
Small Molecule Tyrosine Kinase Inhibitors (TKIs) for Glioblastoma Treatment

 , ,
, ,  ,
,  and
and
Abstract
1. Introduction
2. TK Inhibitors
2.1. ALK Inhibitors
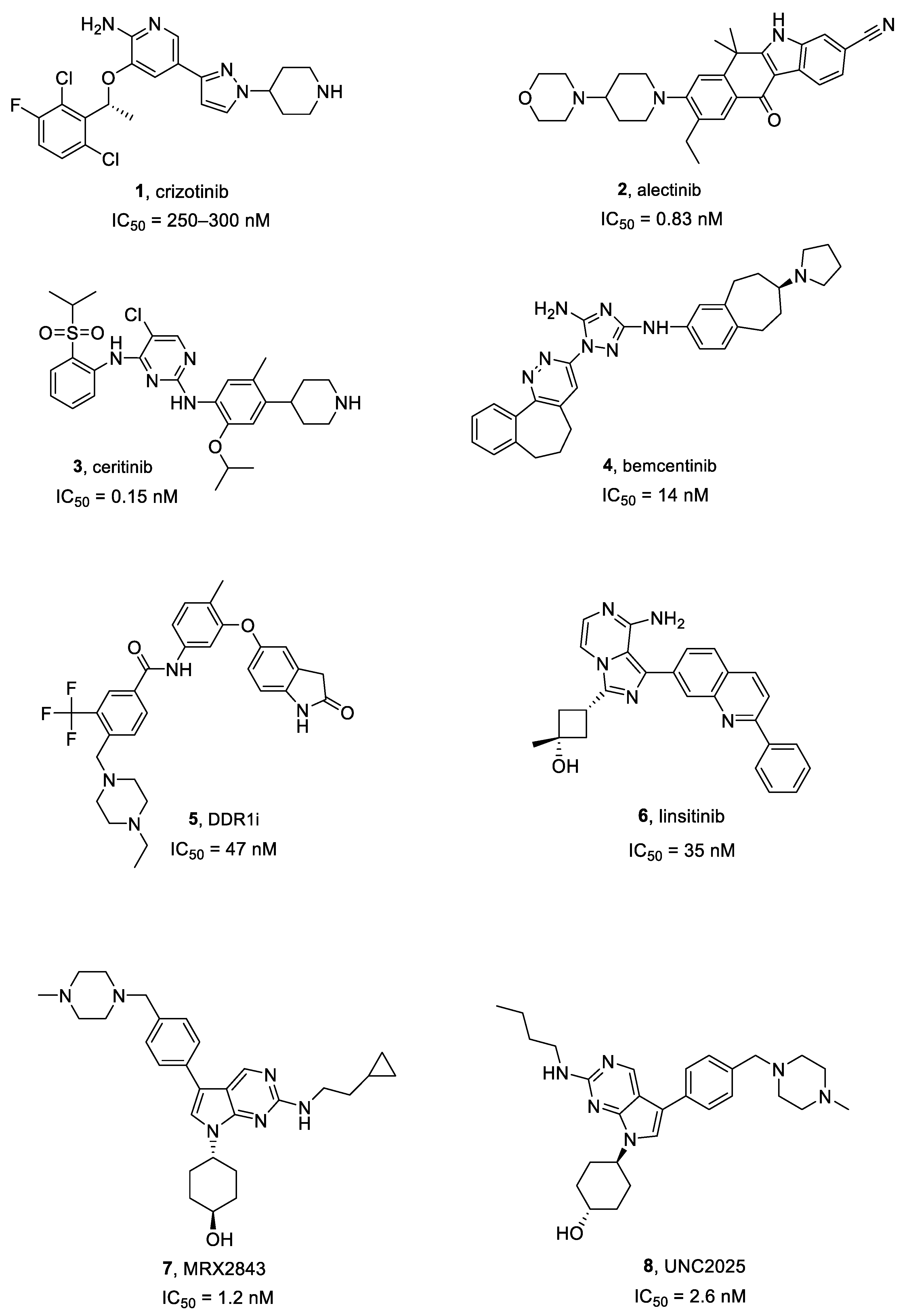
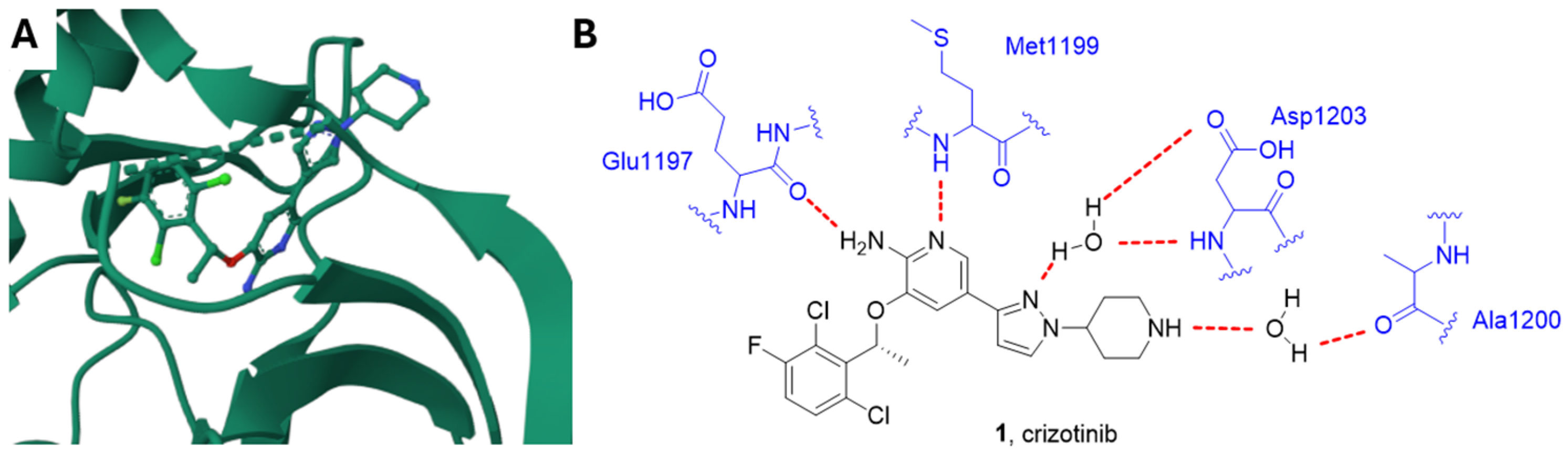
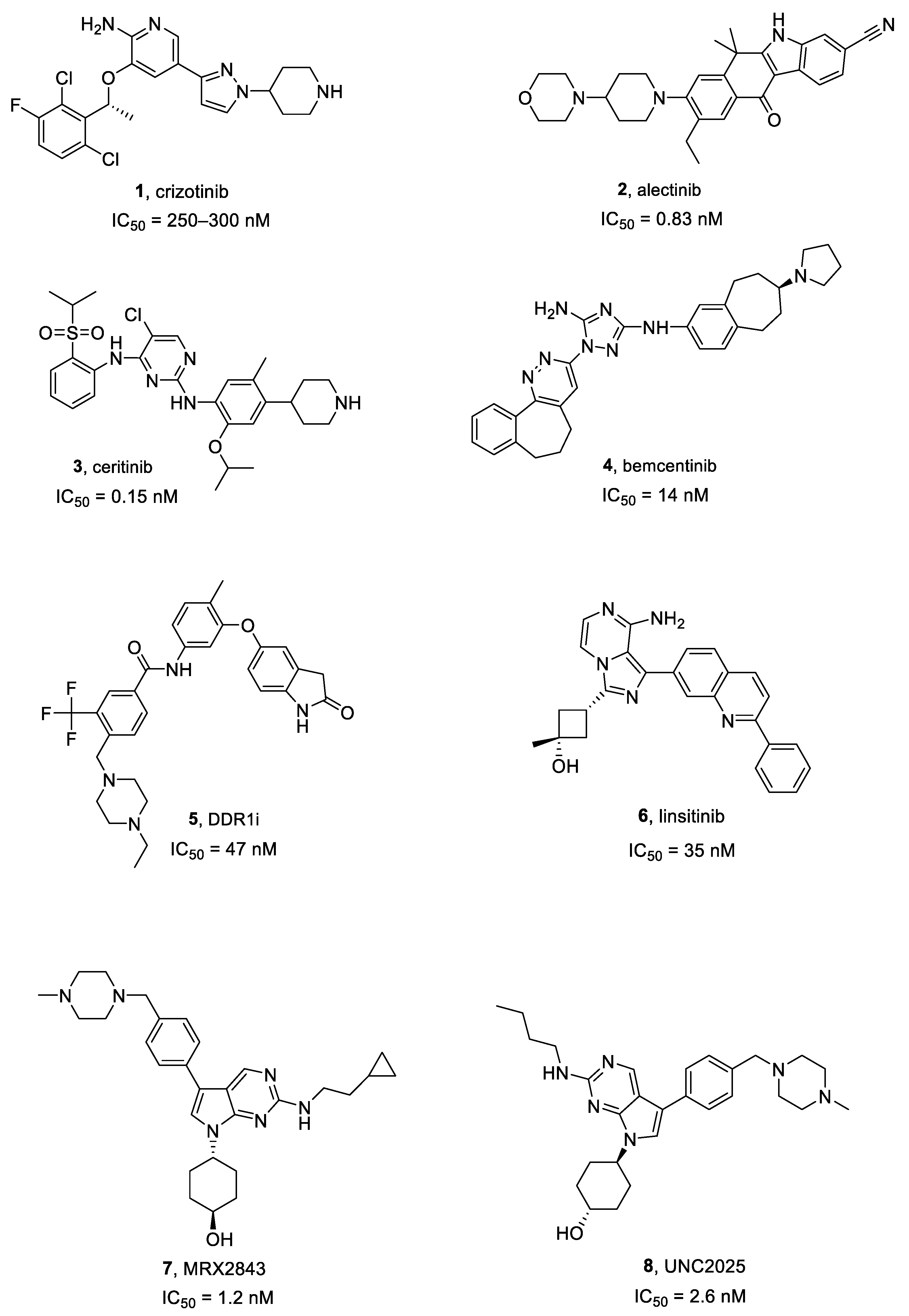
Crizotinib, Alectinib and Ceritinib
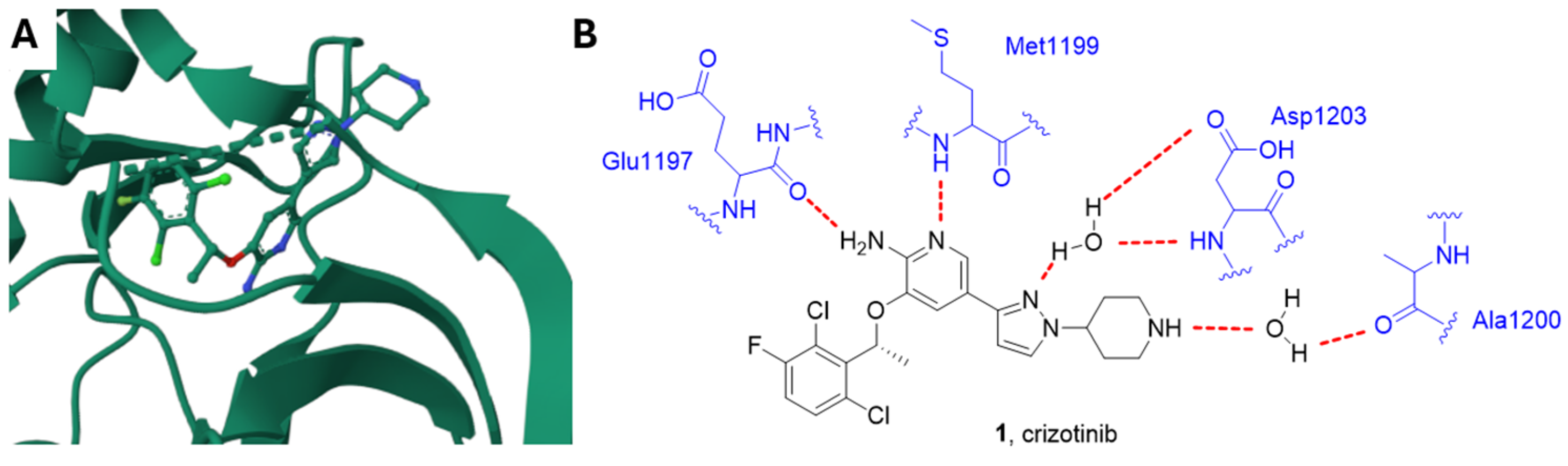
2.2. AXL Inhibitors
Bemcentinib
2.3. Discoidin Domain Receptor 1 (DDR1) Inhibitors
DDR1 Inhibitors
2.4. IGF1R Inhibitors
Linsitinib
2.5. MER Inhibitors
2.5.1. MRX2843
2.5.2. UNC2025
2.6. c-Met Inhibitors
2.6.1. Capmatinib
2.6.2. Tivantinib
2.7. EGFR Inhibitors
2.7.1. Osimertinib
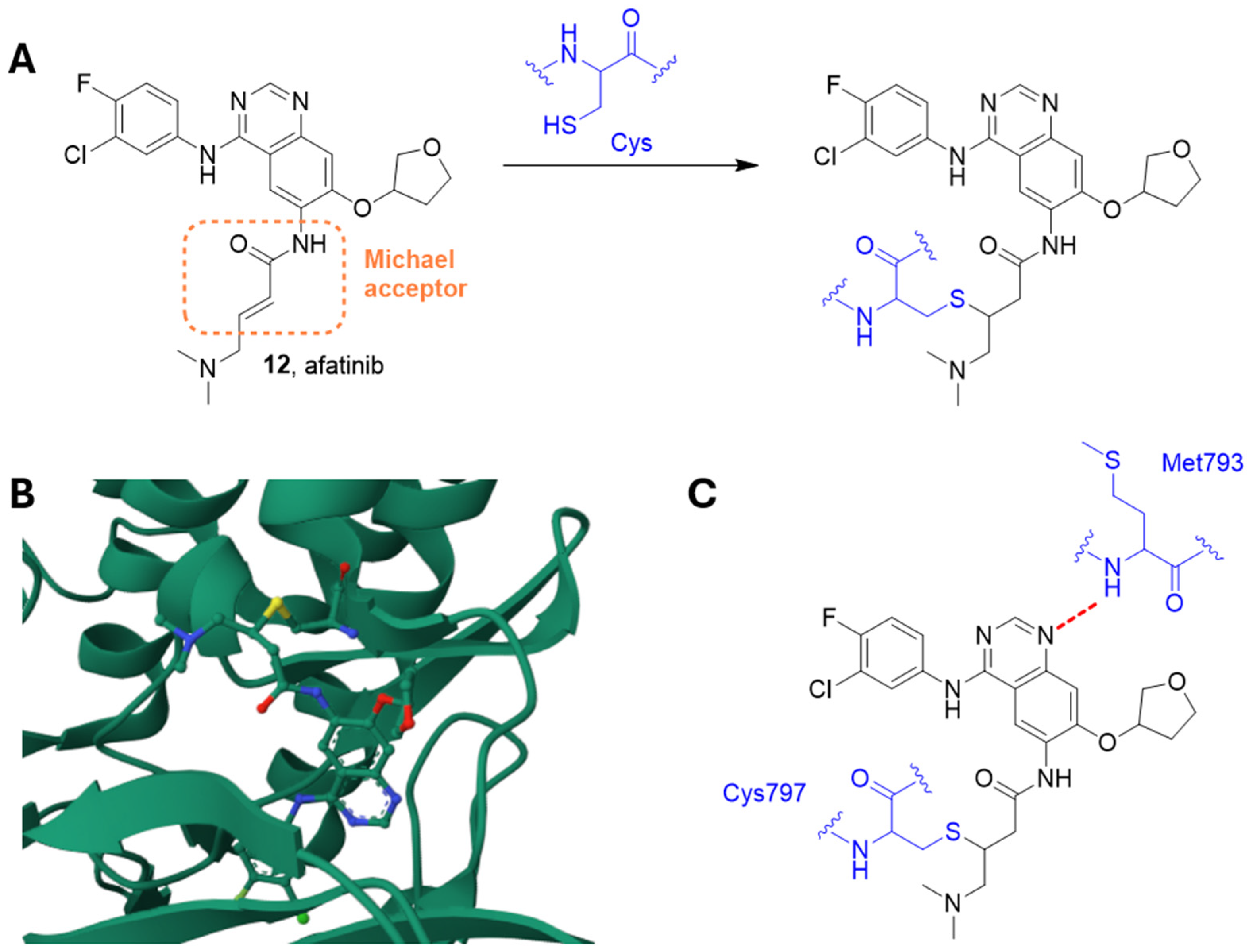
2.7.2. Afatinib
2.7.3. Erlotinib
2.7.4. Gefitinib
2.7.5. Lycorine
2.8. PDGFR Inhibitors
CP-673451
2.9. VEGFR Inhibitors
2.9.1. VGB
2.9.2. Voacangine
2.9.3. Apatinib
2.10. FAK Inhibitors
PF573228
2.11. JAK Inhibitors
2.11.1. AG490
2.11.2. Ruxolitinib
2.12. LCK Inhibitors
LCK-I
2.13. SYK Inhibitors
Bay61-3606, Piceatannol and NVP-QAB205
2.14. Src Inhibitors
2.14.1. KX2-361
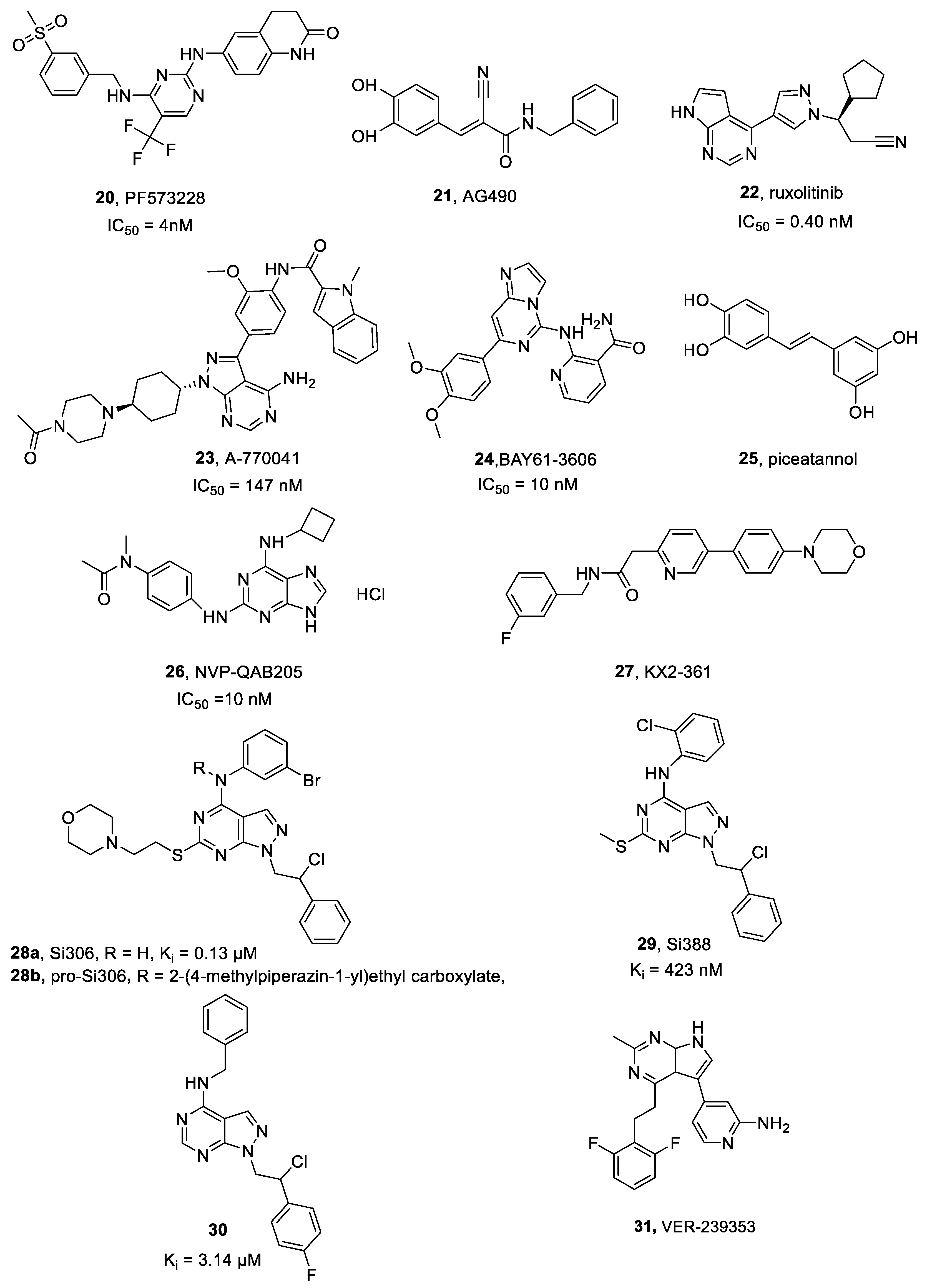
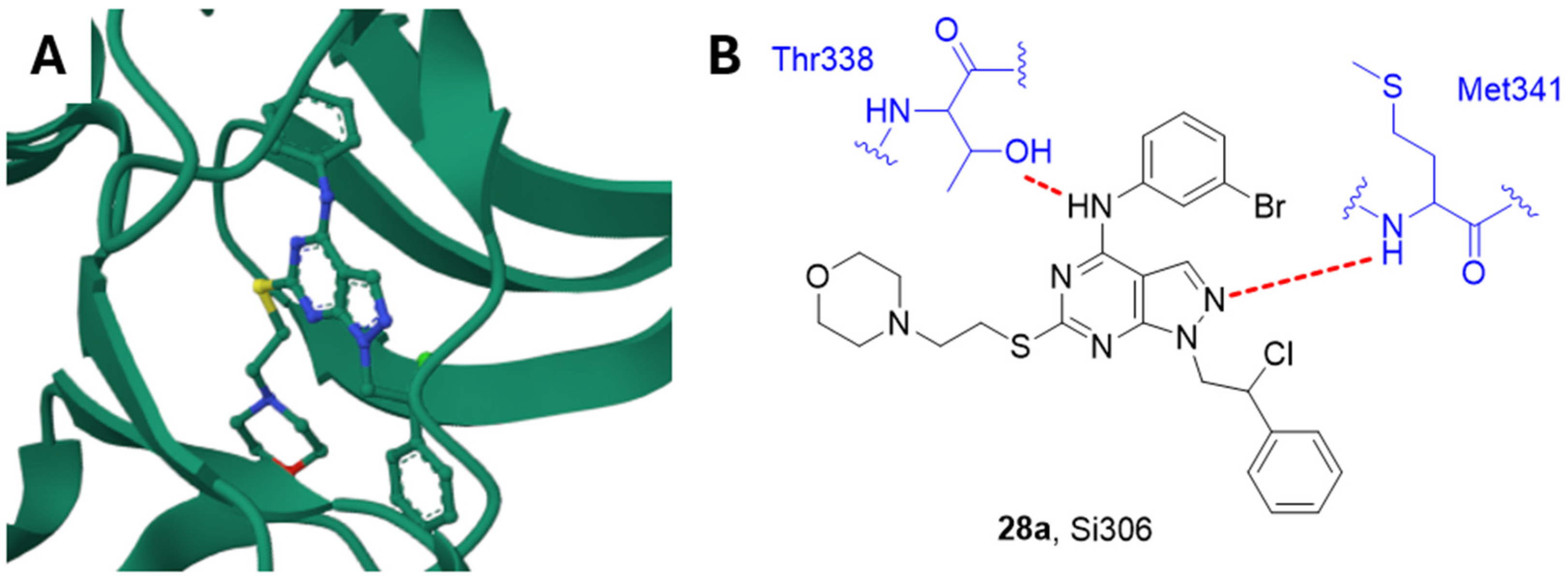
2.14.2. Si306 and Analogue Compounds
2.14.3. TAT-Cx43
2.15. Dual-Specificity Tyrosine Phosphorylation-Regulated Kinase 1A (DYRK1A) Inhibitors
VER-239353
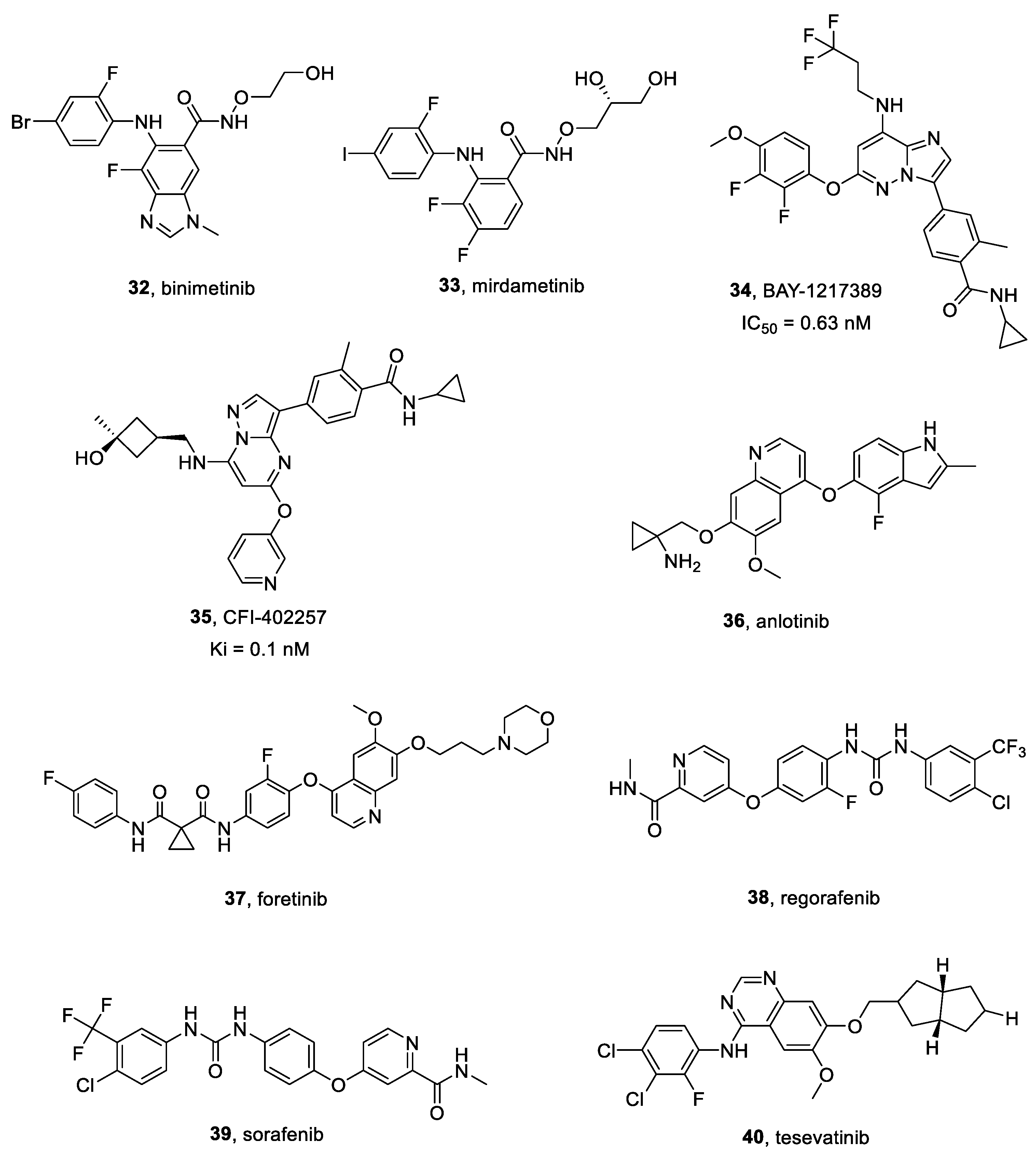
2.16. MEK Inhibitors
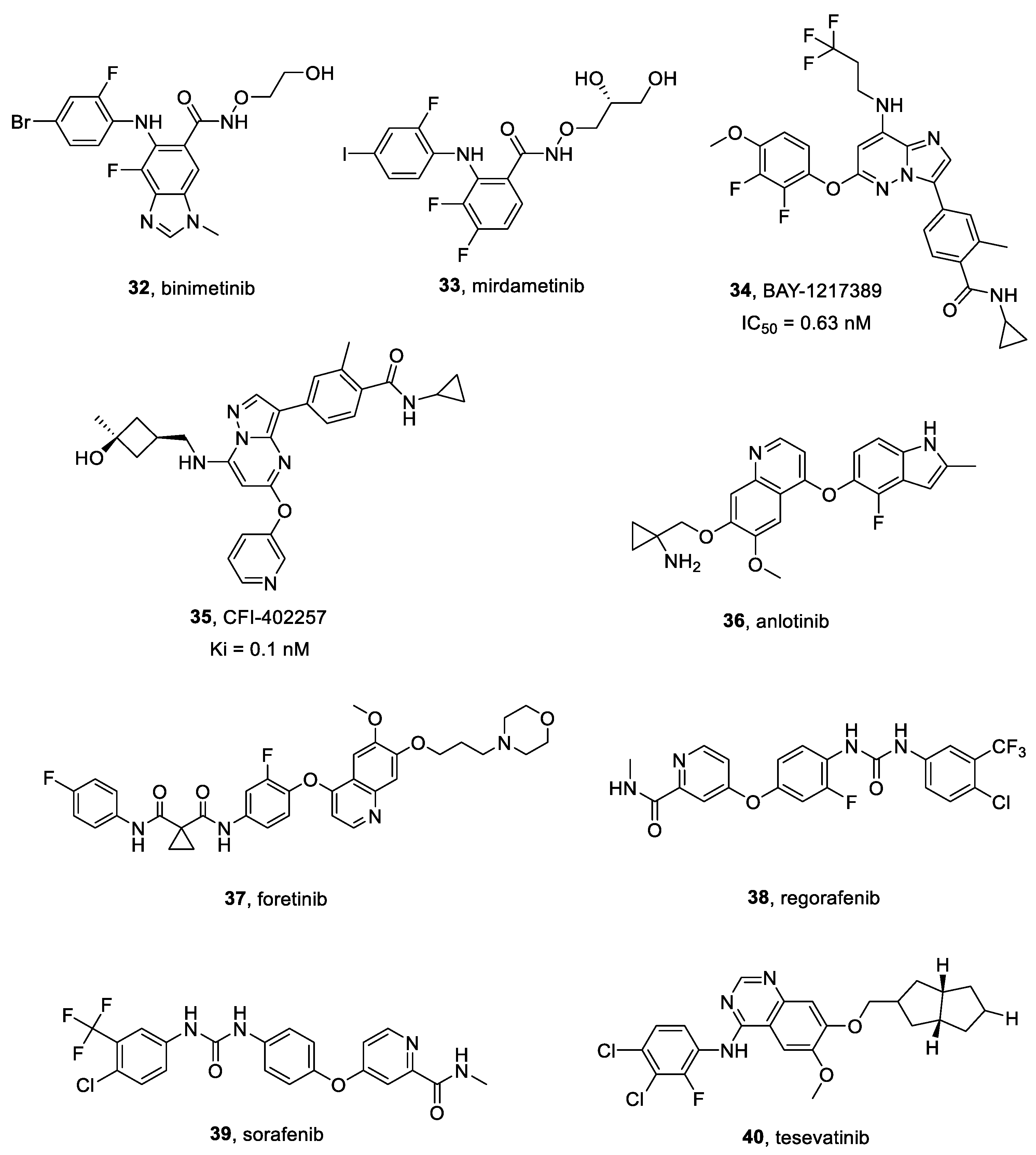
2.16.1. Binimetinib
2.16.2. Mirdametinib
2.17. TTK Inhibitors
BAY-1217389 and CFI-402257
2.18. Multitarget Inhibitors
2.18.1. Anlotinib
2.18.2. CR13626
2.18.3. Foretinib
2.18.4. Regorafenib
2.18.5. Sorafenib
2.18.6. Tesevatinib
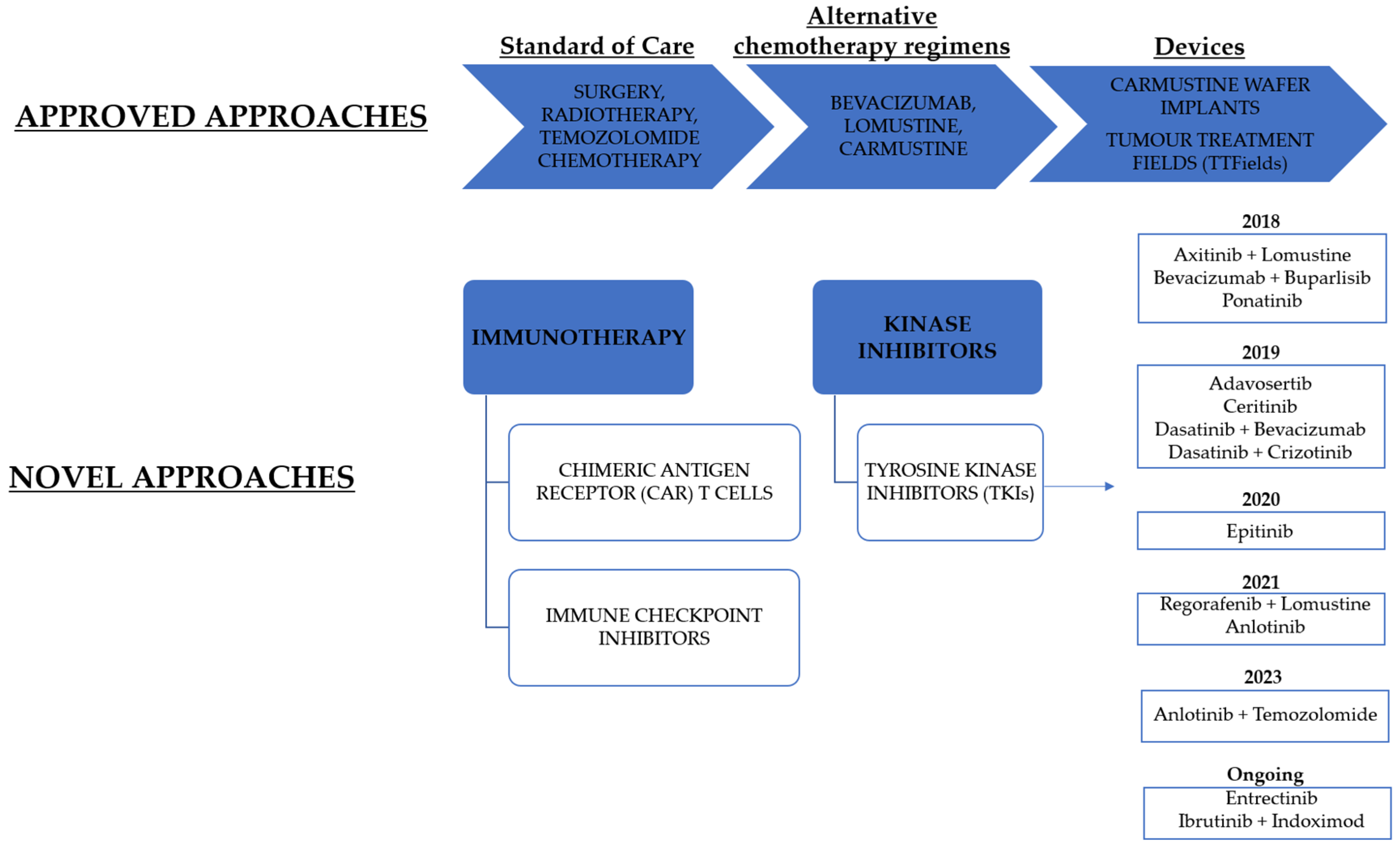
3. Challenges and Opportunities for Drug Delivery of Small Molecules Acting as Tyrosine Kinase Inhibitors
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| A172 | human glioblastoma cells |
| A549 | lung carcinoma cells |
| AC220 | quizartinib |
| ALK | anaplastic lymphoma kinase |
| Akt | protein kinase B |
| BCL-2 | B-cell lymphoma 2 |
| BS-125 | human glioblastoma cell line |
| BS-287 | human glioblastoma cell line |
| Crk-II | CT10 regulator of kinase |
| CXCL | C-X-C motif chemokinel |
| DBTRG | human glioblastoma cell line |
| EBC1 | lung squamous carcinoma cells |
| EGF | epidermal growth factor |
| EML4 | echinoderm microtubule-associated protein-like 4 |
| EOC2 | mouse microglial cell lines |
| ERK | extracellular signal-regulated kinase |
| FGFR | fibroblast growth factor receptor |
| FLT3 | Fms Related Receptor Tyrosine Kinase 3 |
| Fyn | Src family tyrosine kinase |
| GBM6 | human glioblastoma multiforme primary tissue cell line |
| GBM12 | TMZ-resistant GBM cell line |
| GBM8401 | human glioblastoma multiforme cell line |
| GBMSC83 | glioblastoma stem cell line |
| GL261 | glioma stem cell line |
| GSC11 | glioma stem cell line |
| GSC20 | glioma stem cell line |
| GSC23 | glioma stem cell line |
| GSC407 | glioma stem cell line |
| GSC923 | glioma stem cell line |
| GSCD317 | glioma stem cell line |
| HCC | hepatocellular carcinoma |
| HER | human epidermal growth factor receptor |
| HGK | hepatocyte progenitor kinase-like/germinal center kinase-like kinase |
| HUVECs | human umbilical vein endothelial cells |
| IGF | Insulin-like growth factor |
| JAK | Janus kinase |
| Kit | human tyrosine kinase receptor |
| LN-18 | epithelial-like cell line |
| LN-229 | epithelial-like cell line |
| MAPK | mitogen activated protein kinases |
| MCT | mast cell tumor |
| MDA-MB-231 | epithelial, human breast cancer cell line |
| MKN45 | gastric adenocarcinoma cells |
| MMP2 | matrix metalloproteinase-2 |
| mTOR | mammalian target of rapamycin |
| NF-κB | nuclear factor κB |
| PDA | pancreatic ductal adenocarcinoma cells |
| PDGF | platelet-derived growth factor |
| PI3K | phosphoinositide 3-kinase |
| PIK3R3 | phosphoinositide-3-kinase regulatory subunit 3 |
| Raf | rapidly accelerated fibrosarcoma |
| Ras | rat sarcoma |
| RET | rearranged during transfection tyrosine kinase receptor |
| RON | receptor tyrosine kinase |
| RT-PCR | Real time polymerase chain reaction |
| S6K | p70 ribosomal S6 kinase |
| siRNA | small interference RNA |
| SF-126-TR | high-grade human glioma cells temozolomide resistant |
| SF-539 | human glioblastoma cell line |
| SF-767 | human glioblastoma cell line |
| STAT3 | signal transducer and activator of transcription 3 |
| Tie-2 | Angiopoietin-1 receptor tyrosine kinase |
| TGF-α | transforming growth factor-α |
| T98G | fibroblast-like cells |
| T98MG | human glioblastoma cancer cells |
| TGF-α | transforming growth factor alpha |
| U118MG-TR | U118 malignant glioma cells temozolomide resistant |
| U251 | human glioblastoma cancer cells |
| U251-luc | transformed U251 human glioblastoma cancer cells |
| U373 | uppsala human glioblastoma astrocytoma cell line |
| U87MG | uppsala 87 malignant glioma cell line |
| U87TxR | multidrug resistant uppsala 87 glioma cell line |
| uPA | urokinase-type plasminogen activator |
| uPAR | urokinase-type plasminogen activator receptor |
| VEGF | vascular endothelial growth factor |
| Yes | human non-receptor tyrosine kinase |
References
- Le Rhun, E.; Preusser, M.; Roth, P.; Reardon, D.A.; van den Bent, M.; Wen, P.; Reifenberger, G.; Weller, M. Molecular targeted therapy of glioblastoma. Cancer Treat. Rev. 2019, 80, 101896. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Cerdeño, V.; Noctor, S.C. Neural Progenitor Cell Terminology. Front. Neuroanat. 2018, 12, 104. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, Q.; Yang, H.; Mao, Y. The Oncogenesis of Glial Cells in Diffuse Gliomas and Clinical Opportunities. Neurosci. Bull. 2023, 39, 393–408. [Google Scholar] [CrossRef] [PubMed]
- Friedmann-Morvinski, D. Glioblastoma heterogeneity and cancer cell plasticity. Crit. Rev. Oncog. 2014, 19, 327–336. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Holland, E.C.; Cairncross, J.G. Glioma classification: A molecular reappraisal. Am. J. Pathol. 2001, 159, 779–786. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Fan, X.; Zhao, C.; Zhao, Z.; Hu, L.; Wang, D.; Wang, R.; Fang, Z. Molecular subtyping of glioblastoma based on immune-related genes for prognosis. Sci. Rep. 2020, 10, 15495. [Google Scholar] [CrossRef] [PubMed]
- Grochans, S.; Cybulska, A.M.; Simińska, D.; Korbecki, J.; Kojder, K.; Chlubek, D.; Baranowska-Bosiacka, I. Epidemiology of Glioblastoma multiforme-literature review. Cancers 2022, 14, 2412. [Google Scholar] [CrossRef]
- Tykocki, T.; Eltayeb, M. Ten-year survival in glioblastoma. A systematic review. J. Clin. Neurosci. 2018, 54, 7–13. [Google Scholar] [CrossRef]
- Fernandes, C.; Costa, A.; Osório, L.; Costa Lago, R.; Linhares, P.; Carvalho, B.; Caeiro, C. Current Standards of Care in Glioblastoma Therapy. In Glioblastoma; De Vleeschouwer, S., Ed.; Codon Publications: Brisbane, Australia, 2017. [Google Scholar] [CrossRef]
- Minniti, G.; Niyazi, M.; Alongi, F.; Navarria, P.; Belka, C. Current status and recent advances in reirradiation of glioblastoma. Radiat. Oncol. 2021, 16, 36. [Google Scholar] [CrossRef]
- Fisher, J.P.; Adamson, D.C. Current FDA-Approved Therapies for High-Grade Malignant Gliomas. Biomedicines 2021, 9, 324. [Google Scholar] [CrossRef]
- Wismeth, C.; Hau, P.; Fabel, K.; Baumgart, U.; Hirschmann, B.; Koch, H.; Jauch, T.; Grauer, O.; Drechsel, L.; Brawanski, A.; et al. Maintenance therapy with 13-cis retinoid acid in high-grade glioma at complete response after first-line multimodal therapy—A phase-II study. J. Neurooncol. 2004, 68, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.E.; Choi, S.S.; Rogers, J.E.; Lei, X.; De Groot, J.F. Isotretinoin maintenance therapy for glioblastoma: A retrospective review. J. Oncol. Pharm. Pract. 2014, 20, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Kang, X.; Zheng, Y.; Hong, W.; Chen, X.; Li, H.; Huang, B.; Huang, Z.; Tang, H.; Geng, W. Recent Advances in Immune Cell Therapy for Glioblastoma. Front. Immunol. 2020, 11, 544563. [Google Scholar] [CrossRef] [PubMed]
- Guan, X.; Wang, Y.; Sun, Y.; Zhang, C.; Ma, S.; Zhang, D.; Li, D.; Jia, W. CTLA4-Mediated Immunosuppression in Glioblastoma is Associated with the Infiltration of Macrophages in the Tumor Microenvironment. J. Inflamm. Res. 2021, 14, 7315–7329. [Google Scholar] [CrossRef] [PubMed]
