
Diffuse Cutaneous Mastocytosis: A Current Understanding of a Rare Disease

 , ,
, ,  ,
,
Abstract
1. Introduction
2. Genetic Background of Diffuse Cutaneous Mastocytosis
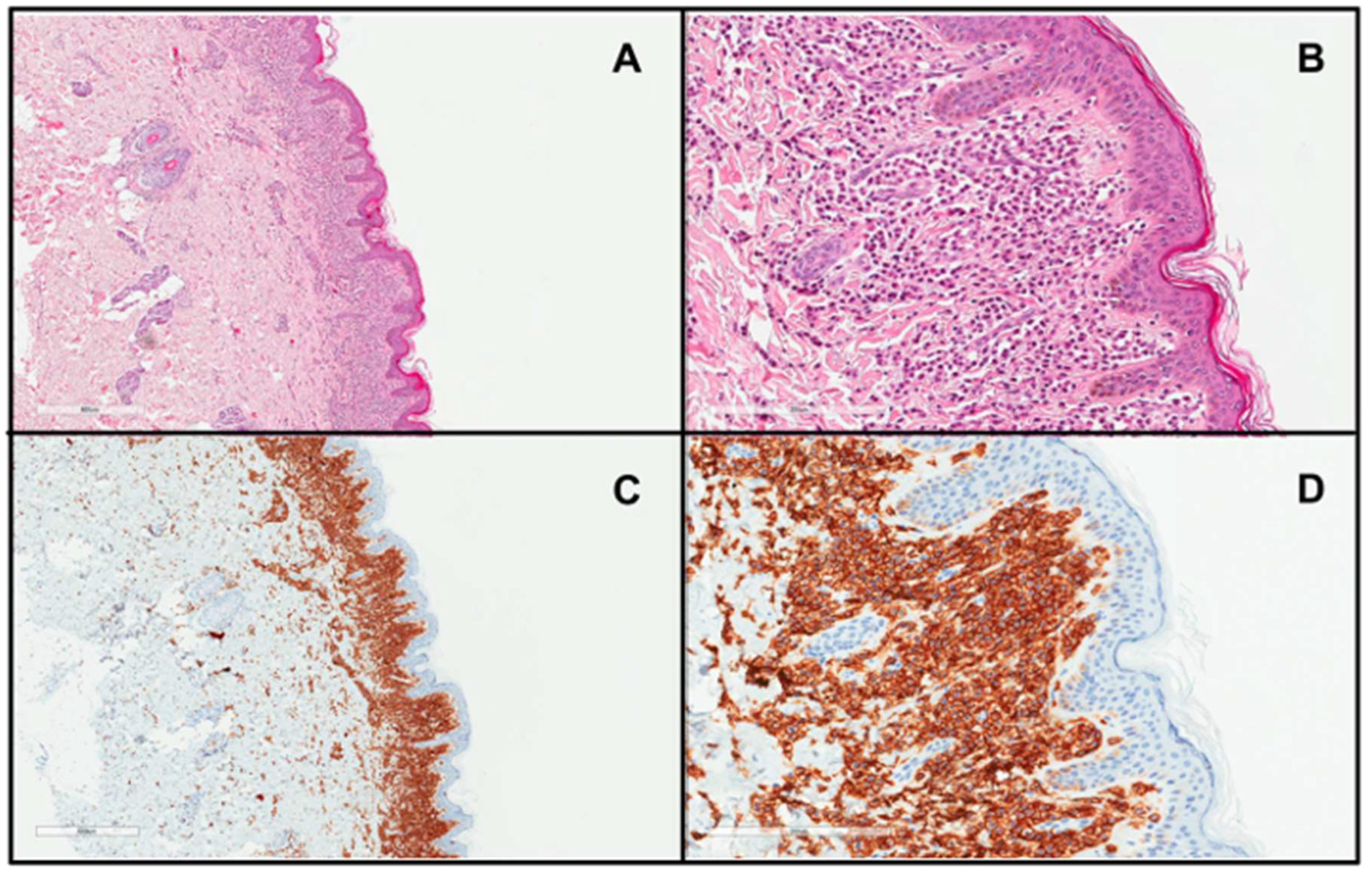
3. Clinical Presentation of Diffuse Cutaneous Mastocytosis
4. Diagnostics and Differential Diagnosis
5. Treatment
5.1. Topical Therapy
5.2. Oral Therapy
5.3. Phototherapy
6. Discussion
7. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Valent, P.; Akin, C.; Sperr, W.R.; Horny, H.-P.; Arock, M.; Metcalfe, D.D.; Galli, S.J. New Insights into the Pathogenesis of Mastocytosis: Emerging Concepts in Diagnosis and Therapy. Annu. Rev. Pathol. Mech. Dis. 2023, 18, 361–386. [Google Scholar] [CrossRef] [PubMed]
- Pardanani, A. Systemic Mastocytosis in Adults: 2023 Update on Diagnosis, Risk Stratification and Management. Am. J. Hematol. 2023, 98, 1097–1116. [Google Scholar] [CrossRef] [PubMed]
- Valent, P.; Akin, C.; Hartmann, K.; Alvarez-Twose, I.; Brockow, K.; Hermine, O.; Niedoszytko, M.; Schwaab, J.; Lyons, J.J.; Carter, M.C.; et al. Updated Diagnostic Criteria and Classification of Mast Cell Disorders: A Consensus Proposal. Hemasphere 2021, 5, e646. [Google Scholar] [CrossRef]
- Valent, P.; Akin, C.; Metcalfe, D.D. Mastocytosis: 2016 Updated WHO Classification and Novel Emerging Treatment Concepts. Blood 2017, 129, 1420–1427. [Google Scholar] [CrossRef] [PubMed]
- Khoury, J.D.; Solary, E.; Abla, O.; Akkari, Y.; Alaggio, R.; Apperley, J.F.; Bejar, R.; Berti, E.; Busque, L.; Chan, J.K.C.; et al. The 5th Edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms. Leukemia 2022, 36, 1703–1719. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, K.; Escribano, L.; Grattan, C.; Brockow, K.; Carter, M.C.; Alvarez-Twose, I.; Matito, A.; Broesby-Olsen, S.; Siebenhaar, F.; Lange, M.; et al. Cutaneous Manifestations in Patients with Mastocytosis: Consensus Report of the European Competence Network on Mastocytosis; The American Academy of Allergy, Asthma & Immunology; And the European Academy of Allergology and Clinical Immunology. J. Allergy Clin. Immunol. 2016, 137, 35–45. [Google Scholar] [CrossRef]
- Méni, C.; Bruneau, J.; Georgin-Lavialle, S.; Le Saché De Peufeilhoux, L.; Damaj, G.; Hadj-Rabia, S.; Fraitag, S.; Dubreuil, P.; Hermine, O.; Bodemer, C. Paediatric Mastocytosis: A Systematic Review of 1747 Cases. Br. J. Dermatol. 2015, 172, 642–651. [Google Scholar] [CrossRef] [PubMed]
- Lange, M.; Hartmann, K.; Carter, M.C.; Siebenhaar, F.; Alvarez-twose, I.; Torrado, I.; Brockow, K.; Renke, J.; Irga-jaworska, N.; Plata-nazar, K.; et al. Molecular Background, Clinical Features and Management of Pediatric Mastocytosis: Status 2021. Int. J. Mol. Sci. 2021, 22, 2586. [Google Scholar] [CrossRef]
- Valent, P.; Akin, C.; Escribano, L.; Födinger, M.; Hartmann, K.; Brockow, K.; Castells, M.; Sperr, W.R.; Kluin-Nelemans, H.C.; Hamdy, N.A.T.; et al. Standards and Standardization in Mastocytosis: Consensus Statements on Diagnostics, Treatment Recommendations and Response Criteria. Eur. J. Clin. Investig. 2007, 37, 435–453. [Google Scholar] [CrossRef]
- Lange, M.; Niedoszytko, M.; Renke, J.; Gleń, J.; Nedoszytko, B. Clinical Aspects of Paediatric Mastocytosis: A Review of 101 Cases. J. Eur. Acad. Dermatol. Venereol. 2011, 27, 97–102. [Google Scholar] [CrossRef]
- Hannaford, R.; Rogers, M. Presentation of Cutaneous Mastocytosis in 173 Children. Australas. J. Dermatol. 2001, 42, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Lange, M.; Zawadzka, A.; Schrörs, S.; Słomka, J.; Ługowska-Umer, H.; Nedoszytko, B.; Nowicki, R. The Role of Serum Tryptase in the Diagnosis and Monitoring of Pediatric Mastocytosis: A Single-Center Experience. Adv. Dermatol. Alergol. 2017, 4, 306–312. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Twose, I.; Vañõ-Galván, S.; Sánchez-Muñoz, L.; Morgado, J.M.; Matito, A.; Torrelo, A.; Jaén, P.; Schwartz, L.B.; Orfao, A.; Escribano, L. Increased Serum Baseline Tryptase Levels and Extensive Skin Involvement Are Predictors for the Severity of Mast Cell Activation Episodes in Children with Mastocytosis. Allergy 2012, 67, 813–821. [Google Scholar] [CrossRef] [PubMed]
- Polivka, L.; Rossignol, J.; Neuraz, A.; Condé, D.; Agopian, J.; Méni, C.; Garcelon, N.; Dubreuil, P.; Maouche-Chrétien, L.; Hadj-Rabia, S.; et al. Criteria for the Regression of Pediatric Mastocytosis: A Long-Term Follow-Up. J. Allergy Clin. Immunol. Pract. 2021, 9, 1695–1704.e5. [Google Scholar] [CrossRef] [PubMed]
- Wiechers, T.; Rabenhorst, A.; Schick, T.; Preussner, L.M.; Förster, A.; Valent, P.; Horny, H.-P.; Sotlar, K.; Hartmann, K. Large Maculopapular Cutaneous Lesions Are Associated with Favorable Outcome in Childhood-Onset Mastocytosis. J. Allergy Clin. Immunol. 2015, 136, 1581–1590.e3. [Google Scholar] [CrossRef]
