
Mode of Metal Ligation Governs Inhibition of Carboxypeptidase A

 , , , ,
, , , ,
Abstract
1. Introduction
2. Results
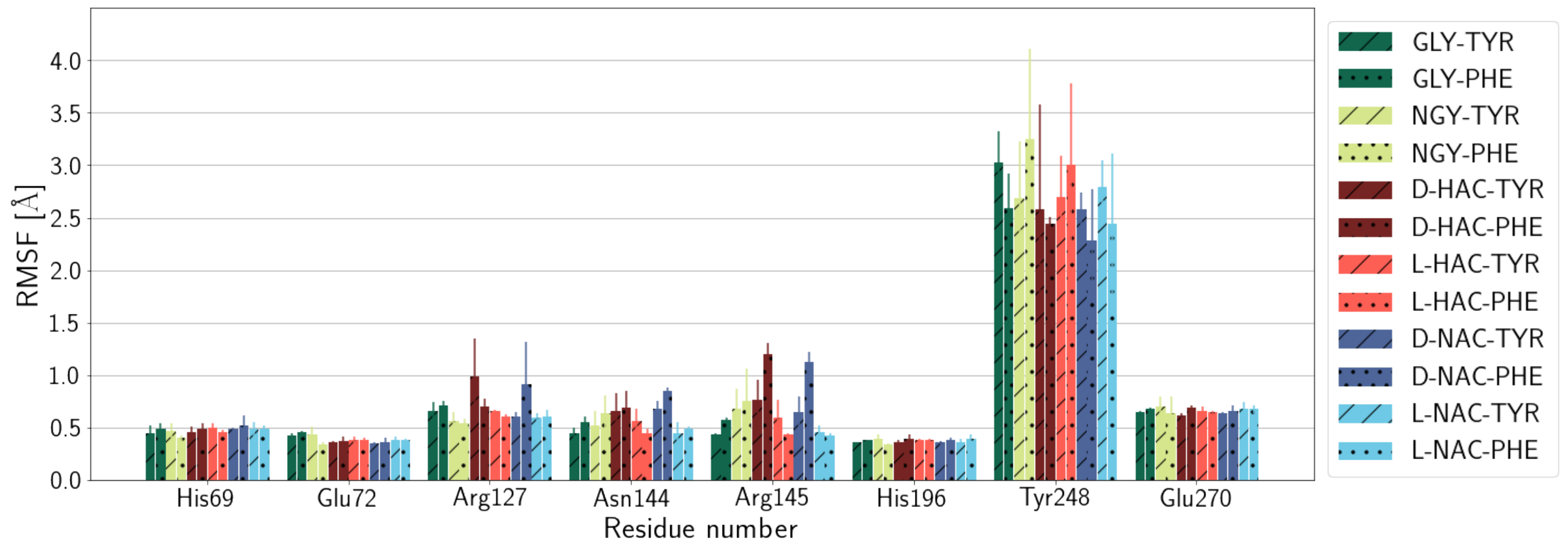
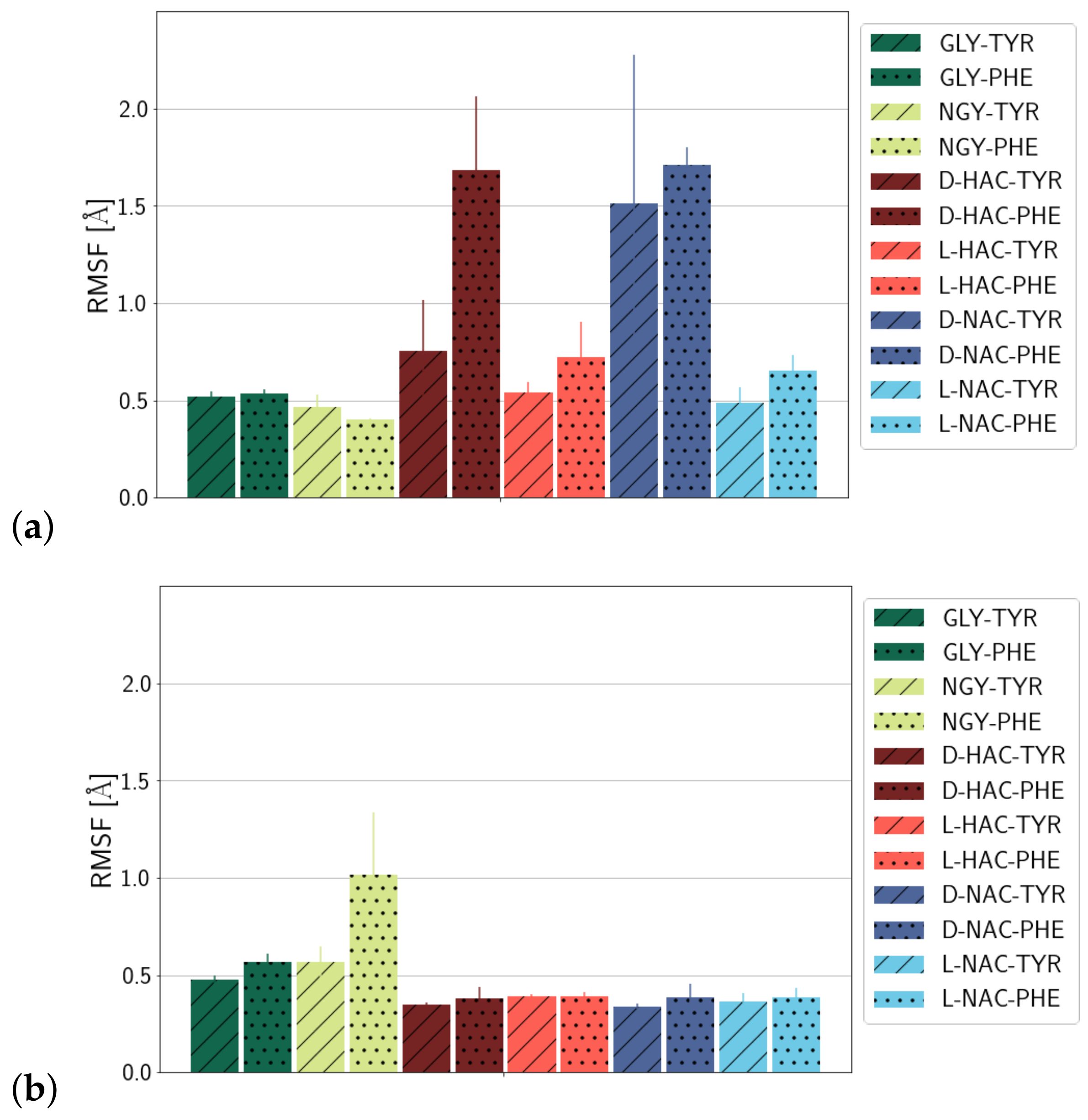
2.1. Fluctuations
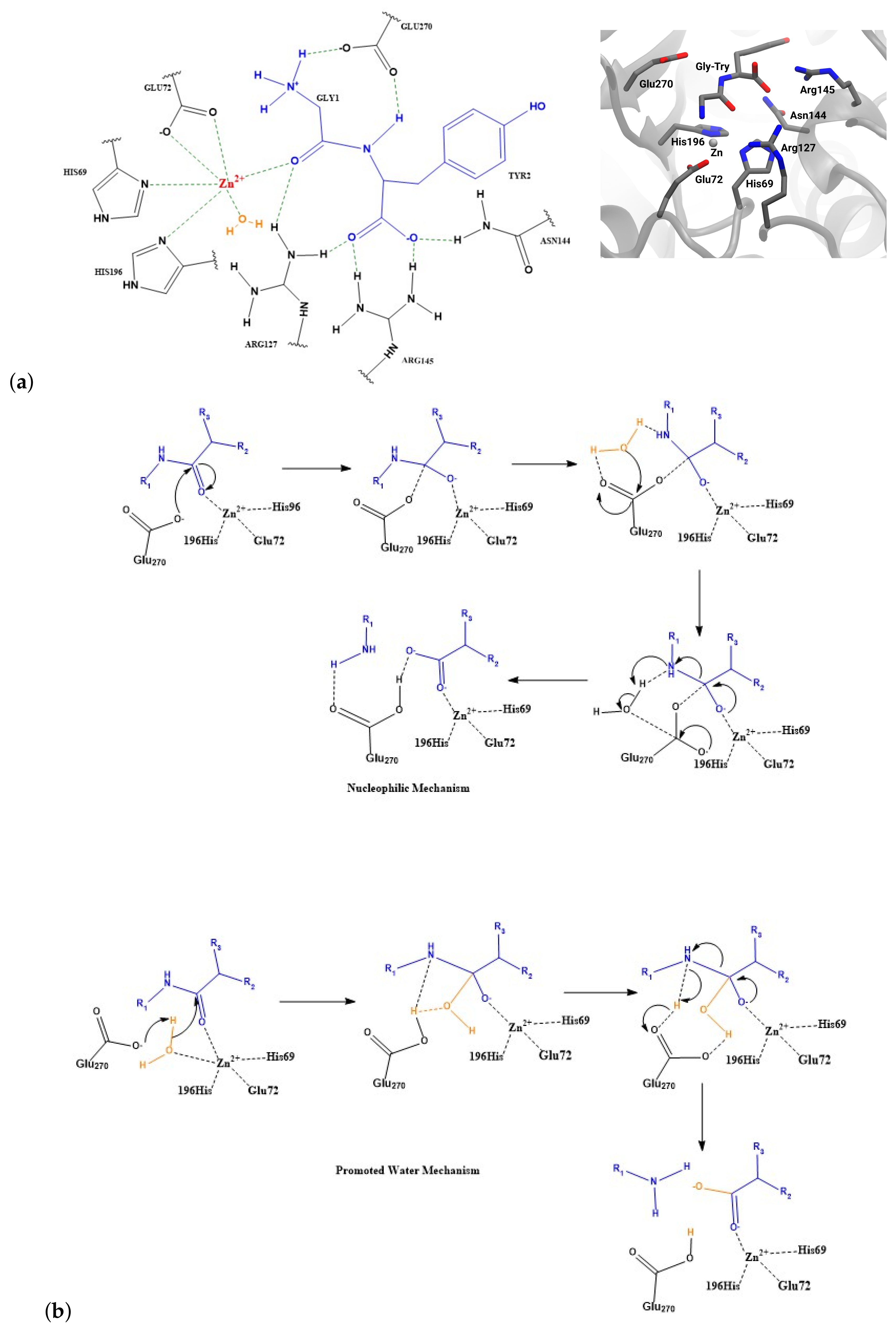
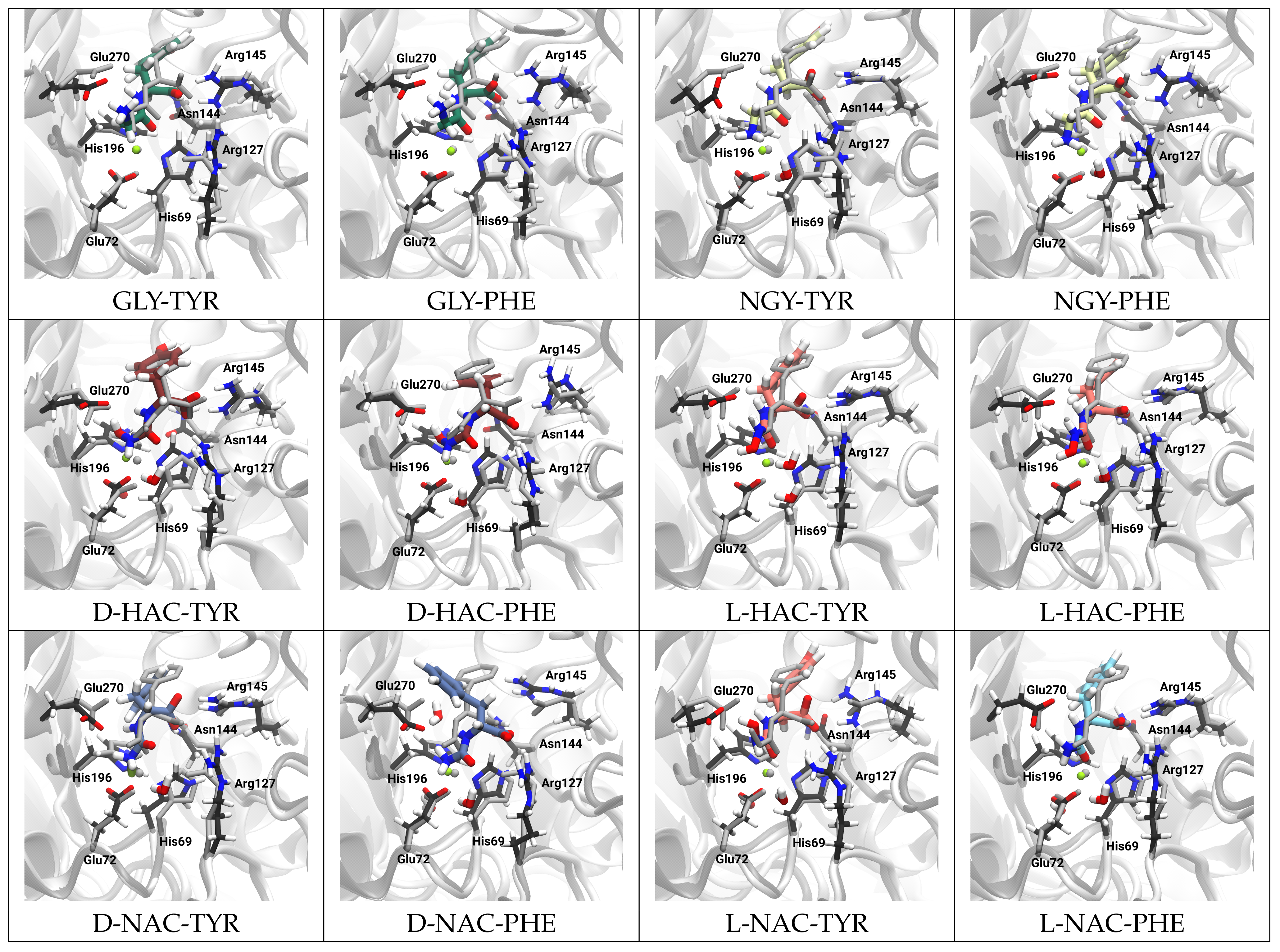
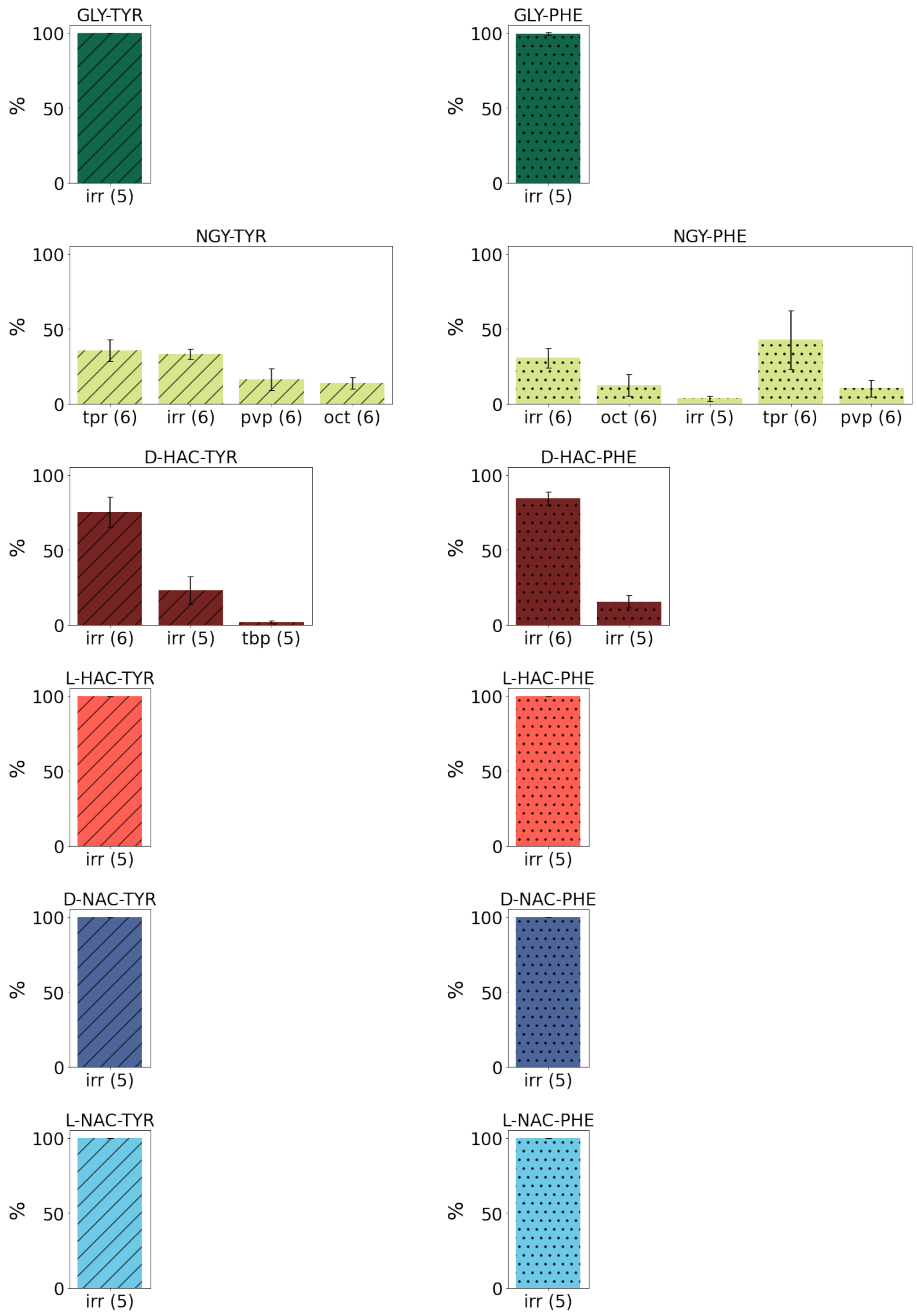
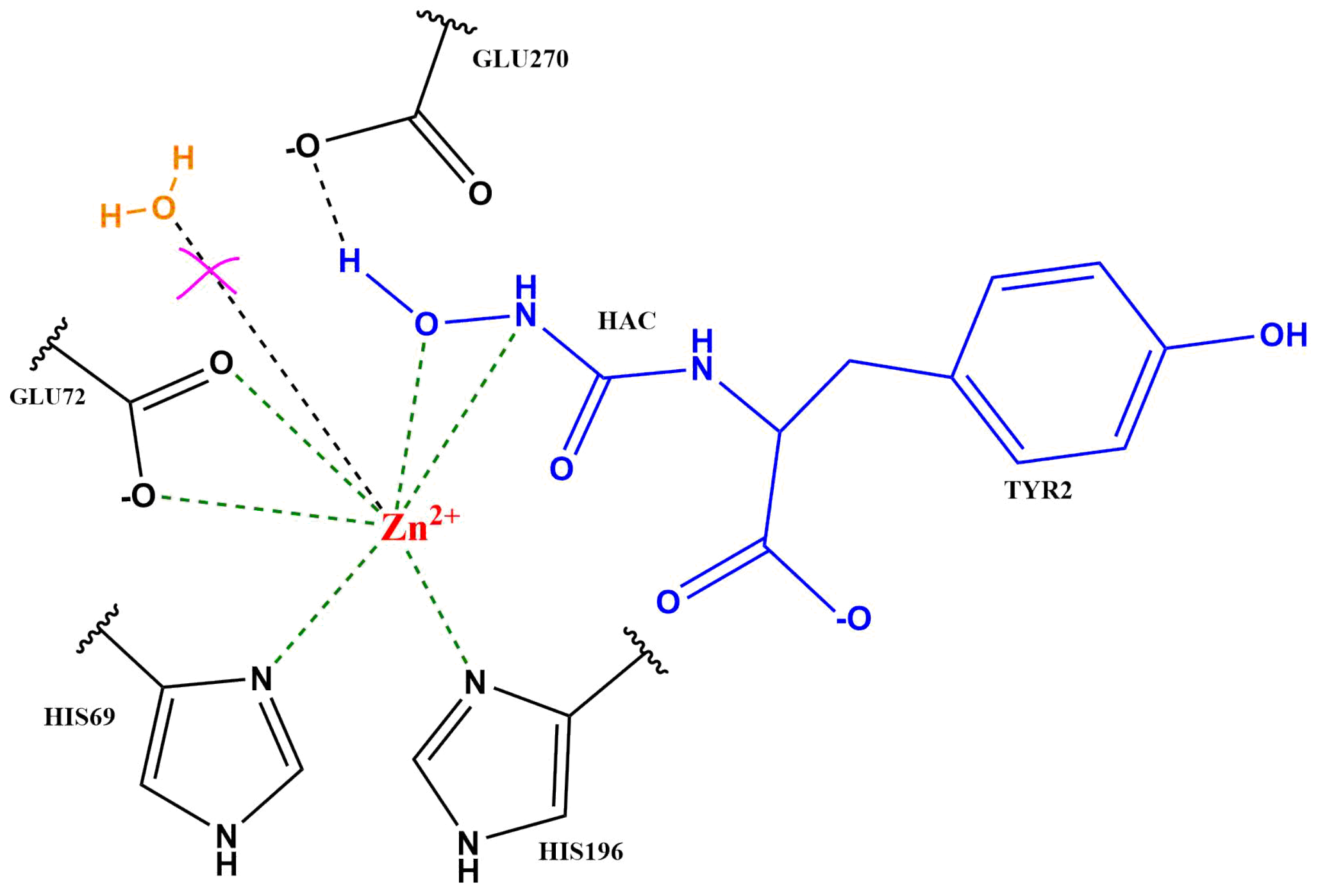
2.2. Zn Ion Coordination
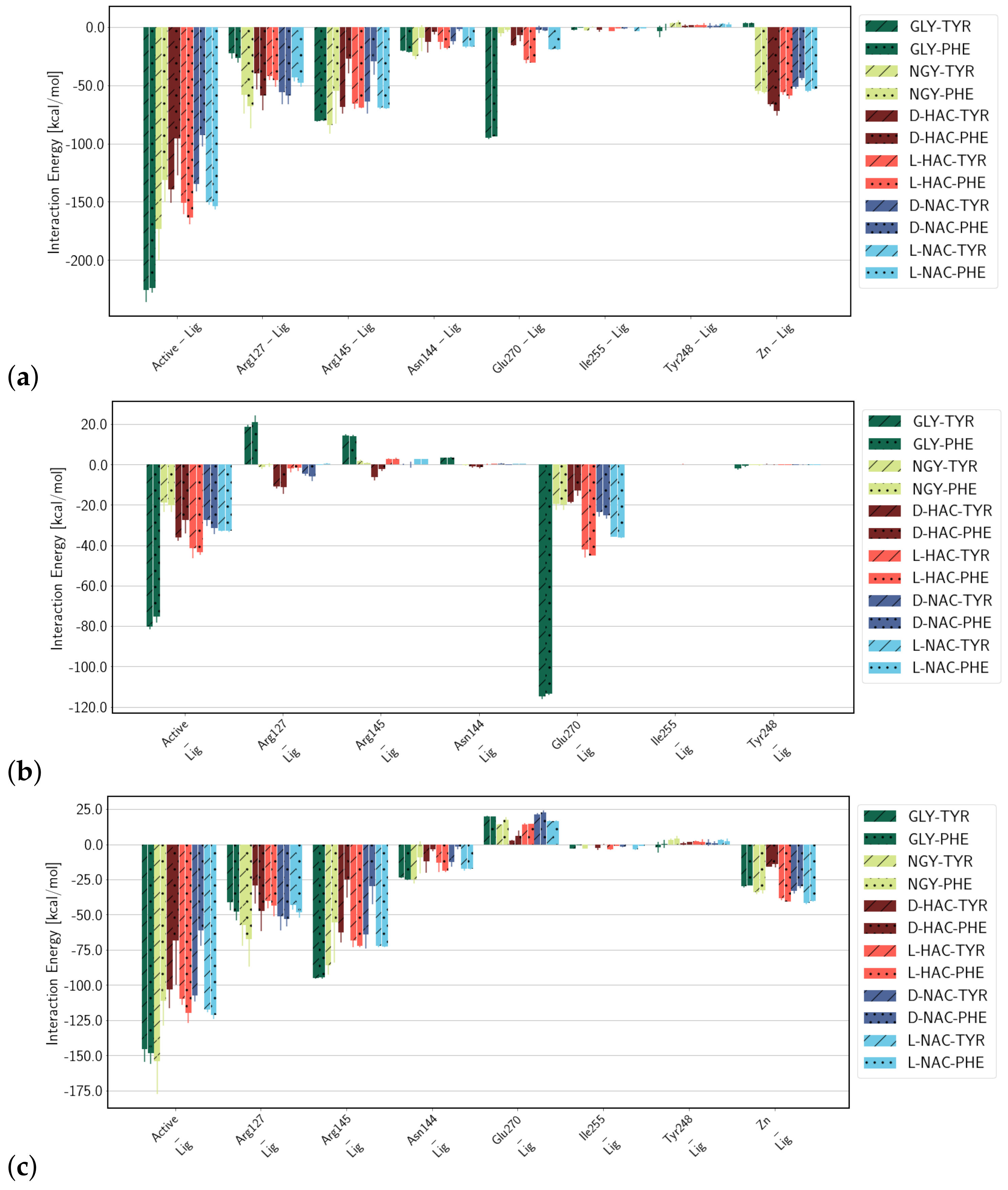
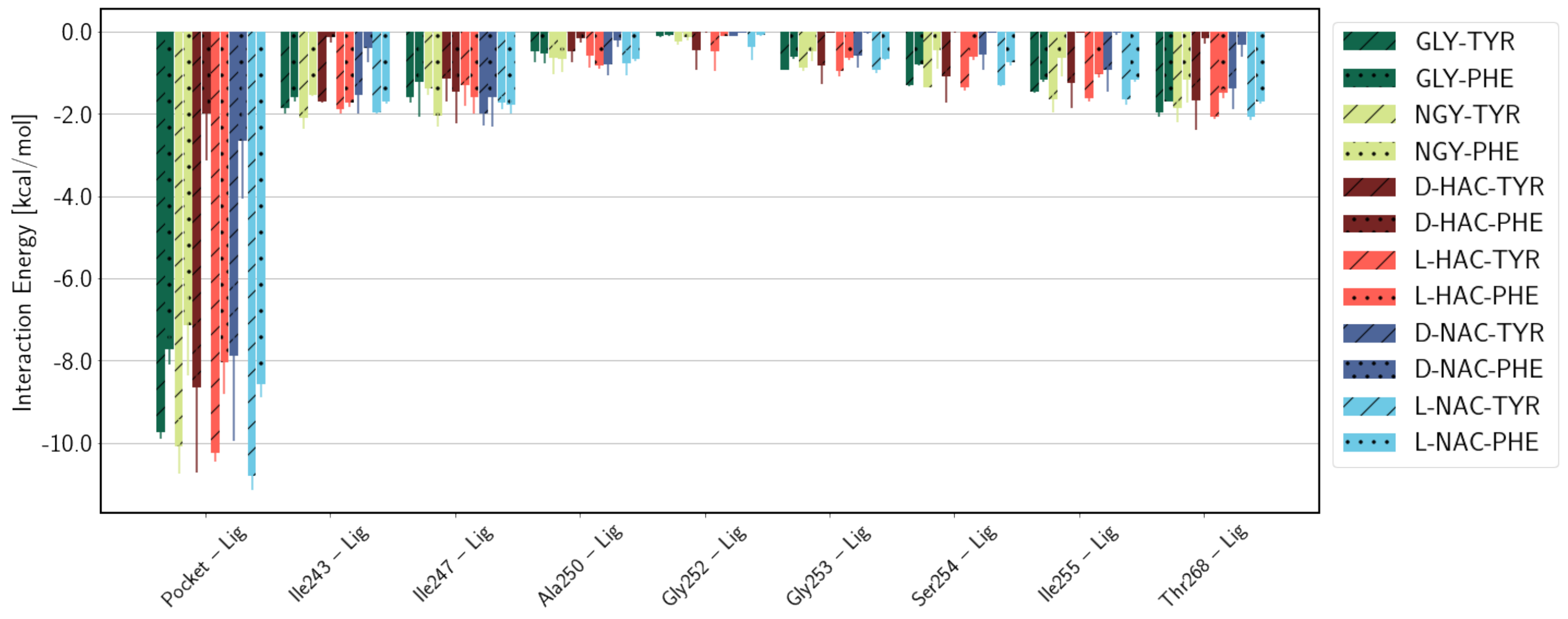
2.3. Interaction Energies
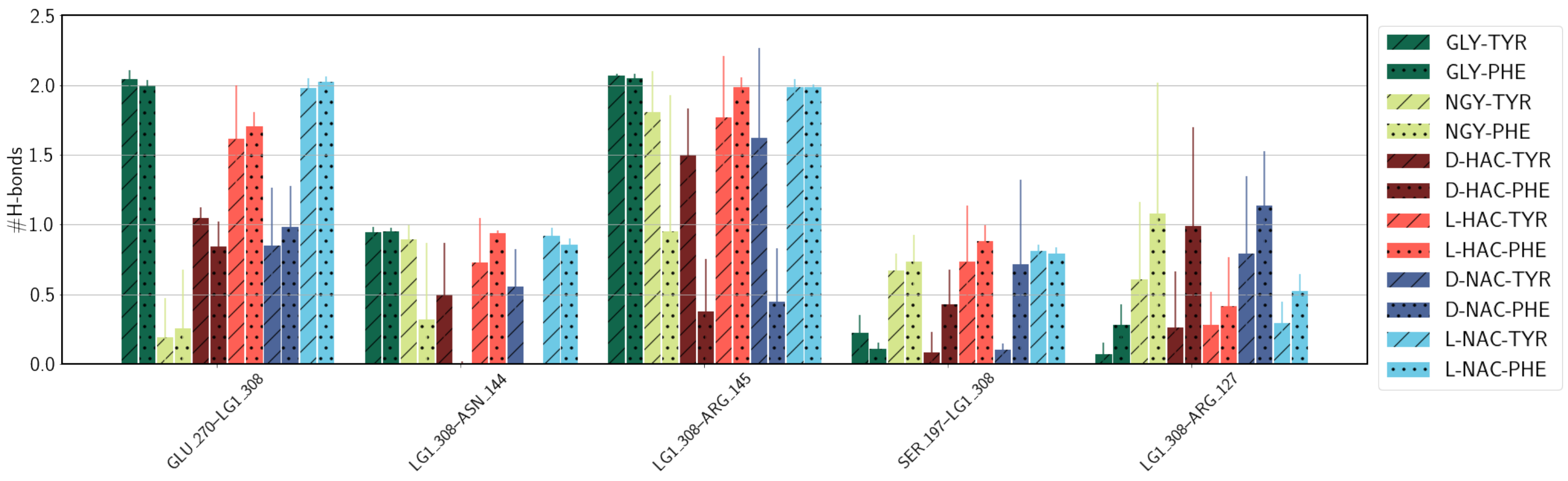
2.4. Hydrogen Bonds
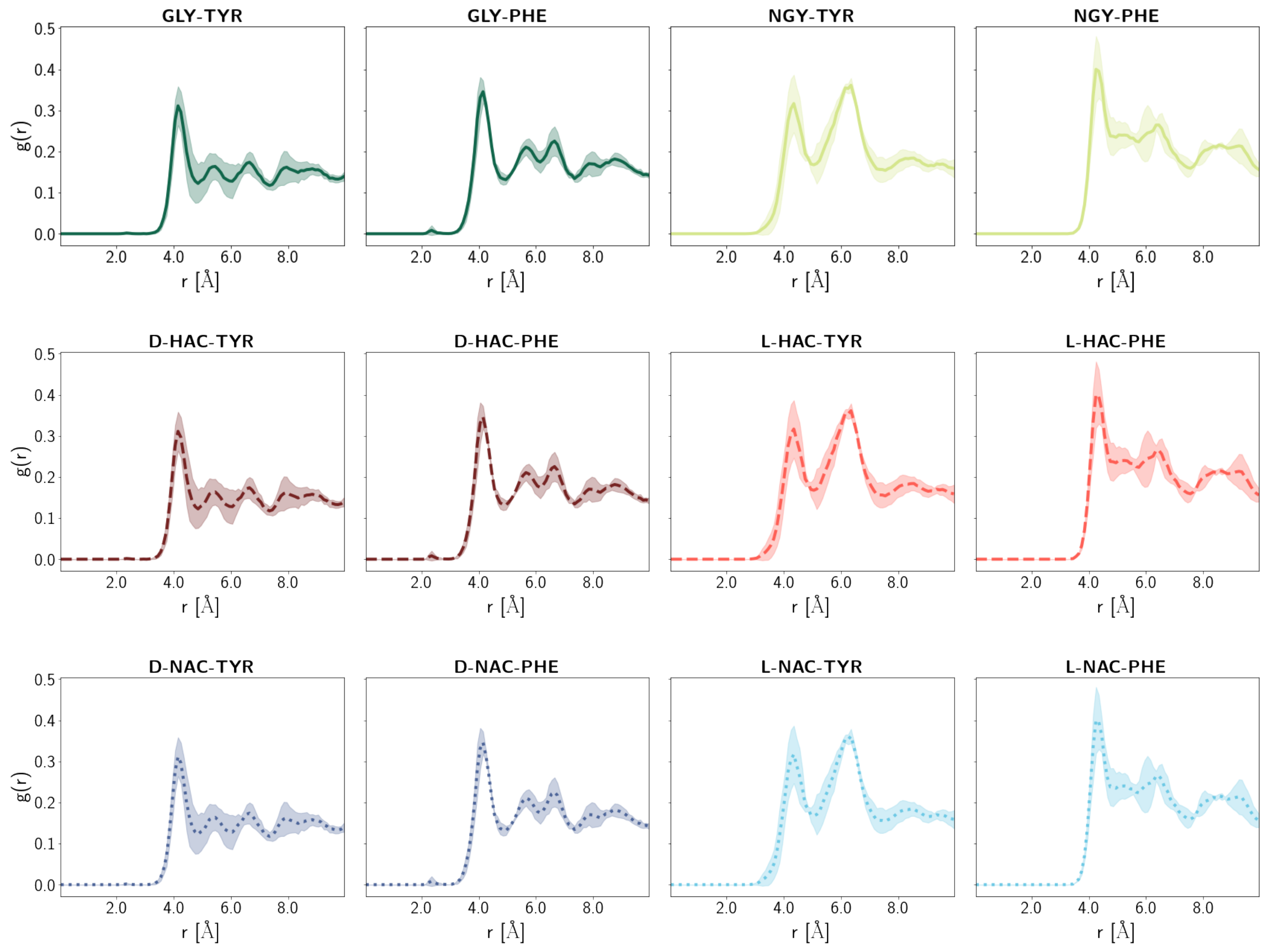
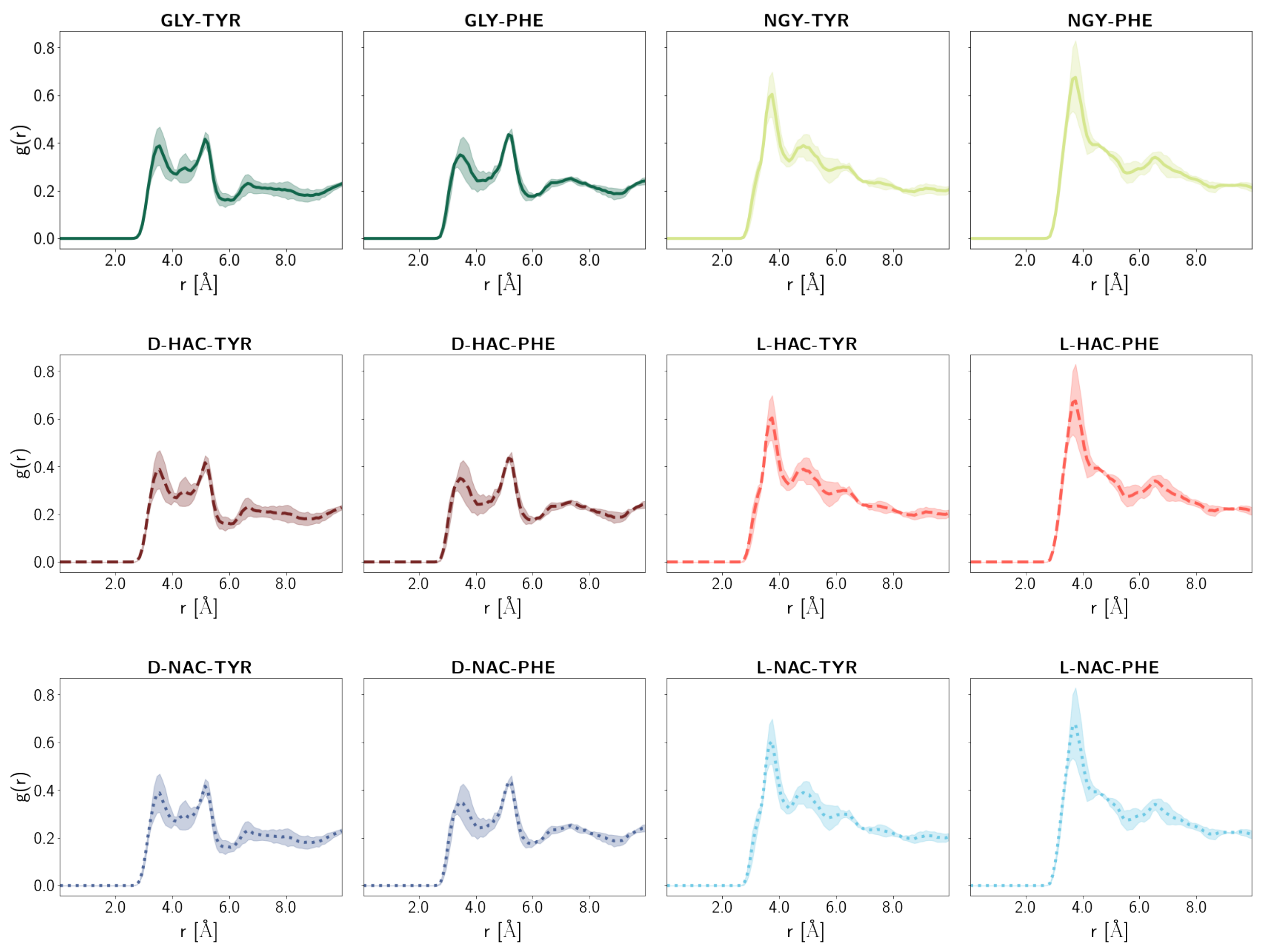
2.5. Water Distribution
3. Discussion
4. Materials and Methods
4.1. Model Setup
4.2. Parameterisation
4.3. Molecular Dynamics Simulations