- Ranjan, S.; Quezado, M.; Garren, N.; Boris, L.; Siegel, C.; Lopes Abath Neto, O.; Theeler, B.J.; Park, D.M.; Nduom, E.; Zaghloul, K.A.; et al. Clinical decision making in the era of immunotherapy for high grade-glioma: Report of four cases. BMC Cancer 2018, 18, 239. [Google Scholar] [CrossRef]
- Duong-Ly, K.C.; Peterson, J.R. The human kinome and kinase inhibition. Curr. Protoc. Pharmacol. 2013. [Google Scholar] [CrossRef]
- Arter, C.; Trask, L.; Ward, S.; Yeoh, S.; Bayliss, R. Structural features of the protein kinase domain and targeted binding by small-molecule inhibitors. J. Biol. Chem. 2022, 298, 102247. [Google Scholar] [CrossRef]
- Du, Z.; Lovly, C.M. Mechanisms of receptor tyrosine kinase activation in cancer. Mol. Cancer 2018, 17, 58. [Google Scholar] [CrossRef]
- Taylor, S.S.; Kornev, A.P. Protein kinases: Evolution of dynamic regulatory proteins. Trends Biochem. Sci. 2011, 36, 65–77. [Google Scholar] [CrossRef]
- Bhullar, K.S.; Lagarón, N.O.; McGowan, E.M.; Parmar, I.; Jha, A.; Hubbard, B.P.; Rupasinghe, H.P.V. Kinase-targeted cancer therapies: Progress, challenges and future directions. Mol. Cancer 2018, 17, 48. [Google Scholar] [CrossRef]
- Li, X.; Wu, C.; Chen, N.; Gu, H.; Yen, A.; Cao, L.; Wang, E.L. PI3K/Akt/mTOR signaling pathway and targeted therapy for glioblastoma. Oncotarget 2016, 7, 33440–33450. [Google Scholar] [CrossRef]
- Liu, H.; Qiu, W.; Sun, T.; Wang, L.; Du, C.; Hu, Y.; Liu, W.; Feng, F.; Chen, Y.; Sun, H. Therapeutic strategies of glioblastoma (GBM): The current advances in the molecular targets and bioactive small molecule compounds. Acta Pharm. Sin. B 2022, 12, 1781–1804. [Google Scholar] [CrossRef] [PubMed]
- Cazes, A.; Lopez-Delisle, L.; Tsarovina, K.; Pierre-Eugène, C.; De Preter, K.; Peuchmaur, M.; Nicolas, A.; Provost, C.; Louis-Brennetot, C.; Daveau, R.; et al. Activated Alk triggers prolonged neurogenesis and Ret upregulation providing a therapeutic target in ALK-mutated neuroblastoma. Oncotarget 2014, 5, 2688–2702. [Google Scholar] [CrossRef] [PubMed]
- Yao, S.; Cheng, M.; Zhang, Q.; Wasik, M.; Kelsh, R.; Winkler, C. Anaplastic lymphoma kinase is required for neurogenesis in the developing central nervous system of zebrafish. PLoS ONE 2013, 8, e63757. [Google Scholar] [CrossRef] [PubMed]
- Chiarle, R.; Voena, C.; Ambrogio, C.; Piva, R.; Inghirami, G. The anaplastic lymphoma kinase in the pathogenesis of cancer. Nat. Rev. Cancer 2008, 8, 11–23. [Google Scholar] [CrossRef]
- Bagci, O.; Tumer, S.; Olgun, N.; Altungoz, O. Copy number status and mutation analyses of anaplastic lymphoma kinase (ALK) gene in 90 sporadic neuroblastoma tumors. Cancer Lett 2012, 317, 72–77. [Google Scholar] [CrossRef]
- Salido, M.; Pijuan, L.; Martínez-Avilés, L.; Galván, A.B.; Cañadas, I.; Rovira, A.; Zanui, M.; Martínez, A.; Longarón, R.; Sole, F.; et al. Increased ALK gene copy number and amplification are frequent in non-small cell lung cancer. J. Thorac. Oncol. 2011, 6, 21–27. [Google Scholar] [CrossRef]
- Karagkounis, G.; Stranjalis, G.; Argyrakos, T.; Pantelaion, V.; Mastoris, K.; Rontogianni, D.; Komaitis, S.; Kalamatianos, T.; Sakas, D.; Tiniakos, D. Anaplastic lymphoma kinase expression and gene alterations in glioblastoma: Correlations with clinical outcome. J. Clin. Pathol. 2017, 70, 593–599. [Google Scholar] [CrossRef]
- Ferguson, S.D.; Xiu, J.; Weathers, S.P.; Zhou, S.; Kesari, S.; Weiss, S.E.; Verhaak, R.G.; Hohl, R.J.; Barger, G.R.; Reddy, S.K.; et al. GBM-associated mutations and altered protein expression are more common in young patients. Oncotarget 2016, 7, 69466–69478. [Google Scholar] [CrossRef]
- Huang, H. Anaplastic Lymphoma Kinase (ALK) Receptor Tyrosine Kinase: A Catalytic Receptor with Many Faces. Int. J. Mol. Sci. 2018, 19, 3448. [Google Scholar] [CrossRef]
- Yi, G.Z.; Xiang, W.; Feng, W.Y.; Chen, Z.Y.; Li, Y.M.; Deng, S.Z.; Guo, M.L.; Zhao, L.; Sun, X.G.; He, M.Y.; et al. Identification of Key Candidate Proteins and Pathways Associated with Temozolomide Resistance in Glioblastoma Based on Subcellular Proteomics and Bioinformatical Analysis. Biomed. Res. Int. 2018, 2018, 5238760. [Google Scholar] [CrossRef] [PubMed]
- Spagnuolo, A.; Maione, P.; Gridelli, C. Evolution in the treatment landscape of non-small cell lung cancer with ALK gene alterations: From the first- to third-generation of ALK inhibitors. Expert. Opin. Emerg. Drugs 2018, 23, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Savooji, J.; Liu, D. Second- and third-generation ALK inhibitors for non-small cell lung cancer. J. Hematol. Oncol. 2016, 9, 19. [Google Scholar] [CrossRef] [PubMed]
- Cheon, S.Y.; Kwon, S. Molecular anatomy of the EML4-ALK fusion protein for the development of novel anticancer drugs. Int. J. Mol. Sci. 2023, 24, 5821. [Google Scholar] [CrossRef]
- Cui, J.J.; Tran-Dubé, M.; Shen, H.; Nambu, M.; Kung, P.P.; Pairish, M.; Jia, L.; Meng, J.; Funk, L.; Botrous, I.; et al. Structure based drug design of crizotinib (PF-02341066), a potent and selective dual inhibitor of mesenchymal-epithelial transition factor (c-MET) kinase and anaplastic lymphoma kinase (ALK). J. Med. Chem. 2011, 54, 6342–6363. [Google Scholar] [CrossRef]
- Martínez-García, M.; Velasco, G.; Pineda, E.; Gil-Gil, M.; Alameda, F.; Capellades, J.; Martín-Soberón, M.C.; López-Valero, I.; Ambel, E.T.; Foro, P.; et al. Safety and Efficacy of Crizotinib in Combination with Temozolomide and Radiotherapy in Patients with Newly Diagnosed Glioblastoma: Phase Ib GEINO 1402 Trial. Cancers 2022, 14, 2393. [Google Scholar] [CrossRef]
- Larkins, E.; Blumenthal, G.M.; Chen, H.; He, K.; Agarwal, R.; Gieser, G.; Stephens, O.; Zahalka, E.; Ringgold, K.; Helms, W.; et al. FDA Approval: Alectinib for the Treatment of Metastatic, ALK-Positive Non-Small Cell Lung Cancer Following Crizotinib. Clin. Cancer Res. 2016, 22, 5171–5176. [Google Scholar] [CrossRef]
- Sakamoto, H.; Tsukaguchi, T.; Hiroshima, S.; Kodama, T.; Kobayashi, T.; Fukami, T.A.; Oikawa, N.; Tsukuda, T.; Ishii, N.; Aoki, Y. CH5424802, a selective ALK inhibitor capable of blocking the resistant gatekeeper mutant. Cancer Cell 2011, 19, 679–690. [Google Scholar] [CrossRef]
- Gadgeel, S.M.; Gandhi, L.; Riely, G.J.; Chiappori, A.A.; West, H.L.; Azada, M.C.; Morcos, P.N.; Lee, R.-M.; Linta Garcia, L.; Yu, L.; et al. Safety and activity of alectinib against systemic disease and brain metastases in patients with crizotinib-resistant ALK-rearranged non-small-cell lung cancer (AF-002JG): Results from the dose-finding portion of a phase ½ study. Lancet Oncol. 2014, 15, 1119–1128. [Google Scholar] [CrossRef]
- Berberich, A.; Schmitt, L.M.; Pusch, S.; Hielscher, T.; Rübmann, P.; Hucke, N.; Latzer, P.; Heßling, B.; Lemke, D.; Kessler, T.; et al. cMyc and ERK activity are associated with resistance to ALK inhibitory treatment in glioblastoma. J. Neurooncol. 2020, 146, 9–23. [Google Scholar] [CrossRef]
- Friboulet, L.; Li, N.; Katayama, R.; Lee, C.C.; Gainor, J.F.; Crystal, A.S.; Michellys, P.Y.; Awad, M.M.; Yanagitani, N.; Kim, S.; et al. The ALK inhibitor ceritinib overcomes crizotinib resistance in non-small cell lung cancer. Cancer Discov. 2014, 4, 662–673. [Google Scholar] [CrossRef] [PubMed]
- Crinò, L.; Ahn, M.J.; De Marinis, F.; Groen, H.J.; Wakelee, H.; Hida, T.; Mok, T.; Spigel, D.; Felip, E.; Nishio, M.; et al. Multicenter phase II study of whole-body and intracranial activity with ceritinib in patients with alk-rearranged non-small-cell lung cancer previously treated with chemotherapy and crizotinib: Results from ASCEND-2. J. Clin. Oncol. 2016, 34, 2866–2873. [Google Scholar] [CrossRef] [PubMed]
- Kawauchi, D.; Takahashi, M.; Satomi, K.; Yamamuro, S.; Kobayashi, T.; Uchida, E.; Honda-Kitahara, M.; Narita, Y.; Iwadate, Y.; Ichimura, K.; et al. The ALK inhibitors, alectinib and ceritinib, induce ALK-independent and STAT3-dependent glioblastoma cell death. Cancer Sci. 2021, 112, 2442–2453. [Google Scholar] [CrossRef]
- Goker Bagca, B.; Ozates, N.P.; Asik, A.; Caglar, H.O.; Gunduz, C.; Biray Avci, C. Temozolomide treatment combined with AZD3463 shows synergistic effect in glioblastoma cells. Biochem. Biophys. Res. Commun. 2020, 533, 1497–1504. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Siemann, D.W. Gas6/Axl signaling pathway in the tumor immune microenvironment. Cancers 2020, 12, 1850. [Google Scholar] [CrossRef] [PubMed]