- Méni, C.; Georgin-Lavialle, S.; Le Saché de Peufeilhoux, L.; Jais, J.P.; Hadj-Rabia, S.; Bruneau, J.; Fraitag, S.; Hanssens, K.; Dubreuil, P.; Hermine, O.; et al. Paediatric Mastocytosis: Long-Term Follow-up of 53 Patients with Whole Sequencing of KIT. A Prospective Study. Br. J. Dermatol. 2018, 179, 925–932. [Google Scholar] [CrossRef]
- Valent, P.; Hartmann, K.; Bonadonna, P.; Niedoszytko, M.; Triggiani, M.; Arock, M.; Brockow, K. Mast Cell Activation Syndromes: Collegium Internationale Allergologicum Update 2022. Int. Arch. Allergy Immunol. 2022, 183, 693–705. [Google Scholar] [CrossRef]
- Bodemer, C.; Hermine, O.; Palmérini, F.; Yang, Y.; Grandpeix-Guyodo, C.; Leventhal, P.S.; Hadj-Rabia, S.; Nasca, L.; Georgin-Lavialle, S.; Cohen-Akenine, A.; et al. Pediatric Mastocytosis Is a Clonal Disease Associated with D816V and Other Activating C-KIT Mutations. J. Investig. Dermatol. 2010, 130, 804–815. [Google Scholar] [CrossRef]
- Heide, R.; Zuidema, E.; Beishuizen, A.; Den Hollander, J.C.; Van Gysel, D.; Seyger, M.M.B.; Pasmans, S.G.M.A.; Kakourou, T.; Oranje, A.P. Clinical Aspects of Diffuse Cutaneous Mastocytosis in Children: Two Variants. Dermatology 2009, 219, 309–315. [Google Scholar] [CrossRef]
- Gülen, T. A Puzzling Mast Cell Trilogy: Anaphylaxis, MCAS, and Mastocytosis. Diagnostics 2023, 13, 3307. [Google Scholar] [CrossRef]
- Valent, P.; Akin, C.; Hartmann, K.; Nilsson, G.; Reiter, A.; Hermine, O.; Sotlar, K.; Sperr, W.R.; Escribano, L.; George, T.I.; et al. Mast Cells as a Unique Hematopoietic Lineage and Cell System: From Paul Ehrlich’s Visions to Precision Medicine Concepts. Theranostics 2020, 10, 10743–10768. [Google Scholar] [CrossRef]
- Valent, P. KIT D816V and the Cytokine Storm in Mastocytosis: Production and Role of Interleukin-6. Haematologica 2020, 105, 5–6. [Google Scholar] [CrossRef] [PubMed]
- Tobío, A.; Bandara, G.; Morris, D.A.; Kim, D.-K.; O’Connell, M.P.; Komarow, H.D.; Carter, M.C.; Smrz, D.; Metcalfe, D.D.; Olivera, A. Oncogenic D816V-KIT Signaling in Mast Cells Causes Persistent IL-6 Production. Haematologica 2020, 105, 124–135. [Google Scholar] [CrossRef] [PubMed]
- Lange, M.; Gleń, J.; Zabłotna, M.; Nedoszytko, B.; Sokołowska-Wojdyło, M.; Rębała, K.; Ługowska-Umer, H.; Niedoszytko, M.; Górska, A.; Sikorska, M.; et al. Interleukin-31 Polymorphisms and Serum IL-31 Level in Patients with Mastocytosis: Correlation with Clinical Presentation and Pruritus. Acta Derm. Venereol. 2017, 97, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Ługowska-Umer, H.; Zabłotna, M.; Lange, M.; Niedoszytko, M.; Nowicki, R.; Nedoszytko, B. -2518A/G Polymorphism of Monocyte Chemotactic Protein 1 (MCP-1/CCL2) Is Associated with Cutaneous Mastocytosis. Adv. Dermatol. Alergol. 2021, 38, 1039–1043. [Google Scholar] [CrossRef] [PubMed]
- Hoermann, G.; Cerny-Reiterer, S.; Perné, A.; Klauser, M.; Hoetzenecker, K.; Klein, K.; Müllauer, L.; Gröger, M.; Nijman, S.M.B.; Klepetko, W.; et al. Identification of Oncostatin M as a STAT5-Dependent Mediator of Bone Marrow Remodeling in KIT D816V-Positive Systemic Mastocytosis. Am. J. Pathol. 2011, 178, 2344–2356. [Google Scholar] [CrossRef] [PubMed]
- Greiner, G.; Witzeneder, N.; Berger, A.; Schmetterer, K.; Eisenwort, G.; Schiefer, A.-I.; Roos, S.; Popow-Kraupp, T.; Müllauer, L.; Zuber, J.; et al. CCL2 Is a KIT D816V–Dependent Modulator of the Bone Marrow Microenvironment in Systemic Mastocytosis. Blood 2017, 129, 371–382. [Google Scholar] [CrossRef] [PubMed]
- Kleewein, K.; Lang, R.; Diem, A.; Vogel, T.; Pohla-Gubo, G.; Bauer, J.W.; Hintner, H.; Laimer, M. Diffuse Cutaneous Mastocytosis Masquerading as Epidermolysis Bullosa. Pediatr. Dermatol. 2011, 28, 720–725. [Google Scholar] [CrossRef]
- Brockow, K.; Jofer, C.; Behrendt, H.; Ring, J. Anaphylaxis in Patients with Mastocytosis: A Study on History, Clinical Features and Risk Factors in 120 Patients. Allergy 2008, 63, 226–232. [Google Scholar] [CrossRef]
- Brockow, K.; Plata-Nazar, K.; Lange, M.; Nedoszytko, B.; Niedoszytko, M.; Valent, P. Mediator-Related Symptoms and Anaphylaxis in Children with Mastocytosis. Int. J. Mol. Sci. 2021, 22, 2684. [Google Scholar] [CrossRef]
- Rama, T.A.; Castells, M. Triggers of Anaphylaxis in Mastocytosis Patients: Evidence of the Current Drug-Avoidance Recommendation. Curr. Treat. Options Allergy 2023, 10, 442–457. [Google Scholar] [CrossRef]
- Gülen, T. Management of Mediator Symptoms, Allergy, and Anaphylaxis in Mastocytosis. Immunol. Allergy. Clin. N. Am. 2023, 43, 681–698. [Google Scholar] [CrossRef] [PubMed]
- Couto, S.; Rama, T.; Martins, C.; Miguel Borrego, L. Mast Cells and Mast Cell Activation Syndrome: What’s New? Mastócitos e Síndrome de Ativação Mastocitária: O Que Há de Novo? Arq. Asma Alerg. Imunol. 2023, 7, 69–77. [Google Scholar] [CrossRef]