4.4. Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| CPA | Carboxypeptidase A |
| MD | Molecular Dynamics |
| HAC | (N-hydroxyamino)carbonyl |
| NAC | (amino)carbonyl |
| PDB | Protein Data Bank |
References
- Christianson, D.W.; Lipscomb, W.N. X-ray crystallographic investigation of substrate binding to carboxypeptidase A at subzero temperature. Proc. Natl. Acad. Sci. USA 1986, 83, 7568–7572. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Lipscomb, W.N. Comparison of the structures of three carboxypeptidase A-phosphonate complexes determined by X-ray crystallography. Biochemistry 1991, 30, 8171–8180. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.H.; Kim, D.H.; Chung, S.J.; Ha, N.C.; Oh, B.H.; Choi, K.Y. Insight into the stereochemistry in the inhibition of carboxypeptidase A with N-(hydroxyaminocarbonyl)phenylalanine: Binding modes of an enantiomeric pair of the inhibitor to carboxypeptidase A. Bioorg. Med. Chem. 2002, 10, 2015–2022. [Google Scholar] [CrossRef] [PubMed]
- Szeto, M.W.Y.; Mujika, J.I.; Zurek, J.; Mulholland, A.J.; Harvey, J.N. QM/MM study on the mechanism of peptide hydrolysis by carboxypeptidase A. Theochem 2009, 898, 106–114. [Google Scholar] [CrossRef]
- Xu, D.; Guo, H. Quantum Mechanical/Molecular Mechanical and Density Functional Theory Studies of a Prototypical Zinc Peptidase (Carboxypeptidase A) Suggest a General Acid-General Base Mechanism. J. Am. Chem. Soc. 2009, 131, 9780–9788. [Google Scholar] [CrossRef]
- Wu, S.; Zhang, C.; Xu, D.; Guo, H. Catalysis of Carboxypeptidase A: Promoted-Water versus Nucleophilic Pathways. J. Chem. Phys. B 2010, 114, 9259–9267. [Google Scholar] [CrossRef]