- Korshunov, V.A. Axl-dependent signalling: A clinical update. Clin. Sci. (Lond.) 2012, 122, 361–368. [Google Scholar] [CrossRef]
- Onken, J.; Vajkoczy, P.; Torka, R.; Hempt, C.; Patsouris, V.; Heppner, F.L.; Radke, J. Phospho-AXL is widely expressed in glioblastoma and associated with significant shorter overall survival. Oncotarget 2017, 8, 50403–50414. [Google Scholar] [CrossRef]
- Scaltriti, M.; Elkabets, M.; Baselga, J. Molecular Pathways: AXL, a Membrane Receptor Mediator of Resistance to Therapy. Clin. Cancer Res. 2016, 22, 1313–1317. [Google Scholar] [CrossRef]
- Myers, S.H.; Brunton, V.G.; Unciti-Broceta, A. AXL inhibitors in cancer: A medicinal chemistry perspective. J. Med. Chem. 2016, 59, 3593–3608. [Google Scholar] [CrossRef]
- Gay, C.M.; Balaji, K.; Byers, L.A. Giving AXL the axe: Targeting AXL in human malignancy. Br. J. Cancer 2017, 116, 415–423. [Google Scholar] [CrossRef]
- Chen, F.; Song, Q.; Yu, Q. Axl inhibitor R428 induces apoptosis of cancer cells by blocking lysosomal acidification and recycling independent of Axl inhibition. Am. J. Cancer Res. 2018, 8, 1466–1482. [Google Scholar] [PubMed]
- Sun, Y.; Bailey, C.P.; Sadighi, Z.; Zaky, W.; Chandra, J. Pediatric high-grade glioma: Aberrant epigenetics and kinase signaling define emerging therapeutic opportunities. J. Neurooncol. 2020, 150, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Scherschinski, L.; Prem, M.; Kremenetskaia, I.; Tinhofer, I.; Vajkoczy, P.; Karbe, A.G.; Onken, J.S. Regulation of the receptor tyrosine kinase AXL in response to therapy and its role in therapy resistance in glioblastoma. Int. J. Mol. Sci. 2022, 23, 982. [Google Scholar] [CrossRef] [PubMed]
- Sadahiro, H.; Kang, K.D.; Gibson, J.T.; Minata, M.; Yu, H.; Shi, J.; Chhipa, R.; Chen, Z.; Lu, S.; Simoni, Y.; et al. Activation of the receptor tyrosine kinase AXL regulates the immune microenvironment in glioblastoma. Cancer Res. 2018, 78, 3002–3013. [Google Scholar] [CrossRef] [PubMed]
- Vehlow, A.; Klapproth, E.; Jin, S.; Hannen, R.; Hauswald, M.; Bartsch, J.W.; Nimsky, C.; Temme, A.; Leitinger, B.; Cordes, N. Interaction of discoidin domain receptor 1 with a 14-3-3-beclin-1-Akt1 complex modulates glioblastoma therapy sensitivity. Cell Rep. 2019, 26, 3672–3683.e7. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.G.; Tan, L.; Weisberg, E.L.; Liu, F.; Canning, P.; Choi, H.G.; Ezell, S.A.; Wu, H.; Zhao, Z.; Wang, J.; et al. Discovery of a potent and selective DDR1 receptor tyrosine kinase inhibitor. ACS Chem. Biol. 2013, 8, 2145–2150. [Google Scholar] [CrossRef]
- Alfaro-Arnedo, E.; López, I.P.; Piñeiro-Hermida, S.; Canalejo, M.; Gotera, C.; Sola, J.J.; Roncero, A.; Peces-Barba, G.; Ruíz-Martínez, C.; Pichel, J.G. IGF1R acts as a cancer-promoting factor in the tumor microenvironment facilitating lung metastasis implantation and progression. Oncogene 2022, 41, 3625–3639. [Google Scholar] [CrossRef]
- Martin, A.; Fernandez, M.C.; Cattaneo, E.R.; Schuster, C.D.; Venara, M.; Clément, F.; Berenstein, A.; Lombardi, M.G.; Bergadá, I.; Gutierrez, M.; et al. Type 1 insulin-like growth factor receptor nuclear localization in high-grade glioma cells enhances motility, metabolism, and in vivo tumorigenesis. Front. Endocrinol. (Lausanne) 2022, 13, 849279. [Google Scholar] [CrossRef]
- Davis, S.L.; Eckhardt, S.G.; Diamond, J.R.; Messersmith, W.A.; Dasari, A.; Weekes, C.D.; Lieu, C.H.; Kane, M.; Choon Tan, A.; Pitts, T.M.; et al. A phase i dose-escalation study of linsitinib (OSI-906), a small-molecule dual insulin-like growth factor-1 receptor/insulin receptor kinase inhibitor, in combination with irinotecan in patients with advanced cancer. Oncologist 2018, 23, 1409-e140. [Google Scholar] [CrossRef]
- Wu, J.; Chen, K.; Zhang, F.; Jin, J.; Zhang, N.; Li, D.; Ying, L.; Chen, W.; Yu, H.; Mao, W.; et al. Overcoming Linsitinib intrinsic resistance through inhibition of nuclear factor-κB signaling in esophageal squamous cell carcinoma. Cancer Med. 2017, 6, 1353–1361. [Google Scholar] [CrossRef]
- Fuentes-Baile, M.; Ventero, M.P.; Encinar, J.A.; García-Morales, P.; Poveda-Deltell, M.; Pérez-Valenciano, E.; Barberá, V.M.; Gallego-Plazas, J.; Rodríguez-Lescure, Á.; Martín-Nieto, J.; et al. Differential effects of IGF-1R small molecule tyrosine kinase inhibitors BMS-754807 and OSI-906 on human cancer cell lines. Cancers 2020, 12, 3717. [Google Scholar] [CrossRef] [PubMed]
- Pipitone, R.M.; Calvaruso, V.; Di Marco, L.; Di Salvo, F.; Gaggianesi, M.; Lupo, G.; Zito, R.; La Mantia, C.; Ramazzotti, M.; Petta, S.; et al. Mer Tyrosine Kinase (MERTK) modulates liver fibrosis progression and hepatocellular carcinoma development. Front. Immunol. 2022, 13, 926236. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Moncayo, G.; Morin, P.; Xue, G.; Grzmil, M.; Lino, M.M.; Clément-Schatlo, V.; Frank, S.; Merlo, A.; Hemmings, B.A. Mer receptor tyrosine kinase promotes invasion and survival in glioblastoma multiforme. Oncogene 2013, 32, 872–882. [Google Scholar] [CrossRef] [PubMed]
- Minson, K.A.; Smith, C.C.; Lee-Sherick, A.B.; DeRyckere, D.; Lasater, E.; Hill, A.A.; Wang, X.; Frye, S.V.; Earp, H.S.; Shah, N.P.; et al. MRX2843, a novel dual MerTK-FLT3 Inhibitor with activity against resistance-conferring FLT3 mutations in acute myeloid leukemia. Blood 2014, 124, 3757. [Google Scholar] [CrossRef]
- Kelvin, J.M.; Chimenti, M.L.; Zhang, D.Y.; Williams, E.K.; Moore, S.G.; Humber, G.M.; Baxter, T.A.; Birnbaum, L.A.; Qui, M.; Zecca, H.; et al. Development of constitutively synergistic nanoformulations to enhance chemosensitivity in T-cell leukemia. J. Control Release 2023, 361, 470–482. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.T.; Butler, M.; Zhang, M.; Zhang, W.; Song, H.; Hwang, L.; Tran, A.D.; Bash, R.E.; Schorzman, A.N.; Pang, Y.; et al. MerTK inhibition decreases immune suppressive glioblastoma-associated macrophages and neoangiogenesis in glioblastoma microenvironment. Neurooncol. Adv. 2020, 2, vdaa065. [Google Scholar] [CrossRef] [PubMed]
- Sufit, A.; Lee-Sherick, A.B.; DeRyckere, D.; Rupji, M.; Dwivedi, B.; Varella-Garcia, M.; Pierce, A.M.; Kowalski, J.; Wang, X.; Frye, S.V.; et al. MERTK inhibition induces polyploidy and promotes cell death and cellular senescence in glioblastoma multiforme. PLoS ONE 2016, 11, e0165107. [Google Scholar] [CrossRef]
- Wu, J.; Frady, L.N.; Bash, R.E.; Cohen, S.M.; Schorzman, A.N.; Su, Y.T.; Irvin, D.M.; Zamboni, W.C.; Wang, X.; Frye, S.V.; et al. MerTK as a therapeutic target in glioblastoma. Neuro-Oncology 2018, 20, 92–102. [Google Scholar] [CrossRef]
- Bladt, F.; Riethmacher, D.; Isenmann, S.; Aguzzi, A.; Birchmeier, C. Essential role for the c-met receptor in the migration of myogenic precursor cells into the limb bud. Nature 1995, 376, 768–771. [Google Scholar] [CrossRef]
- Sennino, B.; Ishiguro-Oonuma, T.; Wei, Y.; Naylor, R.M.; Williamson, C.W.; Bhagwandin, V.; Tabruyn, S.P.; You, W.K.; Chapman, H.A.; Christensen, J.G.; et al. Suppression of tumor invasion and metastasis by concurrent inhibition of c-Met and VEGF signaling in pancreatic neuroendocrine tumors. Cancer Discov. 2012, 2, 270–287. [Google Scholar] [CrossRef]
- Han, Y.; Luo, Y.; Zhao, J.; Li, M.; Jiang, Y. Overexpression of c-Met increases the tumor invasion of human prostate LNCaP cancer cells in vitro and in vivo. Oncol. Lett. 2014, 8, 1618–1624. [Google Scholar] [CrossRef] [PubMed]
- Granito, A.; Guidetti, E.; Gramantieri, L. c-MET receptor tyrosine kinase as a molecular target in advanced hepatocellular carcinoma. J. Hepatocell. Carcinoma 2015, 2, 29–38. [Google Scholar] [CrossRef]
- Park, H.; Kim, D.; Kim, E.; Sa, J.K.; Lee, H.W.; Yu, S.; Oh, J.; Kim, S.H.; Yoon, Y.; Nam, D.H. Tumor inhibitory effect of IRCR201, a novel cross-reactive c-met antibody targeting the PSI domain. Int. J. Mol. Sci. 2017, 18, 1968. [Google Scholar] [CrossRef]
- Miekus, K.; Kijowski, J.; Sekuła, M.; Majka, M. 17AEP-GA, an HSP90 antagonist, is a potent inhibitor of glioblastoma cell proliferation, survival, migration and invasion. Oncol. Rep. 2012, 28, 1903–1909. [Google Scholar] [CrossRef] [PubMed]