- González de Olano, D.; De La Hoz Caballer, B.; Núñez López, R.; Sánchez Muñoz, L.; Cuevas Agustín, M.; Diéguez, M.C.; Álvarez Twose, I.; Castells, M.C.; Escribano Mora, L. Prevalence of Allergy and Anaphylactic Symptoms in 210 Adult and Pediatric Patients with Mastocytosis in Spain: A Study of the Spanish Network on Mastocytosis (REMA). Clin. Exp. Allergy 2007, 37, 1547–1555. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, N.; Shapiro, N.; Bhutada, A.; Rastogi, S. C-KIT-Positive Fatal Diffuse Cutaneous Mastocytosis with Systemic Manifestations in a Neonate. J. Pediatr. Hematol. Oncol. 2019, 41, e338–e340. [Google Scholar] [CrossRef] [PubMed]
- Otani, I.M.; Carroll, R.W.; Yager, P.; Kroshinsky, D.; Murphy, S.; Hornick, J.L.; Akin, C.; Castells, M.; Walter, J.E. Diffuse Cutaneous Mastocytosis with Novel Somatic KIT Mutation K509I and Association with Tuberous Sclerosis. Clin. Case Rep. 2018, 6, 1834–1840. [Google Scholar] [CrossRef] [PubMed]
- Chan, I.J.; Tharp, M.D. Comparison of Lesional Skin C-KIT Mutations with Clinical Phenotype in Patients with Mastocytosis. Clin. Exp. Dermatol. 2018, 43, 416–422. [Google Scholar] [CrossRef]
- Olteanu, E.-G.; Bataneant, M.; Puiu, M.; Chirita-Emandi, A. When Mast Cells Run Amok: A Comprehensive Review and Case Study on Severe Neonatal Diffuse Cutaneous Mastocytosis. Genes 2023, 14, 2021. [Google Scholar] [CrossRef]
- Shah, P.Y.; Sharma, V.; Worobec, A.S.; Metcalfe, D.D.; Zwick, D.C. Congenital Bullous Mastocytosis with Myeloproliferative Disorder and C-Kit Mutation. J. Am. Acad. Dermatol. 1998, 39, 119–121. [Google Scholar] [CrossRef]
- Jenkinson, H.A.; Lundgren, A.D.; Carter, M.C.; Diaz, L.Z.; Levy, M.L. Management of a Neonate with Diffuse Cutaneous Mastocytosis: Case Report and Literature Review. Pediatr. Dermatol. 2019, 36, 486–489. [Google Scholar] [CrossRef]
- Li, Y.; Li, X.; Liu, X.; Kang, L.; Liu, X. Genotypic and Phenotypic Characteristics of Chinese Neonates with Cutaneous Mastocytosis: A Case Report and Literature Review. J. Int. Med. Res. 2020, 48, 0300060520952621. [Google Scholar] [CrossRef] [PubMed]
- Fowler, J.F.; Parsley, W.M.; Cotter, P.G. Familial Urticaria Pigmentosa. Arch. Dermatol. 1986, 122, 80–81. [Google Scholar] [CrossRef] [PubMed]
- Wöhrl, S.; Moritz, K.B.; Bracher, A.; Fischer, G.; Stingl, G.; Loewe, R. A C-Kit Mutation in Exon 18 in Familial Mastocytosis. J. Investig. Dermatol. 2013, 133, 839–841. [Google Scholar] [CrossRef]
- Zanotti, R.; Simioni, L.; Garcia-Montero, A.C.; Perbellini, O.; Bonadonna, P.; Caruso, B.; Jara-Acevedo, M.; Bonifacio, M.; De Matteis, G. Somatic D816V KIT Mutation in a Case of Adult-Onset Familial Mastocytosis. J. Allergy Clin. Immunol. 2013, 131, 605–607. [Google Scholar] [CrossRef]
- de Melo Campos, P.; Machado-Neto, J.A.; Scopim-Ribeiro, R.; Visconte, V.; Tabarroki, A.; Duarte, A.S.S.; Barra, F.F.C.; Vassalo, J.; Rogers, H.J.; Lorand-Metze, I.; et al. Familial Systemic Mastocytosis with Germline KIT K509I Mutation Is Sensitive to Treatment with Imatinib, Dasatinib and PKC412. Leuk. Res. 2014, 38, 1245–1251. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.Y.; Smith, M.L.; Schultheis, B.; Fitzgibbon, J.; Lister, T.A.; Melo, J.V.; Cross, N.C.P.; Cavenagh, J.D. A Novel K509I Mutation of KIT Identified in Familial Mastocytosis—In Vitro and in Vivo Responsiveness to Imatinib Therapy. Leuk. Res. 2006, 30, 373–378. [Google Scholar] [CrossRef] [PubMed]
- Arock, M.; Sotlar, K.; Akin, C.; Broesby-Olsen, S.; Hoermann, G.; Escribano, L.; Kristensen, T.K.; Kluin-Nelemans, H.C.; Hermine, O.; Dubreuil, P.; et al. KIT Mutation Analysis in Mast Cell Neoplasms: Recommendations of the European Competence Network on Mastocytosis. Leukemia 2015, 29, 1223–1232. [Google Scholar] [CrossRef] [PubMed]
- Chan, E.C.; Bai, Y.; Kirshenbaum, A.S.; Fischer, E.R.; Simakova, O.; Bandara, G.; Scott, L.M.; Wisch, L.B.; Cantave, D.; Carter, M.C.; et al. Mastocytosis Associated with a Rare Germline KIT K509I Mutation Displays a Well-Differentiated Mast Cell Phenotype. J. Allergy Clin. Immunol. 2014, 134, 178. [Google Scholar] [CrossRef]
- Ke, H.; Kazi, J.U.; Zhao, H.; Sun, J. Germline Mutations of KIT in Gastrointestinal Stromal Tumor (GIST) and Mastocytosis. Cell Biosci. 2016, 6, 55. [Google Scholar] [CrossRef]
- Hartmann, K.; Wardelmann, E.; Ma, Y.; Merkelbach–Bruse, S.; Preussner, L.M.; Woolery, C.; Baldus, S.E.; Heinicke, T.; Thiele, J.; Buettner, R.; et al. Novel Germline Mutation of KIT Associated With Familial Gastrointestinal Stromal Tumors and Mastocytosis. Gastroenterology 2005, 129, 1042–1046. [Google Scholar] [CrossRef]
- Trevisan, G.; Pauluzzi, P.; Gatti, A.; Semeraro, A. Familial Mastocytosis Associated with Neurosensory Deafness. J. Eur. Acad. Dermatol. Venereol. 2000, 14, 119–122. [Google Scholar] [CrossRef] [PubMed]