- Christianson, D.W.; Lipscomb, W.N. Carboxypeptidase A. Acc. Chem. Res. 1989, 22, 62–69. [Google Scholar] [CrossRef]
- Breslow, R.; Wernick, D.L. Unified picture of mechanism of catalysis by carboxypeptidase A. Proc. Natl. Acad. Sci. USA 1977, 74, 1303–1307. [Google Scholar] [CrossRef]
- Lee, H.C.; Ko, Y.H.; Baek, S.B.; Kim, D.H. Detection of an Anhydride Intermediate in the Carboxypeptidase A catalyzed Hydrolysis of a Peptide Substrate by solid State NMR Spectroscopy and its Mechanistic implication. Bioorg. Med. Chem. Lett. 1998, 8, 3379–3384. [Google Scholar] [CrossRef]
- Makinen, M.W.; Kuo, L.C.; Dymowski, J.J.; Jaffer, S. Catalytic Role of the Metal Ion of Carboxypeptidase A in Ester Hydrolysis. J. Biol. Chem. 1979, 254, 356–366. [Google Scholar] [CrossRef]
- Lee, K.J.; Kim, D.H. Design of mechanism-based carboxypeptidase A inactivators on the basis of the X-ray crystal structure and catalytic reaction pathway. Bioorg. Med. Chem. 1998, 6, 1613–1622. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.J.; Kim, D.H. N-(Hydroxyaminocarbonyl)phenylalanine: A Novel Class of Inhibitor for Carboxypeptidase A. Bioorg. Med. Chem. 2001, 9, 185–189. [Google Scholar] [CrossRef] [PubMed]
- Bukrinsky, J.T.; Bjerrum, M.J.; Kadziola, A. Native Carboxypeptidase A in a New Crystal Environment Reveals a Different Conformation of the Important Tyrosine 248. Biochemistry 1998, 37, 16555–16564. [Google Scholar] [CrossRef] [PubMed]
- Testero, S.A.; Granados, C.; Fernàndez, D.; Gallego, P.; Covaleda, G.; Reverter, D.; Vendrell, J.; Avilé, F.X.; Pallares, I.; Mobashery, S. Discovery of Mechanism-Based Inactivators for Human Pancreatic Carboxypeptidase A from a Focused Synthetic Library. Med. Chem. Lett. 2017, 8, 1122–1127. [Google Scholar] [CrossRef] [PubMed]