- Schuler, M.; Berardi, R.; Lim, W.T.; de Jonge, M.; Bauer, T.M.; Azaro, A.; Gottfried, M.; Han, J.Y.; Lee, D.H.; Wollner, M.; et al. Molecular correlates of response to capmatinib in advanced non-small-cell lung cancer: Clinical and biomarker results from a phase I trial. Ann. Oncol. 2020, 31, 789–797. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wang, Q.; Yang, G.; Marando, C.; Koblish, H.K.; Hall, L.M.; Fridman, J.S.; Behshad, E.; Wynn, R.; Li, Y.; et al. A novel kinase inhibitor, INCB28060, blocks c-MET-dependent signaling, neoplastic activities, and cross-talk with EGFR and HER-3. Clin. Cancer Res. 2011, 17, 7127–7138. [Google Scholar] [CrossRef] [PubMed]
- Baltschukat, S.; Engstler, B.S.; Huang, A.; Hao, H.X.; Tam, A.; Wang, H.Q.; Liang, J.; DiMare, M.T.; Bhang, H.C.; Wang, Y.; et al. Capmatinib (INC280) is active against models of non-small cell lung cancer and other cancer types with defined mechanisms of MET activation. Clin. Cancer Res. 2019, 25, 3164–3175. [Google Scholar] [CrossRef]
- Wagner, A.J.; Goldberg, J.M.; Dubois, S.G.; Choy, E.; Rosen, L.; Pappo, A.; Geller, J.; Judson, I.; Hogg, D.; Senzer, N.; et al. Tivantinib (ARQ 197), a selective inhibitor of MET, in patients with microphthalmia transcription factor-associated tumors: Results of a multicenter phase 2 trial. Cancer 2012, 118, 5894–5902. [Google Scholar] [CrossRef]
- Katayama, R.; Aoyama, A.; Yamori, T.; Qi, J.; Oh-hara, T.; Song, Y.; Engelman, J.A.; Fujita, N. Cytotoxic activity of tivantinib (ARQ 197) is not due solely to c-MET inhibition. Cancer Res 2013, 73, 3087–3096. [Google Scholar] [CrossRef]
- Giannoni, P.; Daniela de Totero, D. The HGF/c-MET axis as a potential target to overcome survival signals and improve therapeutic efficacy in multiple myeloma. Cancer Drug Resist. 2021, 4, 923–933. [Google Scholar] [CrossRef]
- Wu, Y.; Li, Z.; Zhang, L.; Liu, G. Tivantinib hampers the proliferation of glioblastoma cells via PI3K/Akt/mammalian target of rapamycin (mTOR) signaling. Med. Sci. Monit. 2019, 25, 7383–7390. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Zong, H.; Ma, C.; Ming, X.; Shang, M.; Li, K.; He, X.; Du, H.; Cao, L. Epidermal growth factor receptor in glioblastoma. Oncol. Lett. 2017, 14, 512–516. [Google Scholar] [CrossRef] [PubMed]
- Park, K.; Tan, E.H.; O’Byrne, K.; Zhang, L.; Boyer, M.; Mok, T.; Hirsh, V.; Yang, J.C.; Lee, K.H.; Lu, S.; et al. Afatinib versus gefitinib as first-line treatment of patients with EGFR mutation-positive non-small-cell lung cancer (LUX-Lung 7): A phase 2B, open-label, randomised controlled trial. Lancet Oncol. 2016, 17, 577–589. [Google Scholar] [CrossRef]
- Soria, J.C.; Ohe, Y.; Vansteenkiste, J.; Reungwetwattana, T.; Chewaskulyong, B.; Lee, K.H.; Dechaphunkul, A.; Imamura, F.; Nogami, N.; Kurata, T.; et al. Osimertinib in untreated EGFR-mutated advanced non-small-cell lung cancer. N. Engl. J. Med. 2018, 378, 113–125. [Google Scholar] [CrossRef] [PubMed]
- Patel, H.M.; Pawara, R.; Surana, S.J. Chapter 2—Approved and clinical trial third-generation EGFR Inhibitors. In Third Generation EGFR Inhibitors; Elsevier: Amsterdam, The Netherlands, 2019; pp. 25–43. [Google Scholar] [CrossRef]
- Fu, K.; Xie, F.; Wang, F.; Fu, L. Therapeutic strategies for EGFR-mutated non-small cell lung cancer patients with osimertinib resistance. J. Hematol. Oncol. 2022, 15, 173. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Chen, X.; Shi, L.; Shan, Q.; Cao, Q.; Yue, C.; Li, H.; Li, S.; Wang, J.; Gao, S.; et al. The third-generation EGFR inhibitor AZD9291 overcomes primary resistance by continuously blocking ERK signaling in glioblastoma. J. Exp. Clin. Cancer Res. 2019, 38, 219. [Google Scholar] [CrossRef] [PubMed]
- Chagoya, G.; Kwatra, S.G.; Nanni, C.W.; Roberts, C.M.; Phillips, S.M.; Nullmeyergh, S.; Gilmore, S.P.; Spasojevic, I.; Corcoran, D.L.; Young, C.C.; et al. Efficacy of osimertinib against EGFRvIII+ glioblastoma. Oncotarget 2020, 11, 2074–2082. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Shi, J.; Shen, D.; Zhai, X.; Liang, D.; Wang, J.; Xie, C.; Xia, Z.; Cui, J.; Liu, F.; et al. Osimertinib induces paraptosis and TRIP13 confers resistance in glioblastoma cells. Cell Death Discov. 2023, 9, 333. [Google Scholar] [CrossRef]
- Wecker, H.; Waller, C.F. Afatinib. Recent Results Cancer Res. 2018, 211, 199–215. [Google Scholar] [CrossRef]
- Vengoji, R.; Macha, M.A.; Nimmakayala, R.K.; Rachagani, S.; Siddiqui, J.A.; Mallya, K.; Gorantla, S.; Jain, M.; Ponnusamy, M.P.; Batra, S.K.; et al. Afatinib and Temozolomide combination inhibits tumorigenesis by targeting EGFRvIII-cMet signaling in glioblastoma cells. J. Exp. Clin. Cancer Res. 2019, 38, 266. [Google Scholar] [CrossRef]
- Solca, F.; Dahl, G.; Zoephel, A.; Bader, G.; Sanderson, M.; Klein, C.; Kraemer, O.; Himmelsbach, F.; Haaksma, E.; Adolf, G.R. Target binding properties and cellular activity of afatinib (BIBW 2992), an irreversible ErbB family blocker. J. Pharmacol. Exp. Ther. 2012, 343, 342–350. [Google Scholar] [CrossRef] [PubMed]
- Dungo, R.T.; Keating, G.M. Afatinib: First global approval. Drugs 2013, 73, 1503–1515. [Google Scholar] [CrossRef] [PubMed]
- Hidalgo, M. Erlotinib: Preclinical investigations. Oncology (Williston Park) 2003, 17, 11–16. [Google Scholar]
- Stamos, J.; Sliwkowski, M.X.; Eigenbrot, C. Structure of the epidermal growth factor receptor kinase domain alone and in complex with a 4-anilinoquinazoline inhibitor. J. Biol. Chem. 2002, 277, 46265–46272. [Google Scholar] [CrossRef] [PubMed]
- Abdelgalil, A.A.; Al-Kahtani, H.M.; Al-Jenoobi, F.I. Erlotinib. Profiles Drug Subst. Excip. Relat. Methodol. 2020, 45, 93–117. [Google Scholar] [CrossRef]
- Amini, R.; Karami, H.; Bayat, M. Combination Therapy with PIK3R3-siRNA and EGFR-TKI Erlotinib Synergistically Suppresses Glioblastoma Cell Growth In Vitro. Asian Pac. J. Cancer Prev. 2021, 22, 3993–4000. [Google Scholar] [CrossRef]
- Sidorov, M.; Dighe, P.; Woo, R.W.L.; Rodriguez-Brotons, A.; Chen, M.; Ice, R.J.; Vaquero, E.; Jian, D.; Desprez, P.Y.; Nosrati, M.; et al. Dual targeting of EGFR and MTOR pathways inhibits glioblastoma growth by modulating the tumor microenvironment. Cells 2023, 12, 547. [Google Scholar] [CrossRef]
- Dai, Z.; Wang, L.; Wang, X.; Zhao, B.; Zhao, W.; Bhardwaj, S.S.; Ye, J.; Yin, Z.; Zhang, J.; Zhao, S. Oxymatrine induces cell cycle arrest and apoptosis and suppresses the invasion of human glioblastoma cells through the EGFR/PI3K/Akt/mTOR signaling pathway and STAT3. Oncol. Rep. 2018, 40, 867–876. [Google Scholar] [CrossRef]
- Mesbahi, Y.; Zekri, A.; Ahmadian, S.; Alimoghaddam, K.; Ghavamzadeh, A.; Ghaffari, S.H. Targeting of EGFR increase anti-cancer effects of arsenic trioxide: Promising treatment for glioblastoma multiform. Eur. J. Pharmacol. 2018, 820, 274–285. [Google Scholar] [CrossRef]
- Knight, L.A.; Di Nicolantonio, F.; Whitehouse, P.; Mercer, S.; Sharma, S.; Glaysher, S.; Johnson, P.; Cree, I.A. The in vitro effect of gefitinib (‘Iressa’) alone and in combination with cytotoxic chemotherapy on human solid tumours. BMC Cancer 2004, 4, 83. [Google Scholar] [CrossRef]
- Abdel-Aziz, A.A.; El-Azab, A.S.; AlSaif, N.A.; Obaidullah, A.J.; Al-Obaid, A.M.; Al-Suwaidan, I.A. Synthesis, potential antitumor activity, cell cycle analysis, and multitarget mechanisms of novel hydrazones incorporating a 4-methylsulfonylbenzene scaffold: A molecular docking study. J. Enzyme Inhib. Med. Chem. 2021, 36, 1521–1539. [Google Scholar] [CrossRef] [PubMed]
- Karami, A.; Hossienpour, M.; Mohammadi Noori, E.; Rahpyma, M.; Najafi, K.; Kiani, A. Synergistic effect of gefitinib and temozolomide on U87MG glioblastoma angiogenesis. Nutr. Cancer 2022, 74, 1299–1307. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Zhang, T.; Cheng, Z.; Zhu, N.; Wang, H.; Lin, L.; Wang, Z.; Yi, H.; Hu, M. Lycorine inhibits glioblastoma multiforme growth through EGFR suppression. J. Exp. Clin. Cancer Res. 2018, 37, 157. [Google Scholar] [CrossRef] [PubMed]
- Kanaan, R.; Strange, C. Use of multitarget tyrosine kinase inhibitors to attenuate platelet-derived growth factor signalling in lung disease. Eur. Respir. Rev. 2017, 26, 170061. [Google Scholar] [CrossRef] [PubMed]
- Westermark, B. Platelet-derived growth factor in glioblastoma-driver or biomarker. Ups J. Med. Sci. 2014, 119, 298–305. [Google Scholar] [CrossRef]