- Wangberg, H.; Willis, M.J.H.; Lindsey, D.; Schmidgal, E.C.; White, A.A. A Familial Case of Diffuse Cutaneous Mastocytosis. J. Allergy Clin. Immunol. Pract. 2023, 11, 3802–3803. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Boxer, M.; Drummond, A.; Ogston, P.; Hodgins, M.; Burden, A.D. A Germline Mutation in KIT in Familial Diffuse Cutaneous Mastocytosis. J. Med. Genet. 2004, 41, e88. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.J.; Lin, Z.M.; Zhang, J.; Yin, J.H.; Yang, Y. A New Germline Mutation in KIT Associated with Diffuse Cutaneous Mastocytosis in a Chinese Family. Clin. Exp. Dermatol. 2014, 39, 146–149. [Google Scholar] [CrossRef]
- Peters, F.; Fiebig, B.; Lundberg, P.; Jaspers, N.I.; Holzapfel, B.; Ghadimi, M.P.H.; Drebber, U.; Tuchscherer, A.; Ullrich, R.; Hartmann, K.; et al. Detection of the Germline KIT S476I Mutation in a Kindred with Familial Mastocytosis Associated with Gastrointestinal Stromal Tumors. J. Allergy Clin. Immunol. Pract. 2021, 9, 2123–2125.e1. [Google Scholar] [CrossRef]
- Alvarez-Twose, I.; Jara-Acevedo, M.; Morgado, J.M.; García-Montero, A.; Sánchez-Muñoz, L.; Teodósio, C.; Matito, A.; Mayado, A.; Caldas, C.; Mollejo, M.; et al. Clinical, Immunophenotypic, and Molecular Characteristics of Well-Differentiated Systemic Mastocytosis. J. Allergy Clin. Immunol. 2016, 137, 168–178.e1. [Google Scholar] [CrossRef]
- Alvarez-Twose, I.; Matito, A.; Morgado, J.M.; Sánchez-Muñoz, L.; Jara-Acevedo, M.; García-Montero, A.; Mayado, A.; Caldas, C.; Teodósio, C.; Muñoz-González, J.I.; et al. Imatinib in Systemic Mastocytosis: A Phase IV Clinical Trial in Patients Lacking Exon 17 KIT Mutations and Review of the Literature. Oncotarget 2017, 8, 68950–68963. [Google Scholar] [CrossRef]
- Hosking, A.M.; Makdisi, J.; Ortenzio, F.; de Feraudy, S.; Smith, J.; Linden, K. Diffuse Cutaneous Mastocytosis: Case Report and Literature Review. Pediatr. Dermatol. 2018, 35, e348–e352. [Google Scholar] [CrossRef]
- Lange, M.; Niedoszytko, M.; Nedoszytko, B.; Łata, J.; Trzeciak, M.; Biernat, W. Diffuse Cutaneous Mastocytosis: Analysis of 10 Cases and a Brief Review of the Literature. J. Eur. Acad. Dermatol. Venereol. 2012, 26, 1565–1571. [Google Scholar] [CrossRef]
- Willemze, R.; Ruiter, D.J.; Scheffer, E.; van Vloten, W.A. Diffuse Cutaneous Mastocytosis with Multiple Cutaneous Mastocytomas. Br. J. Dermatol. 1980, 102, 601–607. [Google Scholar] [CrossRef]
- Renke, J.; Irga-Jaworska, N.; Lange, M. Pediatric and Hereditary Mastocytosis. Immunol. Allergy Clin. N. Am. 2023, 43, 665–679. [Google Scholar] [CrossRef]
- Park, M.-N.; Kim, G.-A.; Chey, M.J.; Shim, G.H. A Case of Diffuse Cutaneous Mastocytosis in a Newborn. Korean J. Perinatol. 2014, 25, 105. [Google Scholar] [CrossRef]
- Orkin, M.; Good, R.A.; Clawson, C.C.; Fisher, I.; Windhorst, D.B. Bullous Mastocytosis. Arch. Derm. 1970, 101, 547–564. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, T.; Kukita, A. A Fatal Case of Purely Cutaneous Form of Diffuse Mastocytosis. Proc. XII Int. Cong Dermatol. 1962, 2, 1558–1561. [Google Scholar]
- Allison, J. Skin Mastocytosis Presenting as a Neonatal Bullous Eruption. Australas. J. Dermatol. 1967, 9, 83–85. [Google Scholar] [CrossRef]
- Klaber, M.; Pegum, J.S. Diffuse Cutaneous Mastocytosis in Mother and Daughter. Proc. R. Soc. Med. 1976, 69, 16. [Google Scholar] [CrossRef] [PubMed]
- Harrison, P.V.; Cook, L.J.; Lake, H.J.; Shuster, S. Diffuse Cutaneous Mastocytosis: A Report of Neonatal Onset. Acta Derm. Venereol. 1979, 59, 541–543. [Google Scholar] [CrossRef] [PubMed]
- Olgun, N.; Oren, H.; Oren, B.; Irken, G.; Polat, M.; Cevik, N. Diffuse Erythrodermie Cutaneous M Astocytosis with Bone Marrow Infiltration. Dermatology 1993, 187, 127–129. [Google Scholar] [CrossRef]
- Murphy, M.; Walsh, D.; Drumm, B.; Watson, R. Bullous Mastocytosis: A Fatal Outcome. Pediatr. Dermatol. 1999, 16, 452–455. [Google Scholar] [CrossRef]
- Enomoto, U.; Kusakabe, H.; Matsumura, T.; Kuno, T.; Tamai, H.; Kiyokane, K. Diffuse Cutaneous Mastocytosis Responding to Cyproheptadine. Clin. Exp. Dermatol. 1999, 24, 16–18. [Google Scholar] [CrossRef]
- Waxtein, L.M.; Elisa Vega-Memije, M.; Cortés-Franco, R.; Dominguez-Soto, L. Diffuse Cutaneous Mastocytosis with Bone Marrow Infiltration in a Child: A Case Report. Pediatr. Dermatol. 2000, 17, 198–201. [Google Scholar] [CrossRef] [PubMed]
- Kinsler, V.A.; Hawk, J.L.M.; Atherton, D.J. Diffuse Cutaneous Mastocytosis Treated with Psoralen Photochemotherapy: Case Report and Review of the Literature. Br. J. Dermatol. 2005, 152, 179–180. [Google Scholar] [CrossRef] [PubMed]
- Walker, T.; von Komorowski, G.; Scheurlen, W.; Dorn-Beineke, A.; Back, W.; Bayerl, C. Neonatal Mastocytosis with Pachydermic Bullous Skin without C-Kit 816 Mutation. Dermatology 2006, 212, 70–72. [Google Scholar] [CrossRef] [PubMed]