- Breslow, R.; Wernick, D.L. On the mechanism of catalysis by carboxypeptidase A. J. Am. Chem. Soc. 1976, 98, 259–261. [Google Scholar] [CrossRef]
- Adler, M.; Bryant, J.; Buckman, B.; Islam, I.; Larsen, B.; Finster, S.; Kent, L.; May, K.; Mohan, R.; Yuan, S.; et al. Crystal Structures of Potent Thiol-Based Inhibitors Bound to Carboxypeptidase B. Biochemistry 2005, 44, 9339–9347. [Google Scholar] [CrossRef]
- Yoshimoto, N.; Itoh, T.; Inaba, Y.; Ishii, H.; Yamamoto, K. Structural Basis for Inhibition of Carboxypeptidase B by Selenium-Containing Inhibitor: Selenium Coordinates to Zinc in Enzyme. J. Med. Chem. 2013, 56, 7527–7535. [Google Scholar] [CrossRef]
- Adler, M.; Buckman, B.; Bryant, J.; Chang, Z.; Chu, K.; Emayan, K.; Hrvatin, P.; Islam, I.; Morser, J.; Sukovich, D.; et al. Structures of potent selective peptide mimetics bound to carboxypeptidase B. Acta Cryst. 2008, D64, 149–157. [Google Scholar] [CrossRef]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Bernstein, F.C.; Koetzle, T.F.; Williams, G.J.B.; Meyer, E.F., Jr.; Brice, M.D.; Rodgers, J.R.; Kennard, O.; Shimanouchi, T.; Tasumi, M. The protein data bank: A computer-based archival file for macromolecular structures. J. Mol. Biol. 1977, 112, 535–542. [Google Scholar] [CrossRef] [PubMed]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminf. 2012, 4, 17. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Rev. B.01. Gaussian Inc.: Wallingford, CT, USA, 2016; Available online: https://www.gaussian.com (accessed on 2 October 2024).