- Xi, Y.; Chen, M.; Liu, X.; Lu, Z.; Ding, Y.; Li, D. CP-673451, a platelet-derived growth-factor receptor inhibitor, suppresses lung cancer cell proliferation and migration. OncoTargets Ther. 2014, 7, 1215–1221. [Google Scholar] [CrossRef]
- Roberts, W.G.; Whalen, P.M.; Soderstrom, E.; Moraski, G.; Lyssikatos, J.P.; Wang, H.F.; Cooper, B.; Baker, D.A.; Savage, D.; Dalvie, D.; et al. Antiangiogenic and antitumor activity of a selective PDGFR tyrosine kinase inhibitor, CP-673,451. Cancer Res. 2005, 65, 957–966. [Google Scholar] [CrossRef]
- Lane, R.; Cilibrasi, C.; Chen, J.; Shah, K.; Messuti, E.; Mazarakis, N.K.; Stebbing, J.; Critchley, G.; Song, E.; Simon, T.; et al. PDGF-R inhibition induces glioblastoma cell differentiation via DUSP1/p38MAPK signalling. Oncogene 2022, 41, 2749–2763. [Google Scholar] [CrossRef]
- Schenone, S.; Bondavalli, F.; Botta, M. Antiangiogenic agents: An update on small molecule VEGFR inhibitors. Curr. Med. Chem. 2007, 14, 2495–2516. [Google Scholar] [CrossRef]
- Tamura, R.; Morimoto, Y.; Kosugi, K.; Sato, M.; Oishi, Y.; Ueda, R.; Kikuchi, R.; Nagashima, H.; Hikichi, T.; Noji, S.; et al. Clinical and histopathological analyses of VEGF receptors peptide vaccine in patients with primary glioblastoma—A case series. BMC Cancer 2020, 20, 196. [Google Scholar] [CrossRef]
- Sadremomtaz, A.; Mansouri, K.; Alemzadeh, G.; Safa, M.; Rastaghi, A.E.; Asghari, S.M. Dual blockade of VEGFR1 and VEGFR2 by a novel peptide abrogates VEGF-driven angiogenesis, tumor growth, and metastasis through PI3K/AKT and MAPK/ERK1/2 pathway. Biochim. Biophys. Acta Gen. Subj. 2018, 1862, 2688–2700. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Jung, H.J.; Kwon, H.J. A natural small molecule voacangine inhibits angiogenesis both in vitro and in vivo. Biochem. Biophys. Res. Commun. 2012, 417, 330–334. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.M.; Kim, Y.; Jung, Y.; Ko, M.; Marko-Varga, G.; Kwon, H.J. Development of novel VEGFR2 Inhibitors originating from natural product analogues with antiangiogenic impact. J. Med. Chem. 2021, 64, 15858–15867. [Google Scholar] [CrossRef] [PubMed]
- Jang, S.; Strickland, B.; Finis, L.; Kooijman, J.J.; Melis, J.J.T.M.; Zaman, G.J.R.; Van Tornout, J. Comparative biochemical kinase activity analysis identifies rivoceranib as a highly selective VEGFR2 inhibitor. Cancer Chemother. Pharmacol. 2023, 91, 491–499. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Zhou, X.; Sheng, X.; Liang, X. Efficacy and Safety of Apatinib in Patients with Recurrent Glioblastoma. Drugs R&D 2023, 23, 239–244. [Google Scholar] [CrossRef]
- Dawson, J.C.; Serrels, A.; Stupack, D.G.; Schlaepfer, D.D.; Frame, M.C. Targeting FAK in anticancer combination therapies. Nat Rev Cancer 2021, 21, 313–324. [Google Scholar] [CrossRef] [PubMed]
- Slack-Davis, J.K.; Martin, K.H.; Tilghman, R.W.; Iwanicki, M.; Ung, E.J.; Autry, C.; Luzzio, M.J.; Cooper, B.; Kath, J.C.; Roberts, W.G.; et al. Cellular characterization of a novel focal adhesion kinase inhibitor. J. Biol. Chem. 2007, 282, 14845–14852. [Google Scholar] [CrossRef]
- Megison, M.L.; Stewart, J.E.; Nabers, H.C.; Gillory, L.A.; Beierle, E.A. FAK inhibition decreases cell invasion, migration and metastasis in MYCN amplified neuroblastoma. Clin. Exp. Metastasis 2013, 30, 555–568. [Google Scholar] [CrossRef]
- Nguemgo Kouam, P.; Bühler, H.; Hero, T.; Adamietz, I.A. The increased adhesion of tumor cells to endothelial cells after irradiation can be reduced by FAK-inhibition. Radiat. Oncol. 2019, 14, 25. [Google Scholar] [CrossRef]
- Alza, L.; Nàger, M.; Visa, A.; Cantí, C.; Herreros, J. FAK Inhibition Induces Glioblastoma Cell Senescence-Like State through p62 and p27. Cancers 2020, 12, 1086. [Google Scholar] [CrossRef]
- Menet, C.J.; Rompaey, L.V.; Geney, R. Advances in the discovery of selective JAK inhibitors. Prog. Med. Chem. 2013, 52, 153–223. [Google Scholar] [CrossRef] [PubMed]
- Bharadwaj, U.; Kasembeli, M.M.; Robinson, P.; Tweardy, D.J. Targeting Janus kinases and signal transducer and activator of transcription 3 to treat inflammation, fibrosis, and cancer: Rationale, progress, and caution. Pharmacol. Rev. 2020, 72, 486–526. [Google Scholar] [CrossRef] [PubMed]
- Park, A.K.; Kim, P.; Ballester, L.Y.; Esquenazi, Y.; Zhao, Z. Subtype-specific signaling pathways and genomic aberrations associated with prognosis of glioblastoma. Neuro-Oncology 2019, 21, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Sun, Y.; Hou, W.; Ma, L.; Tao, Y.; Li, D.; Xu, C.; Bao, J.; Fan, W. The JAK2/STAT3 pathway inhibitor, AG490, suppresses the abnormal behavior of keloid fibroblasts in vitro. Int. J. Mol. Med. 2020, 46, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Lebedev, T.D.; Vagapova, E.R.; Astashkova, O.O.; Spirin, P.V.; Prassolov, V.S. Inhibition of non-receptor tyrosine kinase JAK2 reduces neuroblastoma cell growth and enhances the action of doxorubicin. Mol. Biol. 2020, 54, 256–261. [Google Scholar] [CrossRef]
- Ajayi, S.; Becker, H.; Reinhardt, H.; Engelhardt, M.; Zeiser, R.; von Bubnoff, N.; Wäsch, R. Ruxolitinib. Recent Results Cancer Res. 2018, 212, 119–132. [Google Scholar] [CrossRef]
- Appeldoorn, T.Y.J.; Munnink, T.H.O.; Morsink, L.M.; Hooge, M.N.L.; Touw, D.J. Pharmacokinetics and pharmacodynamics of ruxolitinib: A review. Clin. Pharmacokinet. 2023, 62, 559–571. [Google Scholar] [CrossRef]
- Delen, E.; Doğanlar, O. The dose dependent effects of ruxolitinib on the invasion and tumorigenesis in gliomas cells via inhibition of interferon gamma-depended JAK/STAT signaling pathway. J. Korean Neurosurg. Soc. 2020, 63, 444–454. [Google Scholar] [CrossRef]
- Goker Bagca, B.; Ozates, N.P.; Biray Avci, C. Ruxolitinib enhances cytotoxic and apoptotic effects of temozolomide on glioblastoma cells by regulating WNT signaling pathway-related genes. Med. Oncol. 2022, 40, 37. [Google Scholar] [CrossRef]
- Ge, L.; Xu, L.; Lu, S.; Yan, H. LCK expression is a potential biomarker for distinguishing primary central nervous system lymphoma from glioblastoma multiforme. FEBS Open Bio 2020, 10, 904–911. [Google Scholar] [CrossRef]
- Stachlewitz, R.F.; Hart, M.A.; Bettencourt, B.; Kebede, T.; Schwartz, A.; Ratnofsky, S.E.; Calderwood, D.J.; Waegell, W.O.; Hirst, G.C. A-770041, a novel and selective small-molecule inhibitor of Lck, prevents heart allograft rejection. J. Pharmacol. Exp. Ther. 2005, 315, 36–41. [Google Scholar] [CrossRef] [PubMed]
- Zepecki, J.P.; Snyder, K.M.; Moreno, M.M.; Fajardo, E.; Fiser, A.; Ness, J.; Sarkar, A.; Toms, S.A.; Tapinos, N. Regulation of human glioma cell migration, tumor growth, and stemness gene expression using a Lck targeted inhibitor. Oncogene 2019, 38, 1734–1750. [Google Scholar] [CrossRef] [PubMed]
- Mansueto, M.S.; Reens, A.; Rakhilina, L.; Chi, A.; Pan, B.S.; Miller, J.R. A reevaluation of the spleen tyrosine kinase (SYK) activation mechanism. J. Biol. Chem. 2019, 294, 7658–7668. [Google Scholar] [CrossRef] [PubMed]
- Moncayo, G.; Grzmil, M.; Smirnova, T.; Zmarz, P.; Huber, R.M.; Hynx, D.; Kohler, H.; Wang, Y.; Hotz, H.R.; Hynes, N.E.; et al. SYK inhibition blocks proliferation and migration of glioma cells and modifies the tumor microenvironment. Neuro-Oncology 2018, 20, 621–631. [Google Scholar] [CrossRef] [PubMed]
- Bhagwat, S.S. Kinase inhibitors for the treatment of inflammatory and autoimmune disorders. Purinergic Signal 2009, 5, 107–115. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Huang, Y.; Liu, Y.; Zhang, X.; Yue, P.; Ma, X.; Miao, Z.; Long, X.; Yang, Y.; Wan, X.; et al. BAY61-3606 attenuates neuroinflammation and neurofunctional damage by inhibiting microglial Mincle/Syk signaling response after traumatic brain injury. Int. J. Mol. Med. 2022, 49, 5. [Google Scholar] [CrossRef]
- Kwon, J.Y.; Seo, S.G.; Heo, Y.S.; Yue, S.; Cheng, J.X.; Lee, K.W.; Kim, K.H. Piceatannol, natural polyphenolic stilbene, inhibits adipogenesis via modulation of mitotic clonal expansion and insulin receptor-dependent insulin signaling in early phase of differentiation. J. Biol. Chem. 2012, 287, 11566–11578. [Google Scholar] [CrossRef]
- MacGlashan, D.; Undem, B.J. Inducing an anergic state in mast cells and basophils without secretion. J. Allergy Clin. Immunol. 2008, 121, 1500–1506.e4. [Google Scholar] [CrossRef]