- Duckworth, A.K.; Bhatti, A.; Barnes, C. Diffuse Cutaneous Mastocytosis in Fraternal Twins. Int. J. Dermatol. 2009, 48, 170–172. [Google Scholar] [CrossRef]
- Ghiasi, M.; Ghanadan, A.; Jesri, S.B.; Sotudeh, S.; Ramyar, A. Diffuse Cutaneous Mastocytosis: Report of a Severe Case with Fatal Outcome. Dermatol. Online J. 2011, 17, 7. [Google Scholar] [CrossRef] [PubMed]
- Koga, H.; Kokubo, T.; Akaishi, M.; Iida, K.; Korematsu, S. Neonatal Onset Diffuse Cutaneous Mastocytosis: A Case Report and Review of the Literature. Pediatr. Dermatol. 2011, 28, 542–546. [Google Scholar] [CrossRef] [PubMed]
- Wawrzycki, B.; Pietrzak, A.; Chodorowska, G.; Kanitakis, J. Diffuse Cutaneous Bullous Mastocytosis in a Newborn. Dermatol. Ther. 2013, 26, 176–179. [Google Scholar] [CrossRef]
- Gupta, M.; Akin, C.; Sanders, G.M.; Chan, M.P.; Ross, C.W.; Castells, M.C. Blisters, Vaccines, and Mast Cells: A Difficult Case of Diffuse Cutaneous Mastocytosis. J. Allergy Clin. Immunol. Pract. 2019, 7, 1370–1372. [Google Scholar] [CrossRef]
- Cardoso, J.M.; Cabral, C.A.S.; Lellis, R.F.; Ravelli, F.N. Bullous Congenital Diffuse Cutaneous Mastocytosis. An. Bras. Dermatol. 2020, 95, 255–256. [Google Scholar] [CrossRef]
- Turnbull, L.; Calhoun, D.A.; Agarwal, V.; Drehner, D.; Chua, C. Congenital Mastocytosis: Case Report and Review of the Literature. Cureus 2020, 12, e10565. [Google Scholar] [CrossRef]
- Rayinda, T.; Mira Oktarina, D.A.; Danarti, R. Diffuse Cutaneous Mastocytosis Masquerading as Linear IgA Bullous Dermatosis of Childhood. Dermatol. Rep. 2021, 13, 9021. [Google Scholar] [CrossRef]
- Özdemir, Ö. Cutaneous Mastocytosis in Childhood: An Update from the Literature. J. Clin. Pract. Res. 2023, 45, 311–320. [Google Scholar] [CrossRef]
- Tiano, R.; Krase, I.Z.; Sacco, K. Updates in Diagnosis and Management of Paediatric Mastocytosis. Curr. Opin. Allergy Clin. Immunol. 2023, 23, 158–163. [Google Scholar] [CrossRef]
- Golitz, L.E.; Weston, W.L.; Lane, A.T. Bullous Mastocytosis: Diffuse Cutaneous Mastocytosis with Extensive Blisters Mimicking Scalded Skin Syndrome or Erythema Multiforme. Pediatr. Dermatol. 1984, 1, 288–294. [Google Scholar] [CrossRef] [PubMed]
- Oranje, A.P.; Soekanto, W.; Sukardi, A.; Vuzevski, V.D.; Willigen, A.V.D.; Afiani, H.M. Diffuse Cutaneous Mastocytosis Mimicking Staphylococcal Scalded-Skin Syndrome: Report of Three Cases. Pediatr. Dermatol. 1991, 8, 147–151. [Google Scholar] [CrossRef]
- Salas-Alanis, J.C.; Rosales-Mendoza, C.E.; Ocampo-Candiani, J. Bullous Mastocytosis Mimicking Congenital Epidermolysis Bullosa. Case Rep. Dermatol. 2014, 6, 129–133. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J. Bullous Cutaneous Mastocytosis, a Rarely Reported Disease in Asian Children. Asian Pac. J. Allergy Immunol. 2014, 32, 354–357. [Google Scholar] [CrossRef]
- Jezierska, M.; Stefanowicz, J.; Romanowicz, G.; Kosiak, W.; Lange, M. Langerhans Cell Histiocytosis in Children—A Disease with Many Faces. Recent Advances in Pathogenesis, Diagnostic Examinations and Treatment. Adv. Dermatol. Alergol. 2018, 35, 6–17. [Google Scholar] [CrossRef]
- Abuhay, H.; Clark, A.S.; Carter, M.C. Occurrence of Unexpected Adverse Reactions to Vaccines in Children with Mastocytosis. J. Pediatr. Res. 2020, 7, 81–86. [Google Scholar] [CrossRef]
- Parente, R.; Pucino, V.; Magliacane, D.; Petraroli, A.; Loffredo, S.; Marone, G.; Triggiani, M. Evaluation of Vaccination Safety in Children with Mastocytosis. Pediatr. Allergy Immunol. 2017, 28, 93–95. [Google Scholar] [CrossRef]
- González-González, O.; Leal, E.; Martín-Martínez, M.; Bautista, L.; Ballesteros, M.P.; Torrado, J.J.; Serrano, D.R. Guiding Clinical Prescription of Topical Extemporaneous Formulations of Sodium Cromoglycate Based on Pharmaceutical Performance. Pharmaceutics 2023, 15, 1609. [Google Scholar] [CrossRef] [PubMed]
- Siebenhaar, F.; Akin, C.; Bindslev-Jensen, C.; Maurer, M.; Broesby-Olsen, S. Treatment Strategies in Mastocytosis. Immunol. Allergy Clin. N. Am. 2014, 34, 433–447. [Google Scholar] [CrossRef] [PubMed]
- Sandru, F.; Petca, R.C.; Costescu, M.; Dumitrașcu, M.C.; Popa, A.; Petca, A.; Miulescu, R.G. Cutaneous Mastocytosis in Childhood—Update from the Literature. J. Clin. Med. 2021, 10, 1474. [Google Scholar] [CrossRef] [PubMed]
- Muraro, A.; Roberts, G.; Worm, M.; Bilò, M.B.; Brockow, K.; Fernández Rivas, M.; Santos, A.F.; Zolkipli, Z.Q.; Bellou, A.; Beyer, K.; et al. Anaphylaxis: Guidelines from the European Academy of Allergy and Clinical Immunology. Allergy 2014, 69, 1026–1045. [Google Scholar] [CrossRef] [PubMed]
- Morren, M.-A.; Hoppé, A.; Renard, M.; Debiec Rychter, M.; Uyttebroeck, A.; Dubreuil, P.; Martin, L. Imatinib Mesylate in the Treatment of Diffuse Cutaneous Mastocytosis. J. Pediatr. 2013, 162, 205–207. [Google Scholar] [CrossRef] [PubMed]
