G | - | 0.0 | Hom | AD/AR | Splicing Loss | Pathogenic | FSGS |
| 89 | COL4A5 | X | g.107911582C>G | c.3638C>G | - | 0.0 | Het | AD/AR | Missense | Pathogenic | Unknown ESRD |
| 103 | PODXL | 7 | g.131241029_131241030insGGCGGC | c.89_90insGCCGCC | - | 0.0 | Het | AD/AR | In-frame insertion | VUS/Likely Pathogenic | Unknown ESRD |
| 164 | LAMB2 | 3 | g.49162783G>C | c.2623C>G | - | 0.0 | Hom | AR | Missense | VUS/Likely Pathogenic | FSGS |
| 168 | LMNA | 1 | g.156107470G>A | c.1634G>A | rs142191737 | 0.0005141 | Hom | AR | Missense | Likely Pathogenic | Unknown ESRD |
| 169 | NPHS2 | 1 | g.179526214C>T | c.686G>A | rs61747728 | 0.03601 | Hom | AR | Missense | VUS/Likely Pathogenic/Pathogenic | Unknown ESRD |
| 176 | COL4A4 | 2 | g.227967540C>T | c.895G>A | rs757578262 | 0.0 | Het | AD/AR | Missense | Likely Pathogenic | Unknown ESRD |
| 2 | g.227985764C>T | c.293G>A | - | 0.0 | Het | AD/AR | Missense | Likely Pathogenic | |||
| 182 | NPHS1 | 19 | g.36336621G>T | c.1707C>A | - | 0.0 | Hom | AR | Missense | Pathogenic | Unknown ESRD |
| 190 | INF2 | 14 | g.105169734G>A | c.610G>A | rs1049200069 | 0.0 | Het | AD | Missense | VUS | FSGS |
| 195 | TRPC6 | 11 | g.101323799G>A | c.2683C>T | rs121434394 | 0.0 | Het | AD | Missense | Pathogenic | FSGS |
| 196 | EMP2 | 16 | g.10641396C>G | c.78+1G>C | rs747072310 | 0.00003103 | Hom | AR | Splicing Variant | Likely Pathogenic | FSGS |
| 198 | PLCE1 | 10 | g.96013971G>A | c.3304G>A | rs763011760 | 0.000008838 | Hom | AR | Missense | VUS/Likely Pathogenic | FSGS |
| 212 | WT1 | 11 | g.32413530T>C | c.1435A>G | - | 0.0 | Het | AD | Missense | VUS/Likely Pathogenic | Unknown ESRD |
| 219 | CUBN | 10 | g.16918949T>G | c.9053A>C | rs370778353 | 0.0001936 | Hom | AR | Missense | VUS/Likely Pathogenic | FSGS |
| 223 | PAX2 | 10 | g.102509528_102509529insG | c.76dup | rs768607170 rs77453353 | 0.00003590 | Het | AD | Frameshift | Pathogenic | FSGS |
| 232 | COL4A4 | 2 | g.227896862_227896870del | c.3699_3706+1del | - | 0.0 | Hom | AD/AR | Splice junction loss | Pathogenic | Unknown CKD |
| 235 | ADCK4 | 19 | g.41206037_41206038insCA | c.1077_1078insTG | - | 0.0 | Hom | AR | Frameshift | Pathogenic | Unknown CKD |
| 237 | COL4A4 | 2 | g.227968749C>T | c.755G>A | - | 0.0 | Het | AD/AR | Missense | VUS/Likely Pathogenic | FSGS |
| 243 | EMP2 | 16 | g.10641396C>G | c.78+1G>C | rs747072310 | 0.00003103 | Hom | AR | Splicing Variant | Likely Pathogenic | Unknown CKD |
| 244 | PODXL | 7 | g.131241029_131241030insGGGGAC | c.89_90insGTCCCC | - | 0.0 | Hom | AD/AR | In-frame insertion | VUS/Likely Pathogenic | FSGS |
| Sample | Candidate Gene Name | Chromosome | Genomic Variation | cDNA Variation | dbSNP | Allele Frequency | Inheritance | Genotype | Function | ACMG | Phenotype * |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 48 | PLCE1 | 10 | g.95987105A>G | c.1852A>G | rs200180170 | 0.0004975 | AR | Het | Missense | VUS | Unknown ESRD |