- Patou, J.; Holtappels, G.; Affleck, K.; van Cauwenberge, P.; Bachert, C. Syk-kinase inhibition prevents mast cell activation in nasal polyps. Rhinology 2011, 49, 100–106. [Google Scholar] [CrossRef]
- Caner, A.; Asik, E.; Ozpolat, B. SRC signaling in cancer and tumor microenvironment. Adv. Exp. Med. Biol. 2021, 1270, 57–71. [Google Scholar] [CrossRef]
- Musumeci, F.; Schenone, S.; Brullo, C.; Botta, M. An update on dual Src/Abl inhibitors. Future Med. Chem. 2012, 4, 799–822. [Google Scholar] [CrossRef] [PubMed]
- Cirotti, C.; Contadini, C.; Barilà, D. SRC kinase in glioblastoma news from an old acquaintance. Cancers 2020, 12, 1558. [Google Scholar] [CrossRef] [PubMed]
- Smolinski, M.P.; Bu, Y.; Clements, J.; Gelman, I.H.; Hegab, T.; Cutler, D.L.; Fang, J.W.S.; Fetterly, G.; Kwan, R.; Barnett, A.; et al. Discovery of Novel Dual Mechanism of Action Src Signaling and Tubulin Polymerization Inhibitors (KX2-391 and KX2-361). J. Med. Chem. 2018, 61, 4704–4719. [Google Scholar] [CrossRef]
- Ciesielski, M.J.; Bu, Y.; Munich, S.A.; Teegarden, P.; Smolinski, M.P.; Clements, J.L.; Lau, J.Y.N.; Hangauer, D.G.; Fenstermaker, R.A. KX2-361: A novel orally bioavailable small molecule dual Src/tubulin inhibitor that provides long term survival in a murine model of glioblastoma. J. Neurooncol. 2018, 140, 519–527. [Google Scholar] [CrossRef] [PubMed]
- Tintori, C.; Fallacara, A.L.; Radi, M.; Zamperini, C.; Dreassi, E.; Crespan, E.; Maga, G.; Schenone, S.; Musumeci, F.; Brullo, C.; et al. Combining X-ray crystallography and molecular modeling toward the optimization of pyrazolo[3,4-d]pyrimidines as potent c-Src inhibitors active in vivo against neuroblastoma. J. Med. Chem. 2015, 58, 347–361. [Google Scholar] [CrossRef] [PubMed]
- Rango, E.; Pastorino, F.; Brignole, C.; Mancini, A.; Poggialini, F.; Di Maria, S.; Zamperini, C.; Iovenitti, G.; Fallacara, A.L.; Sabetta, S.; et al. The pyrazolo[3,4-d]pyrimidine derivative Si306 encapsulated into anti-GD2-immunoliposomes as therapeutic treatment of neuroblastoma. Biomedicines 2022, 10, 659. [Google Scholar] [CrossRef] [PubMed]
- Greco, C.; Taresco, V.; Pearce, A.K.; Vasey, C.E.; Smith, S.; Rahman, R.; Alexander, C.; Cavanagh, R.J.; Musumeci, F.; Schenone, S. Development of pyrazolo[3,4-d]pyrimidine kinase inhibitors as potential clinical candidates for glioblastoma multiforme. ACS Med. Chem. Lett. 2020, 11, 657–663. [Google Scholar] [CrossRef]
- Vignaroli, G.; Iovenitti, G.; Zamperini, C.; Coniglio, F.; Calandro, P.; Molinari, A.; Fallacara, A.L.; Sartucci, A.; Calgani, A.; Colecchia, D.; et al. Prodrugs of pyrazolo[3,4-d]pyrimidines: From library synthesis to evaluation as potential anticancer agents in an orthotopic glioblastoma model. J. Med. Chem. 2017, 60, 6305–6320. [Google Scholar] [CrossRef]
- Nešović, M.; Divac Rankov, A.; Podolski-Renić, A.; Nikolić, I.; Tasić, G.; Mancini, A.; Schenone, S.; Pešić, M.; Dinić, J. Src Inhibitors Pyrazolo[3,4-d]pyrimidines, Si306 and Pro-Si306, inhibit focal adhesion kinase and suppress human glioblastoma invasion in vitro and in vivo. Cancers 2020, 12, 1570. [Google Scholar] [CrossRef]
- Kostić, A.; Jovanović Stojanov, S.; Podolski-Renić, A.; Nešović, M.; Dragoj, M.; Nikolić, I.; Tasić, G.; Schenone, S.; Pešić, M.; Dinić, J. Pyrazolo[3,4-d]pyrimidine tyrosine kinase inhibitors induce oxidative stress in patient-derived glioblastoma cells. Brain Sci. 2021, 11, 884. [Google Scholar] [CrossRef]
- Jovanović Stojanov, S.; Kostić, A.; Ljujić, M.; Lupšić, E.; Schenone, S.; Pešić, M.; Dinić, J. Autophagy inhibition enhances anti-glioblastoma effects of pyrazolo[3,4-d]pyrimidine tyrosine kinase inhibitors. Life 2022, 12, 1503. [Google Scholar] [CrossRef] [PubMed]
- Contadini, C.; Cirotti, C.; Carbone, A.; Norouzi, M.; Cianciusi, A.; Crespan, E.; Perini, C.; Maga, G.; Barilà, D.; Musumeci, F.; et al. Identification and biological characterization of the pyrazolo[3,4-d]pyrimidine derivative SI388 active as src inhibitor. Pharmaceuticals 2023, 16, 958. [Google Scholar] [CrossRef] [PubMed]
- Poggialini, F.; Vagaggini, C.; Brai, A.; Pasqualini, C.; Crespan, E.; Maga, G.; Perini, C.; Cabella, N.; Botta, L.; Musumeci, F.; et al. Biological evaluation and in vitro characterization of adme profile of in-house pyrazolo[3,4-d]pyrimidines as dual tyrosine kinase inhibitors active against glioblastoma multiforme. Pharmaceutics 2023, 15, 453. [Google Scholar] [CrossRef] [PubMed]
- Jaraíz-Rodríguez, M.; Rocío Talaverón, R.; García-Vicente, L.; Pelaz, S.G.; Domínguez-Prieto, M.; Álvarez-Vázquez, A.; Flores-Hernández, R.; Sin, W.C.; Bechberger, J.; Medina, J.M.; et al. Connexin43 peptide, TAT-Cx43266–283, selectively targets glioma cells, impairs malignant growth, and enhances survival in mouse models in vivo. Neuro-Oncology 2020, 15, 493–504. [Google Scholar] [CrossRef] [PubMed]
- Pelaz, S.G.; Jaraíz-Rodríguez, M.; Álvarez-Vázquez, A.; Talaverón, R.; García-Vicente, L.; Flores-Hernández, R.; Gómez de Cedrón, M.; Tabernero, M.; Ramírez de Molina, A.; Lillo, C.; et al. Targeting metabolic plasticity in glioma stem cells in vitro and in vivo through specific inhibition of c-Src by TAT-Cx43266-283. EBioMedicine 2020, 62, 103134. [Google Scholar] [CrossRef] [PubMed]
- Abbassi, R.; Johns, T.G.; Kassiou, M.; Munoz, L. DYRK1A in neurodegeneration and cancer: Molecular basis and clinical implications. Pharmacol. Ther. 2015, 151, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Litovchick, L.; Florens, L.A.; Swanson, S.K.; Washburn, M.P.; DeCaprio, J.A. DYRK1A protein kinase promotes quiescence and senescence through DREAM complex assembly. Genes Dev. 2011, 25, 801–813. [Google Scholar] [CrossRef]
- Recasens, A.; Humphrey, S.J.; Ellis, M.; Hoque, M.; Abbassi, R.H.; Chen, B.; Longworth, M.; Needham, E.J.; James, D.E.; Johns, T.G.; et al. Global phosphoproteomics reveals DYRK1A regulates CDK1 activity in glioblastoma cells. Cell Death Discov. 2021, 7, 81. [Google Scholar] [CrossRef]
- Massey, A.J.; Benwell, K.; Burbridge, M.; Kotschy, A.; Walmsley, D.L. Targeting DYRK1A/B kinases to modulate p21-cyclin D1-p27 signalling and induce anti-tumour activity in a model of human glioblastoma. J. Cell Mol. Med. 2021, 25, 10650–10662. [Google Scholar] [CrossRef]
- Akinleye, A.; Furqan, M.; Mukhi, N.; Ravella, P.; Liu, D. MEK and the inhibitors: From bench to bedside. J. Hematol. Oncol. 2013, 6, 27. [Google Scholar] [CrossRef]
- Selvasaravanan, K.D.; Wiederspohn, N.; Hadzalic, A.; Strobel, H.; Payer, C.; Schuster, A.; Karpel-Massler, G.; Siegelin, M.D.; Halatsch, M.E.; Debatin, K.M.; et al. The limitations of targeting MEK signalling in Glioblastoma therapy. Sci. Rep. 2020, 10, 7401. [Google Scholar] [CrossRef] [PubMed]
- Monk, B.J.; Grisham, R.N.; Banerjee, S.; Kalbacher, E.; Mirza, M.R.; Romero, I.; Vuylsteke, P.; Coleman, R.L.; Hilpert, F.; Oza, A.M.; et al. MILO/ENGOT-ov11: Binimetinib versus physician’s choice chemotherapy in recurrent or persistent low-grade serous carcinomas of the ovary, fallopian tube, or primary peritoneum. J. Clin. Oncol. 2020, 38, 3753–3762. [Google Scholar] [CrossRef] [PubMed]
- Woodfield, S.E.; Zhang, L.; Scorsone, K.A.; Liu, Y.; Zage, P.E. Binimetinib inhibits MEK and is effective against neuroblastoma tumor cells with low NF1 expression. BMC Cancer 2016, 16, 172. [Google Scholar] [CrossRef] [PubMed]
- Bikhezar, F.; de Kruijff, R.M.; van der Meer, A.J.G.M.; Torrelo Villa, G.; van der Pol, S.M.A.; Becerril Aragon, G.; Gasol Garcia, A.; Narayan, R.S.; de Vries, H.E.; Slotman, B.J.; et al. Preclinical evaluation of binimetinib (MEK162) delivered via polymeric nanocarriers in combination with radiation and temozolomide in glioma. J. Neurooncol. 2020, 146, 239–246. [Google Scholar] [CrossRef] [PubMed]
- Narayan, R.S.; Gasol, A.; Slangen, P.L.G.; Cornelissen, F.M.G.; Lagerweij, T.; Veldman, H.Y.Y.E.; Dik, R.; van den Berg, J.; Slotman, B.J.; Würdinger, T.; et al. Identification of MEK162 as a radiosensitizer for the treatment of glioblastoma. Mol. Cancer Ther. 2018, 17, 347–354. [Google Scholar] [CrossRef]
- Vinitsky, A.; Chiang, J.; Bag, A.K.; Campagne, O.; Stewart, C.F.; Dunphy, P.; Shulkin, B.; Li, Q.; Lin, T.; Hoehn, M.E.; et al. LGG-22. SJ901: Phase I/II evaluation of single agent mirdametinib (PD-0325901), a brain-penetrant MEK1/2 inhibitor, for the treatment of children, adolescents, and young adults with low-grade glioma (LGG). Neuro-Oncology 2022, 24, i92. [Google Scholar] [CrossRef]