- Hughes, J.D.M.; Olynyc, T.; Chapdelaine, H.; Segal, L.; Miedzybrodzki, B.; Ben-Shoshan, M. Effective Management of Severe Cutaneous Mastocytosis in Young Children with Omalizumab (Xolair®). Clin. Exp. Dermatol. 2018, 43, 573–576. [Google Scholar] [CrossRef]
- Calzavara-Pinton, P.; Bettolini, L.; Tonon, F.; Rossi, M.; Venturini, M. The Realistic Positioning of UVA1 Phototherapy after 25 Years of Clinical Experience and the Availability of New Biologics and Small Molecules: A Retrospective Clinical Study. Front. Med. 2023, 10, 1295145. [Google Scholar] [CrossRef]
- Czarny, J.; Żuk, M.; Żawrocki, A.; Plata-Nazar, K.; Biernat, W.; Niedoszytko, M.; Ługowska-Umer, H.; Nedoszytko, B.; Wasąg, B.; Nowicki, R.; et al. New Approach to Paediatric Mastocytosis: Implications of KIT D816V Mutation Detection in Peripheral Blood. Acta Derm. Venereol. 2020, 100, adv00149. [Google Scholar] [CrossRef]
- Le, M.; Miedzybrodzki, B.; Olynych, T.; Chapdelaine, H.; Ben-Shoshan, M. Natural History and Treatment of Cutaneous and Systemic Mastocytosis. Postgrad. Med. 2017, 129, 896–901. [Google Scholar] [CrossRef]
- Czarny, J.; Renke, J.; Żawrocki, A.; Nowicki, R.J.; Lange, M. Natural Evolution in Pediatric Cutaneous Mastocytosis: 10-year Follow-up. Int. J. Dermatol. 2021, 60, 1253–1257. [Google Scholar] [CrossRef]
- Huang, A.; Fiadorchanka, N.; Brar, K.; Balderacchi, J.L.; Glick, S.A. In Utero Presentation of Aggressive Systemic Mastocytosis in a Neonate. Br. J. Dermatol. 2017, 177, 1439–1441. [Google Scholar] [CrossRef] [PubMed]
- Angus, J.; Leach, I.H.; Grant, J.; Ravenscroft, J.C. Systemic Mastocytosis with Diffuse Cutaneous Involvement and Haematological Disease Presenting in Utero Treated Unsuccessfully with Vincristine. Clin. Exp. Dermatol. 2008, 33, 36–39. [Google Scholar] [CrossRef] [PubMed]
- Waters, W.; Lacson, P. Mast Cell Leukemia as Urticaria Pigmentosa: Report of a Case. Pediatrics 1957, 19, 1033–1042. [Google Scholar] [CrossRef] [PubMed]
- Meneghini, C.L.; Angelini, G. Systemic Mastocytosis with Diffuse Crocodile-like Pachydermic Skin, Pedunculated Pseudofibromas and Comedones. Br. J. Dermatol. 1980, 103, 329–334. [Google Scholar] [CrossRef]
- Rayner, J.E.; Laino, A.M.; Nufer, K.L.; Adams, L.; Raphael, A.P.; Menzies, S.W.; Soyer, H.P. Clinical Perspective of 3D Total Body Photography for Early Detection and Screening of Melanoma. Front. Med. 2018, 5, 152. [Google Scholar] [CrossRef] [PubMed]
- An, X.; Al-Muriesh, M.; Abdul-Fattah, B.; Gao, Y.; Tao, J.; Yang, J.; Huang, C. Reflectance Confocal Microscopy as a Diagnostic Tool for Mastocytoma in Children. J. Am. Acad. Dermatol. 2020, 83, 1781–1784. [Google Scholar] [CrossRef] [PubMed]
- Kröger, M.; Scheffel, J.; Nikolaev, V.V.; Shirshin, E.A.; Siebenhaar, F.; Schleusener, J.; Lademann, J.; Maurer, M.; Darvin, M.E. In Vivo Non-Invasive Staining-Free Visualization of Dermal Mast Cells in Healthy, Allergy and Mastocytosis Humans Using Two-Photon Fluorescence Lifetime Imaging. Sci. Rep. 2020, 10, 14930. [Google Scholar] [CrossRef]
- Siebenhaar, F.; Altrichter, S.; Bonnekoh, H.; Hawro, T.; Hawro, M.; Michaelis, E.G.; Kantor, A.M.; Chang, A.T.; Youngblood, B.A.; Singh, B.; et al. Safety and Efficacy of Lirentelimab in Patients with Refractory Indolent Systemic Mastocytosis: A First-in-Human Clinical Trial. Br. J. Dermatol. 2023, 189, 511–519. [Google Scholar] [CrossRef]
- Metz, M.; Kolkhir, P.; Altrichter, S.; Siebenhaar, F.; Levi-Schaffer, F.; Youngblood, B.A.; Church, M.K.; Maurer, M. Mast Cell Silencing: A Novel Therapeutic Approach for Urticaria and Other Mast Cell-mediated Diseases. Allergy 2023, 79, 37–51. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
Cutaneous mastocytosis (CM)
|
| Systemic mastocytosis (SM) Non-advanced forms of SM
|
| Mast cell sarcoma (MCS) |
| Disease Onset | Follow-Up Time | Anaphylaxis | Clinical Features and Outcome | |
|---|---|---|---|---|
| Yasuda T. and Kukita A. 1962 [64] | At birth | 3 months | NA | Hemorrhagic bullous lesions, diffuse brown pigmentation of the skin Episodes of severe fever and dyspnea No evidence of SM Death at 3 months of age |
| Allison J. 1967 [65] | At birth | 8 days | NA | Generalized blistering, erythema Death at 8 days of age |
| Orkin et al., 1970 [63] | 5 months | 3 years | Once, 30 min after a meal | Generalized erythema, blistering, thickened skin No evidence of SM Partial improvement of skin lesions |
| Klaber M. et al., 1976 [66] | 6 months and 2 months | 25 years and 3 months | NA | Two cases Persistently thickened skin and pronounced Dermographism Erythematous, leathery skin |
| Harrison P. et al., 1979 [67] | 20 days | 4 years | NA | In the infantile period, erythema, blistering, and diffuse thickening of the skin with a leather-like appearance At the age of 4 years, there is slightly erythematous and thickened skin in some areas |