| 60 | MYO1E | 15 | g.59450575G>A | c.2789C>T | - | 0.0 | AR | Het | Missense | VUS | Unknown ESRD |
| 92 | COQ6 | 14 | g.74428075T>C | c.1091T>C | rs747211443 | 0.00001548 | AR | Het | Missense | VUS | Unknown ESRD |
| 170 | PLCE1 | 10 | g.96013948G>C | c.3281G>C | rs61732523 | 0.0002567 | AR | Het | Missense | Pathogenic (VUS meaning) | Unknown ESRD |
| g.95931087G>T | c.1643G>T | rs17417407 | 0.1667 | AR | Het | Missense | Benign (VUS meaning) | ||||
| 191 | NPHS1 | 19 | g.36340506A>C | c.658T>G | rs115333628 | 0.001827 | AR | Het | Missense | VUS/conflicting interpretation of pathogenicity based on ClinVar | Unknown ESRD |
| g.36342212C>T | c.349G>A | rs3814995 | 0.3102 | AR | Het | Missense | Benign |
| Sample | Gene | Chrom | Genomic Variation | cDNA | dbSNP | Allele Frequency | Genotype | Inheritance | Functional Effect | ACMG Interpretation | Phenotype |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 223 | PAX2 | 10 | g.102509528_102509529insG | c.76dup | rs768607170 | 0.00003590 | Het | AD | Frameshift | Pathogenic | FSGS |
| 224 | PAX2 | 10 | g.102509528_102509529insG | c.76dup | rs768607170 | 0.00003590 | Het | AD | Frameshift | Pathogenic | MPGN II |
| 225 | PAX2 | 10 | g.102509528_102509529insG | c.76dup | rs768607170 | 0.00003590 | Het | AD | Frameshift | Pathogenic | Unknown ESRD |
| ID Sample | GLA Genomic Variation (Chromosome X) | Genotype | GLA cDNA Variation | ACMG Interpretation | Notes | Phenotype * |
|---|---|---|---|---|---|---|
| 5 | g.100653420C>A | HET | c.937G>T; p.Asp313Tyr | Pathogenic | exon 6 of 7 position 136 of 198 (coding) | Unknown CKD |
| 7 | g.100662901G>A | HET | c.-10C>T | Benign | exon 1 of 7 (5′UTR) position 101 of 304 | FSGS |
| 11 | g.100662901G>A | HET | c.-10C>T | Benign | exon 1 of 7 (5′UTR) position 101 of 304 | Unknown CKD |
| 38 | g.100662901G>A | HOM | c.-10C>T | Benign | exon 1 of 7 (5′UTR) position 101 of 304 | FSGS |
| 41 | g.100662901G>A | HET | c.-10C>T | Benign | exon 1 of 7 (5′UTR) position 101 of 304 | Unknown ESRD (TX) |
| 43 | g.100662901G>A | HOM | c.-10C>T | Benign | exon 1 of 7 (5′UTR) position 101 of 304 | Unknown ESRD (TX) |
| 46 | g.100662901G>A | HET | c.-10C>T | Benign | exon 1 of 7 (5′UTR) position 101 of 304 | Unknown ESRD (TX) |
| 65 | g.100662903C>T | HET | c.-12G>A | Benign | exon 1 of 7 (5′UTR) position 11 of 216 | FSGS |
| 93 | g.100662903C>T | HET | c.-12G>A | Benign | exon 1 of 7 (5′UTR) position 11 of 216 | FSGS |
| 133 | g.100662901G>A | HOM | c.-10C>T | Benign | exon 1 of 7 (5′UTR) position 101 of 304 | Unknown ESRD (TX) |
| 148 | g.100662901G>A | HOM | c.-10C>T | Benign | exon 1 of 7 (5′UTR) position 101 of 304 | Unknown ESRD (TX) |
| 152 | g.100662901G>A | HOM | c.-10C>T | Benign | exon 1 of 7 (5′UTR) position 101 of 304 | Unknown ESRD (TX) |
| 153 | g.100662903C>T | HOM | c.-12G>A | Benign | exon 1 of 7 (5′UTR) position 11 of 216 | Unknown ESRD (TX) |