- Houweling, M.; Abdul, U.K.; Brahm, C.; Lagerweij, T.; Heukelom, S.; Koken, P.W.; Honeywell, R.; Wedekind, L.E.; Peters, G.J.; Verheul, H.; et al. Radio-sensitizing effect of MEK inhibition in glioblastoma in vitro and in vivo. J. Cancer Res. Clin. Oncol. 2023, 149, 297–305. [Google Scholar] [CrossRef]
- Xie, Y.; Wang, A.; Lin, J.; Wu, L.; Zhang, H.; Yang, X.; Wan, X.; Miao, R.; Sang, X.; Zhao, H. Mps1/TTK: A novel target and biomarker for cancer. J. Drug Target 2017, 25, 112–118. [Google Scholar] [CrossRef]
- Wang, J.; Xie, Y.; Bai, X.; Wang, N.; Yu, H.; Deng, Z.; Lian, M.; Yu, S.; Liu, H.; Xie, W.; et al. Targeting dual specificity protein kinase TTK attenuates tumorigenesis of glioblastoma. Oncotarget 2018, 9, 3081–3088. [Google Scholar] [CrossRef]
- Atrafi, F.; Boix, O.; Subbiah, V.; Diamond, J.R.; Chawla, S.P.; Tolcher, A.W.; LoRusso, P.M.; Eder, J.P.; Gutierrez, M.; Sankhala, K.; et al. A phase I study of an MPS1 inhibitor (BAY 1217389) in combination with paclitaxel using a novel randomized continual reassessment method for dose escalation. Clin. Cancer Res. 2021, 27, 6366–6375. [Google Scholar] [CrossRef]
- Yu, J.; Gao, G.; Wei, X.; Wang, Y. TTK Protein Kinase promotes temozolomide resistance through inducing autophagy in glioblastoma. BMC Cancer 2022, 22, 786. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Laufer, R.; Patel, N.K.; Ng, G.; Sampson, P.B.; Li, S.W.; Lang, Y.; Feher, M.; Brokx, R.; Beletskaya, I.; et al. Discovery of pyrazolo[1,5-a]pyrimidine TTK Inhibitors: CFI-402257 is a potent, selective, bioavailable anticancer agent. ACS Med. Chem. Lett. 2016, 7, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Mason, J.M.; Wei, X.; Fletcher, G.C.; Kiarash, R.; Brokx, R.; Hodgson, R.; Beletskaya, I.; Bray, M.R.; Mak, T.W. Functional characterization of CFI-402257, a potent and selective Mps1/TTK kinase inhibitor, for the treatment of cancer. Proc. Natl. Acad. Sci. USA 2017, 114, 3127–3132. [Google Scholar] [CrossRef] [PubMed]
- Garuti, L.; Roberti, M.; Bottegoni, G. Multi-kinase inhibitors. Curr. Med. Chem. 2015, 22, 695–712. [Google Scholar] [CrossRef] [PubMed]
- Shen, G.; Zheng, F.; Ren, D.; Du, F.; Dong, Q.; Wang, Z.; Zhao, F.; Ahmad, R.; Zhao, J. Anlotinib: A novel multi-targeting tyrosine kinase inhibitor in clinical development. J. Hematol. Oncol. 2018, 11, 120. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Zhou, A.; Zhang, W.; Jiang, Z.; Chen, B.; Zhao, J.; Li, Z.; Wang, L.; Bi, X.; Zhao, H.; et al. Anlotinib in the treatment of advanced hepatocellular carcinoma: An open-label phase II study (ALTER-0802 study). Hepatol. Int. 2021, 15, 621–629. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; Wang, H.; Pan, H.; Chen, J.; Deng, C. Anlotinib combined with temozolomide suppresses glioblastoma growth via mediation of JAK2/STAT3 signaling pathway. Cancer Chemother. Pharmacol. 2022, 89, 183–196. [Google Scholar] [CrossRef]
- Sun, Y.; Niu, W.; Du, F.; Du, C.; Li, S.; Wang, J.; Li, L.; Wang, F.; Hao, Y.; Li, C.; et al. Safety, pharmacokinetics, and antitumor properties of anlotinib, an oral multi-target tyrosine kinase inhibitor, in patients with advanced refractory solid tumors. J. Hematol. Oncol. 2016, 9, 105. [Google Scholar] [CrossRef]
- Galimberti, C.; Piepoli, T.; Letari, O.; Artusi, R.; Persiani, S.; Caselli, G.; Rovati, L.C. CR13626: A novel oral brain penetrant tyrosine kinase inhibitor that reduces tumor growth and prolongs survival in a mouse model of glioblastoma. Am. J. Cancer Res. 2021, 11, 3558–3574. [Google Scholar]
- Chen, H.M.; Tsai, C.H.; Hung, W.C. Foretinib inhibits angiogenesis, lymphangiogenesis and tumor growth of pancreatic cancer in vivo by decreasing VEGFR-2/3 and TIE-2 signaling. Oncotarget 2015, 6, 14940–14952. [Google Scholar] [CrossRef]
- Gortany, N.K.; Panahi, G.; Ghafari, H.; Shekari, M.; Ghazi-Khansari, M. Foretinib induces G2/M cell cycle arrest, apoptosis, and invasion in human glioblastoma cells through c-MET inhibition. Cancer Chemother. Pharmacol. 2021, 87, 827–842. [Google Scholar] [CrossRef] [PubMed]
- Han, K.M.; Kang, R.J.; Jeon, H.; Lee, H.J.; Lee, J.S.; Park, H.; Gak Jeon, S.; Suk, K.; Seo, J.; Hoe, H.S. Regorafenib regulates ad pathology, neuroinflammation, and dendritic spinogenesis in cells and a mouse model of AD. Cells 2020, 9, 1655. [Google Scholar] [CrossRef] [PubMed]
- Chiang, I.T.; Liu, Y.C.; Liu, H.S.; Ali, A.A.A.; Chou, S.Y.; Hsu, T.I.; Hsu, F.T. Regorafenib Reverses Temozolomide-Induced CXCL12/CXCR4 signaling and triggers apoptosis mechanism in glioblastoma. Neurotherapeutics 2022, 19, 616–634. [Google Scholar] [CrossRef] [PubMed]
- Zeiner, P.S.; Kinzig, M.; Divé, I.; Maurer, G.D.; Filipski, K.; Harter, P.N.; Senft, C.; Bähr, O.; Hattingen, E.; Steinbach, J.P.; et al. Regorafenib CSF penetration, efficacy, and mri patterns in recurrent malignant glioma patients. J. Clin. Med. 2019, 8, 2031. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhan, Y.; Wu, Z.; Lin, M.; Jin, X.; Jiang, L.; Qiu, Y. A novel multitarget kinase inhibitor BZG with potent anticancer activity in vitro and vivo enhances efficacy of sorafenib through PI3K pathways in hepatocellular carcinoma cells. Biomed. Pharmacother. 2020, 125, 110033. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Jo, Y.; Oh, H.K.; Kim, E.H. Sorafenib increases tumor treating fields-induced cell death in glioblastoma by inhibiting STAT3. Am. J. Cancer Res. 2020, 10, 3475–3486. [Google Scholar] [PubMed]
- Zajak, A.; Sumorek-Wiadro, J.; Maciejczik, A.; Langner, E.; Wertel, I.; Rzeski, W.; Jakubowicz-Gil, J. LY294002 and sorafenib as inhibitors of intracellular survival pathways in the elimination of human glioma cells by programmed cell death. Cell Tissue Res. 2021, 386, 17–28. [Google Scholar] [CrossRef]
- Sweeney, W.E.; Frost, P.; Avner, E.D. Tesevatinib ameliorates progression of polycystic kidney disease in rodent models of autosomal recessive polycystic kidney disease. World J. Nephrol. 2017, 6, 188–200. [Google Scholar] [CrossRef]
- Kizilbash, S.H.; Gupta, S.K.; Parrish, K.E.; Laramy, J.K.; Kim, M.; Gampa, G.; Carlson, B.L.; Bakken, K.K.; Mladek, A.C.; Schroeder, M.A.; et al. In vivo efficacy of Tesevatinib in EGFR-amplified patient-derived xenograft glioblastoma models may be limited by tissue binding and compensatory signaling. Mol. Cancer Ther. 2021, 20, 1009–1018. [Google Scholar] [CrossRef]
- Brar, H.K.; Jose, J.; Wu, Z.; Sharma, M. Tyrosine kinase inhibitors for glioblastoma multiforme: Challenges and opportunities for drug delivery. Pharmaceutics 2022, 15, 59. [Google Scholar] [CrossRef]
- Cooper, E.; Choi, P.J.; Denny, W.A.; Jose, J.; Dragunow, M.; Park, T.I. The use of heptamethine cyanine dyes as drug-conjugate systems in the treatment of primary and metastatic brain tumors. Front. Oncol. 2021, 11, 654921. [Google Scholar] [CrossRef] [PubMed]
- Bozzuto, G.; Molinari, A. Liposomes as nanomedical devices. Int. J. Nanomed. 2015, 10, 975–999. [Google Scholar] [CrossRef] [PubMed]
- Lakkadwala, S.; Singh, J. Co-delivery of doxorubicin and erlotinib through liposomal nanoparticles for glioblastoma tumor regression using an in vitro brain tumor model. Colloids Surf. B Biointerfaces 2019, 173, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Rehman, U.; Parveen, N.; Sheikh, A.; Abourehab, M.A.S.; Sahebkar, A.; Kesharwani, P. Polymeric nanoparticles-siRNA as an emerging nano-polyplexes against ovarian cancer. Colloids Surf. B Biointerfaces 2022, 218, 112766. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.M.; Ahmad, F.J.; Panda, A.K.; Talegaonkar, S. Investigation of imatinib loaded surface decorated biodegradable nanocarriers against glioblastoma cell lines: Intracellular uptake and cytotoxicity studies. Int. J. Pharm. 2016, 507, 61–71. [Google Scholar] [CrossRef] [PubMed]
- Sahoo, S.K.; Labhasetwar, V. Nanotech approaches to drug delivery and imaging. Drug Discov. Today 2003, 8, 1112–1120. [Google Scholar] [CrossRef]
- Xu, W.; Ye, C.; Qing, X.; Liu, S.; Lv, X.; Wang, W.; Dong, X.; Zhang, Y. Multi-target tyrosine kinase inhibitor nanoparticle delivery systems for cancer therapy. Mater. Today Bio 2022, 16, 100358. [Google Scholar] [CrossRef]