| Willemze R. et al., 1980 [60] | At birth | 25 years | None | Persistent generalized erythema since birth Cutaneous tumors with MC infiltration on the arms and legs No evidence of SM |
| Olgun N. et al., 1993 [68] | 5 months | NA | NA | Widespread bullous eruptions, small papules over the entire skin surface, and erythematous rash Intermittent flushing and fever; intense itching; MC infiltrates in BM After 7 months, some improvement |
| Shah P. et al., 1998 [39] | At birth | 4 months | NA | Diffuse bullae Death at the age of 5 months due to bacterial endocarditis, pneumonia, and ascites |
| Murphy M. et al., 1999 [69] | At birth | 17 months | NA | Generalized erythema, blistering Generalized lymphadenopathy Death at the age of 17 months |
| Enomoto U. et al., 1999 [70] | At birth | 3 years | NA | Generalized brown, thickened leathery skin, and blistering No evidence of SM At the age of 36, there was slight blistering and hyperpigmentation |
| Waxtein L. et al., 2000 [71] | 3 months | NA | NA | At the age of 3 months, generalized hyperpigmented skin with intense pruritus At the age of 10, diffuse infiltrated and hyperpigmented plaques on the trunk and extremities, and papular and yellowish nodules on the scalp and ears MC infiltrates BM |
| Tang X. et al., 2004 [53] | 4 months | 2 years | NA | Familial DCM with the A533D mutation Generalized pruritus of the skin and blistering on the scalp At the age of 2 years, the patient had only a solitary yellow-brown papule The patient’s father, paternal uncle, paternal grandfather, and paternal great aunt were diagnosed with DCM None of the family members had SM |
| Kinsler V. et al., 2005 [72] | 10 weeks | 6 years | NA | At the age of 10 weeks, there was an erythematous rash on the head, neck, and shoulders, thickened skin, and marked dermographism At the age of 6 years, small patches of urticaria |
| Walker T. et al., 2006 [73] | At birth | 6 months | NA | Pachydermia, erythrodermia, dry skin and blisters |
| Duckworth A. et al., 2009 [74] | At birth | 5 years | NA | Male fraternal twins Erythema, pink-brown macules and papules, diffusely thickened skin No systemic involvement Flushing, pruritus, and abdominal pain |
| Ghiasi M. et al., 2011 [75] | At birth | 1 month | None | Generalized erythema, thickened skin, scattered nodules, and hemorrhagic bullae at birth Serum tryptase level: 6 ng/mL Hepatosplenomegaly Death at the age of 1 month due to an unknown reason |
| Kleewein K. et al., 2011 [28] | 3 months | NA | NA | Generalized bullous eruptions and hemorrhagic crusts on the scalp, face, ear, and trunk Serum tryptase level: 58.9 ng/mL |
| Koga H. et al., 2011 [76] | At birth | 14 months | None | Diffuse erythema and blisters on the scalp, face, and extremities At the age of 14 months, erythrodermic rash No evidence of SM. |
| Wawrzycki B. et al., 2013 [77] | At birth | 7 months | NA | Tense blisters on thickened, erythematous skin on the face, trunk, and limbs No evidence of SM |
| Wang H. et al., 2014 [54] | 5 months | 35 years | NA | Familial DCM witch germline p.S451C mutation In the infantile period, generalized blistering At the age of 2 years, there is diffuse thickening of the skin and no blistering |
| Park M. et al., 2014 [62] | At birth | 12 months | Once due to the unknown trigger | Diffuse leathery, erythrodermic rush, blisters on the head, neck, and trunk, with erosions on the face, scalp, and trunk Hepatosplenomegaly and mesenteric lymphadenopathy At the age of 12 months, hyperpigmented skin lesions |
| Otani I. et al., 2018 [36] | 4 months | 3 years | NA | Bullous skin lesions covering a third of the body surface, chest, and abdomen with almost complete reattached epidermis, back with healing, clean-based erosions Thickened skin with a yellowish color at the age of 5 months No further remission after 3 years of KIT K509I mutation Association with Tuberous Sclerosis |
| Hosking A. et al., 2018 [58] | 6 months | NA | NA | Generalized bullous lesions Fevers and emesis No evidence of SM Partial improvement with time |
| Gupta M. et al., 2019 [78] | 3 months | NA | NA | Blisters on the scalp and on the back Thickening of the skin on the abdomen and back No evidence of SM |
| Chaudhary N. et al., 2019 [35] | At birth | 7 weeks | None | Generalized thickening of the skin with folds on the neck and inguinal regions, widespread hyperpigmented crusted lesions, some hypopigmented nodular lesions Hepatosplenomegaly (SM excluded) Death at 7 weeks of age due to acute respiratory distress and worsening cardiac function |
| Jenkinson H. et al., 2019 [40] | 11 days | 8 months | NA | Diffusely infiltrated red-brown skin with a marked leathery appearance and large, tense blisters on both hands No evidence of SM At the age of 8 months, some improvement |