| 167 | g.100652828G>T | HET | c.1259C>A; p.Thr420Lys | VUS/Likely Pathogenic | exon 7 of 7 position 260 of 297 (coding) | Unknown ESRD (TX) |
| 169 | g.100662901G>A | HET | c.-10C>T | Benign | exon 1 of 7 (5′UTR) position 101 of 304 | Unknown ESRD (TX) |
| 176 | g.100662901G>A | HET | c.-10C>T | Benign | exon 1 of 7 (5′UTR) position 101 of 304 | Unknown ESRD (TX) |
| 190 | g.100662901G>A | HET | c.-10C>T | Benign | exon 1 of 7 (5′UTR) position 101 of 304 | FSGS |
| 192 | g.100662901G>A | HOM | c.-10C>T | Benign | exon 1 of 7 (5′UTR) position 101 of 304 | Unknown CKD |
| 211 | g.100653420C>A | HET | c.937G>T; p.Asp313Tyr | Pathogenic | exon 6 of 7 position 136 of 198 (coding) | Unknown ESRD (TX) |
| g.100662901G>A | HET | c.-10C>T | Benign | exon 1 of 7 (5′UTR) position 101 of 304 | Unknown ESRD (TX) | |
| 217 | g.100662903C>T | HOM | c.-12G>A | Benign | exon 1 of 7 (5′UTR) position 11 of 216 | Unknown ESRD (TX) |
| 223 | g.100662903C>T | HOM | c.-12G>A | Benign | exon 1 of 7 (5′UTR) position 11 of 216 | FSGS |
| 224 | g.100662903C>T | HET | c.-12G>A | Benign | exon 1 of 7 (5′UTR) position 11 of 216 | Unknown ESRD (TX) |
| 226 | g.100662903C>T | HET | c.-12G>A | Benign | exon 1 of 7 (5′UTR) position 11 of 216 | Unknown CKD |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mitrotti, A.; Di Bari, I.; Giliberti, M.; Franzin, R.; Conserva, F.; Chiusolo, A.; Gigante, M.; Accetturo, M.; Cafiero, C.; Ricciato, L.; et al. What Is Hidden in Patients with Unknown Nephropathy? Genetic Screening Could Be the Missing Link in Kidney Transplantation Diagnosis and Management. Int. J. Mol. Sci. 2024, 25, 1436. https://doi.org/10.3390/ijms25031436
Mitrotti A, Di Bari I, Giliberti M, Franzin R, Conserva F, Chiusolo A, Gigante M, Accetturo M, Cafiero C, Ricciato L, et al. What Is Hidden in Patients with Unknown Nephropathy? Genetic Screening Could Be the Missing Link in Kidney Transplantation Diagnosis and Management. International Journal of Molecular Sciences. 2024; 25(3):1436. https://doi.org/10.3390/ijms25031436
Chicago/Turabian StyleMitrotti, Adele, Ighli Di Bari, Marica Giliberti, Rossana Franzin, Francesca Conserva, Anna Chiusolo, Maddalena Gigante, Matteo Accetturo, Cesira Cafiero, Luisa Ricciato, and et al. 2024. "What Is Hidden in Patients with Unknown Nephropathy? Genetic Screening Could Be the Missing Link in Kidney Transplantation Diagnosis and Management" International Journal of Molecular Sciences 25, no. 3: 1436. https://doi.org/10.3390/ijms25031436
APA StyleMitrotti, A., Di Bari, I., Giliberti, M., Franzin, R., Conserva, F., Chiusolo, A., Gigante, M., Accetturo, M., Cafiero, C., Ricciato, L., Stea, E. D., Forleo, C., Gallone, A., Rossini, M., Fiorentino, M., Castellano, G., Pontrelli, P., & Gesualdo, L. (2024). What Is Hidden in Patients with Unknown Nephropathy? Genetic Screening Could Be the Missing Link in Kidney Transplantation Diagnosis and Management. International Journal of Molecular Sciences, 25(3), 1436. https://doi.org/10.3390/ijms25031436