- Greish, K.; Jasim, A.; Parayath, N.; Abdelghany, S.; Alkhateeb, A.; Taurin, S.; Nehoff, H. Micellar formulations of Crizotinib and Dasatinib in the management of glioblastoma multiforme. J. Drug Target 2018, 26, 692–708. [Google Scholar] [CrossRef]
- Kratz, F. Albumin as a drug carrier: Design of prodrugs, drug conjugates and nanoparticles. J. Control Release 2008, 132, 171–183. [Google Scholar] [CrossRef]
- Yang, Z.; Du, Y.; Lei, L.; Xia, X.; Wang, X.; Tong, F.; Li, Y.; Gao, H. Co-delivery of ibrutinib and hydroxychloroquine by albumin nanoparticles for enhanced chemotherapy of glioma. Int. J. Pharm. 2023, 630, 122436. [Google Scholar] [CrossRef]
- Zhou, X.; Shi, K.; Hao, Y.; Yang, C.; Zha, R.; Yi, C.; Qian, Z. Advances in nanotechnology-based delivery systems for EGFR tyrosine kinases inhibitors in cancer therapy. Asian J. Pharm. Sci. 2020, 15, 26–41. [Google Scholar] [CrossRef] [PubMed]
- Moore, T.L.; Grimes, S.W.; Lewis, R.L.; Alexis, F. Multilayered polymer-coated carbon nanotubes to deliver dasatinib. Mol. Pharm. 2014, 11, 276–282. [Google Scholar] [CrossRef] [PubMed]
- Saliou, B.; Thomas, O.; Lautram, N.; Clavreul, A.; Hureaux, J.; Urban, T.; Benoit, J.P.; Lagarce, F. Development and in vitro evaluation of a novel lipid nanocapsule formulation of etoposide. Eur. J. Pharm. Sci. 2013, 50, 172–180. [Google Scholar] [CrossRef]
- Clavreul, A.; Roger, E.; Pourbaghi-Masouleh, M.; Lemaire, L.; Tétaud, C.; Menei, P. Development and characterization of sorafenib-loaded lipid nanocapsules for the treatment of glioblastoma. Drug Deliv. 2018, 25, 1756–1765. [Google Scholar] [CrossRef]
- Sanai, N.; Li, J.; Boerner, J.; Stark, K.; Wu, J.; Kim, S.; Derogatis, A.; Mehta, S.; Dhruv, H.D.; Heilbrun, L.K.; et al. Phase 0 Trial of AZD1775 in first-recurrence glioblastoma patients. Clin. Cancer Res. 2018, 24, 3820–3828. [Google Scholar] [CrossRef] [PubMed]
- Duerinck, J.; Du Four, S.; Bouttens, F.; Andre, C.; Verschaeve, V.; Van Fraeyenhove, F.; Chaskis, C.; D’Haene, N.; Le Mercier, M.; Rogiers, A.; et al. Randomized phase II trial comparing axitinib with the combination of axitinib and lomustine in patients with recurrent glioblastoma. J. Neurooncol. 2018, 136, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Hainsworth, J.D.; Becker, K.P.; Mekhail, T.; Chowdhary, S.A.; Eakle, J.F.; Wright, D.; Langdon, R.M.; Yost, K.J.; Padula, G.D.A.; West-Osterfield, K.; et al. Phase I/II study of bevacizumab with BKM120, an oral PI3K inhibitor, in patients with refractory solid tumors (phase I) and relapsed/refractory glioblastoma (phase II). J. Neurooncol. 2019, 144, 303–311. [Google Scholar] [CrossRef]
- Mehta, S.; Fiorelli, R.; Bao, X.; Pennington-Krygier, C.; Derogatis, A.; Kim, S.; Yoo, W.; Li, J.; Sanai, N. A Phase 0 trial of ceritinib in patients with brain metastases and recurrent glioblastoma. Clin. Cancer Res. 2022, 28, 289–297. [Google Scholar] [CrossRef]
- Desai, A.V.; Robinson, G.W.; Gauvain, K.; Basu, E.M.; Macy, M.E.; Maese, L.; Whipple, N.S.; Sabnis, A.J.; Foster, J.H.; Shusterman, S.; et al. Entrectinib in children and young adults with solid or primary CNS tumors harboring NTRK, ROS1, or ALK aberrations (STARTRK-NG). Neuro-Oncology 2022, 24, 1776–1789. [Google Scholar] [CrossRef]
- Lee, E.Q.; Muzikansky, A.; Duda, D.G.; Gaffey, S.; Dietrich, J.; Nayak, L.; Chukwueke, U.N.; Beroukhim, R.; Doherty, L.; Laub, C.K.; et al. Phase II trial of ponatinib in patients with bevacizumab-refractory glioblastoma. Cancer Med. 2019, 8, 5988–5994. [Google Scholar] [CrossRef]
- Lombardi, G.; De Salvo, G.L.; Brandes, A.A.; Eoli, M.; Rudà, R.; Faedi, M.; Lolli, I.; Pace, A.; Daniele, B.; Pasqualetti, F.; et al. Regorafenib compared with lomustine in patients with relapsed glioblastoma (REGOMA): A multicentre, open-label, randomised, controlled, phase 2 trial. Lancet Oncol. 2019, 20, 110–119. [Google Scholar] [CrossRef] [PubMed]
- Galanis, E.; Anderson, S.K.; Twohy, E.L.; Carrero, X.W.; Dixon, J.G.; Tran, D.D.; Jeyapalan, S.A.; Anderson, D.M.; Kaufmann, T.J.; Feathers, R.W.; et al. A phase 1 and randomized, placebo-controlled phase 2 trial of bevacizumab plus dasatinib in patients with recurrent glioblastoma: Alliance/North Central Cancer Treatment Group N0872. Cancer 2019, 125, 3790–3800. [Google Scholar] [CrossRef] [PubMed]
- Kato, S.; Jardim, D.L.; Johnson, F.M.; Subbiah, V.; Piha-Paul, S.; Tsimberidou, A.M.; Falchook, G.S.; Karp, D.; Zinner, R.; Wheler, J.; et al. Phase I study of the combination of crizotinib (as a MET inhibitor) and dasatinib (as a c-SRC inhibitor) in patients with advanced cancer. Invest. New Drugs 2018, 36, 416–423. [Google Scholar] [CrossRef] [PubMed]
- HMPL-813 in Treating Patients with Glioblastoma. Available online: https://clinicaltrials.gov/study/NCT03231501?cond=Glioblastoma&term=Tyrosine%20Kinase%20Inhibitor&limit=50&page=1&rank=2 (accessed on 17 January 2024).
- Han, B.; Li, K.; Zhao, Y.; Li, B.; Cheng, Y.; Zhou, J.; Lu, Y.; Shi, Y.; Wang, Z.; Jiang, L.; et al. Anlotinib as a third-line therapy in patients with refractory advanced non-small-cell lung cancer: A multicentre, randomised phase II trial (ALTER0302). Br. J. Cancer 2018, 118, 654–661. [Google Scholar] [CrossRef]
- Anlotinib Combined with Dose-Dense Temozolomide for the First Recurrent or Progressive Glioblastoma after STUPP Regimen. Available online: https://clinicaltrials.gov/study/NCT04547855?cond=Glioblastoma&term=Tyrosine%20Kinase%20Inhibitor&rank=10#publications (accessed on 17 January 2024).
- Johnson, T.S.; MacDonald, T.J.; Pacholczyk, R.; Aguilera, D.; Al-Basheer, A.; Bajaj, M.; Bandopadhayay, P.; Berrong, Z.; Bouffet, E.; Castellino, R.C.; et al. Indoximod-based chemo-immunotherapy for pediatric brain tumors: A first-in-children phase 1 trial. Neuro-Oncology 2023. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Drug Combined with | Pharmaceutical Company | Targeted Kinase | NCT Number | Phase | Status |
|---|---|---|---|---|---|---|
| Adavosertib [207] | - | - | WEE1 | NCT02207010 | 0/1 | Completed |
| Axitinib [208] | Lomustine | Pfizer | VEGFR | NCT01562197 | 2 | Completed |
| Bevacizumab [209] | Buparlisib | Novartis | VEGFR, Src | NCT01349660 | 1/2 | Completed, has results |
| Ceritinib [210] | - | Novartis | ALK, IGFR1, FAK | NCT02605746 | 0/1 | Completed |
| Entrectinib [211] | - | Hoffmann-La Roche | TRK, ALK, ROS1 | NCT02650401 | 1/2 | Ongoing |
| Ponatinib [212] | - | - | VEGFR, PDGFR, FGFR, Src | NCT02478164 | 2 | Completed, has results |
| Regorafenib [213] | Lomustine | Bayer | VEGFR, TIE-2, PDGFR, FGFR, KIT, RET, RAF | NCT02926222 | 2 | Completed |
| Dasatinib [214] | Bevacizumab | - | VEGFR, Src | NCT00892177 | 1/2 | Completed, has results |
| Dasatinib [215] | Crizotinib | Pfizer | Src, c-MET | NCT01744652 | 1 | Completed |
| Epitinib [216] | - | - | EGFR | NCT03231501 | 1 | Completed |
| Anlotinib [217] | - | - | VEGFR, FGFR, PDGFR, c-Kit | NCT04004975 | 1/2 | Completed |
| Anlotinib [218] | Temozolomide | - | VEGFR, FGFR, PDGFR, c-Kit | NCT04547855 | 2 | Completed |
| Ibrutinib [219] | Indoximod | - | Btk | NCT05106296 | 1 | Ongoing |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Frumento, D.; Grossi, G.; Falesiedi, M.; Musumeci, F.; Carbone, A.; Schenone, S. Small Molecule Tyrosine Kinase Inhibitors (TKIs) for Glioblastoma Treatment. Int. J. Mol. Sci. 2024, 25, 1398. https://doi.org/10.3390/ijms25031398
Frumento D, Grossi G, Falesiedi M, Musumeci F, Carbone A, Schenone S. Small Molecule Tyrosine Kinase Inhibitors (TKIs) for Glioblastoma Treatment. International Journal of Molecular Sciences. 2024; 25(3):1398. https://doi.org/10.3390/ijms25031398
Chicago/Turabian StyleFrumento, Davide, Giancarlo Grossi, Marta Falesiedi, Francesca Musumeci, Anna Carbone, and Silvia Schenone. 2024. "Small Molecule Tyrosine Kinase Inhibitors (TKIs) for Glioblastoma Treatment" International Journal of Molecular Sciences 25, no. 3: 1398. https://doi.org/10.3390/ijms25031398
APA StyleFrumento, D., Grossi, G., Falesiedi, M., Musumeci, F., Carbone, A., & Schenone, S. (2024). Small Molecule Tyrosine Kinase Inhibitors (TKIs) for Glioblastoma Treatment. International Journal of Molecular Sciences, 25(3), 1398. https://doi.org/10.3390/ijms25031398