| Li Y et al., 2020 [41] | At birth | NA | NA | Diffused red-brown papules and plaques on the scalp, face, trunk, and extremities Death at the age of one month due to severe infection, respiratory failure, and circulatory failure |
| Cardoso J. et al., 2020 [79] | At birth | NA | NA | Diffuse erythematous leathery plaques and tense bullaes No evidence of SM |
| Turnbull L. et al., 2020 [80] | At birth | NA | NA | Purpuric papules, macules, and bullae on the scalp, face, neck, chest, abdomen, and extremities No evidence of SM Some improvement with time |
| Rayinda T. et al., 2021 [81] | 1.5 years | 2 weeks | NA | Flaccid blisters on the face and body, papules, skin erosion, and erythematous wheals on the back of the face and chest |
| Wangberg H. et al., 2023 [52] | 6 months | Ongoing | 3 reactions provoked by peripheral intravenous placement and unknown triggers | Familial DCM with germline mutation A533D Diffuse blistering rash No evidence of SM At the age of 3 years, some improvement |
| Olteanu E. et. Al, 2023 [38] | At birth | 2 years | NA | Thickened skin, generalized subcutaneous nodules on the face, scalp, trunk, back, hands, and feet At the age of 2 years, nodular lesions on the scalp and face |
| Disease | Skin Lesions Resembling DCM | Main Clinical Features of the Disease |
|---|---|---|
| Staphylococcal scalded skin syndrome (SSSS) | Blistering Redness of the entire skin Desquamation of the skin | Denudation of the skin caused by exotoxin produced by phage group II strains of Staphylococcus species Usually presents 48 h after birth (rare in children older than six years) Culture from the site of the suspected primary infection is warranted |
| Epidermolysis bullosa (EB) | Generalized bullous eruptions | Genetic collagen disorder is characterized by skin fragility leading to blistering, wounds, and scarring Identification of typical gene mutations |
| Impetigo bullosa (IB) | Small vesicles that can grow into tense bullae and erosions | Superficial, highly contagious bacterial (Staphylococcus aureus and Streptococcus pyogenes) skin infection Pustules, blisters, and honey-colored crusted erosions Bacterial cultures can be used for confirmation of a diagnosis |
| Erythema multiforme (EM) | Blisters based on erythematous skin lesions | Target-like lesions present symmetrically on the extremities (especially on extensor surfaces) and spread centripetally Precipitating factors: infections, especially the herpes simplex virus, and medications Histology: vacuolar interface dermatitis with marked infiltration with lymphocytes along the dermo-epidermal junction |
| Atopic dermatitis | Pruritic rash, erythroderma in severe cases | A defect in the skin barrier causes xerosis. Severe pruritus In infants, edematous papules and plaques that may have vesicles or crust on the scalp, face, and extensor extremities |
| Langerhans cell histiocytosis | Extensive rush and blistering in infants | Clonal disease of the monocyte-macrophage system A wide spectrum of skin lesions Histology with immunophenotyping: accumulation of CD1a-positive and/or CD207-positive dendritic cells |
| Linear IgA bullous dermatosis | Plaques and papules with blistering | Widespread annular blisters that exhibit a predilection for the lower abdomen, thighs, and groin Direct immunofluorescence: linear IgA deposits on the basement membrane zone |
| Incontinentia pigment | Blistering rash | Blistering, present in the early stages of infancy, heals spontaneously Blistering stage, followed by the development of verrucous lesions verrucous lesions and hyperpigmentation Coexisting signs: hair loss (alopecia) and dental abnormalities |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rydz, A.; Lange, M.; Ługowska-Umer, H.; Sikorska, M.; Nowicki, R.J.; Morales-Cabeza, C.; Alvarez-Twose, I. Diffuse Cutaneous Mastocytosis: A Current Understanding of a Rare Disease. Int. J. Mol. Sci. 2024, 25, 1401. https://doi.org/10.3390/ijms25031401
Rydz A, Lange M, Ługowska-Umer H, Sikorska M, Nowicki RJ, Morales-Cabeza C, Alvarez-Twose I. Diffuse Cutaneous Mastocytosis: A Current Understanding of a Rare Disease. International Journal of Molecular Sciences. 2024; 25(3):1401. https://doi.org/10.3390/ijms25031401
Chicago/Turabian StyleRydz, Agnieszka, Magdalena Lange, Hanna Ługowska-Umer, Monika Sikorska, Roman J. Nowicki, Cristina Morales-Cabeza, and Iván Alvarez-Twose. 2024. "Diffuse Cutaneous Mastocytosis: A Current Understanding of a Rare Disease" International Journal of Molecular Sciences 25, no. 3: 1401. https://doi.org/10.3390/ijms25031401
APA StyleRydz, A., Lange, M., Ługowska-Umer, H., Sikorska, M., Nowicki, R. J., Morales-Cabeza, C., & Alvarez-Twose, I. (2024). Diffuse Cutaneous Mastocytosis: A Current Understanding of a Rare Disease. International Journal of Molecular Sciences, 25(3), 1401. https://doi.org/10.3390/ijms25031401