- Binning, R.C., Jr.; Curtiss, L.A. Compact contracted basis-sets for 3rd-row atoms - Ga-Kr. J. Comput. Chem. 1990, 11, 1206–1216. [Google Scholar] [CrossRef]
- Blaudeau, J.P.; McGrath, M.P.; Curtiss, L.A.; Radom, L. Extension of Gaussian-2 (G2) theory to molecules containing third-row atoms K and Ca. J. Chem. Phys. 1997, 107, 5016–5021. [Google Scholar] [CrossRef]
- Ditchfield, R.; Hehre, W.J.; Pople, J.A. Self-Consistent Molecular-Orbital Methods. IX. An Extended Gaussian-Type Basis for Molecular-Orbital Studies of Organic Molecules. J. Chem. Phys. 1971, 54, 724–728. [Google Scholar] [CrossRef]
- Francl, M.M.; Pietro, W.J.; Hehre, W.J.; Binkley, J.S.; DeFrees, D.J.; Pople, J.A.; Gordon, M.S. Self-Consistent Molecular Orbital Methods. 23. A polarization-type basis set for 2nd-row elements. J. Chem. Phys. 1982, 77, 3654–3665. [Google Scholar] [CrossRef]
- Gordon, M.S. The isomers of silacyclopropane. Chem. Phys. Lett. 1980, 76, 163–168. [Google Scholar] [CrossRef]
- Hariharan, P.C.; Pople, J.A. Influence of polarization functions on molecular-orbital hydrogenation energies. Theor. Chem. Acc. 1973, 28, 213–222. [Google Scholar] [CrossRef]
- Hariharan, P.C.; Pople, J.A. Accuracy of AHn equilibrium geometries by single determinant molecular-orbital theory. Mol. Phys. 1974, 27, 209–214. [Google Scholar] [CrossRef]
- Hehre, W.J.; Ditchfield, R.; Pople, J.A. Self-Consistent Molecular Orbital Methods. XII. Further Extensions of Gaussian-Type Basis Sets for Use in Molecular Orbital Studies of Organic Molecules. J. Chem. Phys. 1972, 56, 2257–2261. [Google Scholar] [CrossRef]
- Rassolov, V.A.; Pople, J.A.; Ratner, M.A.; Windus, T.L. 6-31G* basis set for atoms K through Zn. J. Chem. Phys. 1998, 109, 1223–1229. [Google Scholar] [CrossRef]
- Rassolov, V.A.; Ratner, M.A.; Pople, J.A.; Redfern, P.C.; Curtiss, L.A. 6-31G* Basis Set for Third-Row Atoms. J. Comput. Chem. 2001, 22, 976–984. [Google Scholar] [CrossRef]
- Bayly, C.I.; Cieplak, P.; Cornell, W.; Kollman, P.A. A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: The RESP model. J. Phys. Chem. 1993, 97, 10269–10280. [Google Scholar] [CrossRef]
- Cieplak, P.; Cornell, W.D.; Bayly, C.; Kollman, P.A. Application of the multimolecule and multiconformational RESP methodology to biopolymers: Charge derivation for DNA, RNA, and proteins. J. Comput. Chem. 1995, 16, 1357–1377. [Google Scholar] [CrossRef]
- Dupradeau, F.Y.; Pigache, A.; Zaffran, T.; Savineau, C.; Lelong, R.; Grivel, N.; Lelong, D.; Rosanski, W.; Cieplak, P. The R.E.D. tools: Advances in RESP and ESP charge derivation and force field library building. Phys. Chem. Chem. Phys. 2010, 12, 7821–7839. [Google Scholar] [CrossRef]
- Vanquelef, E.; Simon, S.; Marquant, G.; Garcia, E.; Klimerak, G.; Delepine, J.C.; Cieplak, P.; Dupradeau, F.Y. R.E.D. Server: A web service for deriving RESP and ESP charges and building force field libraries for new molecules and molecular fragments. Nucleic Acids Res. 2011, 39, W511–W517. [Google Scholar] [CrossRef]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and Testing of a General Amber Force Field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Wang, J.; Wang, W.; Kollman, P.A.; Case, D.A. Automatic atom type and bond type perception in molecular mechanical calculations. J. Mol. Graph. Model. 2006, 25, 247–260. [Google Scholar] [CrossRef]
- Li, P.; Merz, K.M.J. MCPB.py: A Python Based Metal Center Parameter Builder. J. Chem. Inf. Model. 2016, 56, 599–604. [Google Scholar] [CrossRef]
- Li, P.; Merz, K.M. Building Bonded Model for A Ligand Binding Metalloprotein with MCPB.py. 2015. Available online: https://ambermd.org/tutorials/advanced/tutorial20/mcpbpy.php (accessed on 2 October 2024).
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Peters, M.B.; Yang, Y.; Wang, B.; Füsti-Molnár, L.; Weaver, M.N.; Merz, K.M. Structural Survey of Zinc Containing Proteins and the Development of the Zinc AMBER Force Field (ZAFF). J. Chem. Theory Comput. 2010, 6, 2935–2947. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Joung, I.S.; Cheatham, T.E., III. Determination of Alkali and Halide Monovalent Ion Parameters for Use in Explicitly Solvated Biomolecular Simulations. J. Phys. Chem. B 2008, 112, 9020–9041. [Google Scholar] [CrossRef]
- Yu, Z.; Li, P.; Merz, K.M.J. Extended Zinc AMBER Force Field (EZAFF). J. Chem. Theory Comput. 2018, 14, 242–254. [Google Scholar] [CrossRef]
- Volkenandt, S.; Imhof, P. Comparison of Empirical Zn2+ Models in Protein–DNA Complexes. Biophysica 2023, 3, 214–230. [Google Scholar] [CrossRef]
- Ryckaert, J.P.; Ciccotti, G.; Berendsen, H.J.C. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef]
- Roe, D.R.; Cheatham, T.E., III. PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef]
- Van Rossum, G.; Drake, F.L. Python 3 Reference Manual; CreateSpace: Scotts Valley, CA, USA, 2009. [Google Scholar]
- Harris, C.R.; Millman, K.J.; van der Walt, S.J.; Gommers, R.; Virtanen, P.; Cournapeau, D.; Wieser, E.; Taylor, J.; Berg, S.; Smith, N.J.; et al. Array programming with NumPy. Nature 2020, 585, 357–362. [Google Scholar] [CrossRef]
- Hunter, J.D. Matplotlib: A 2D Graphics Environment. Comput. Sci. Eng. 2007, 9, 90–95. [Google Scholar] [CrossRef]
- Hassan, I.; Ferraro, F.; Imhof, P. Effect of the Hydration Shell on the Carbonyl Vibration in the Ala-Leu-Ala-Leu Peptide. Molecules 2021, 26, 2148. [Google Scholar] [CrossRef] [PubMed]
- Andreini, C.; Cavallaro, G.; Lorenzini, S. FindGeo: A tool for determining metal coordination geometry. Bioinformatics 2012, 28, 1658–1660. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD—Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
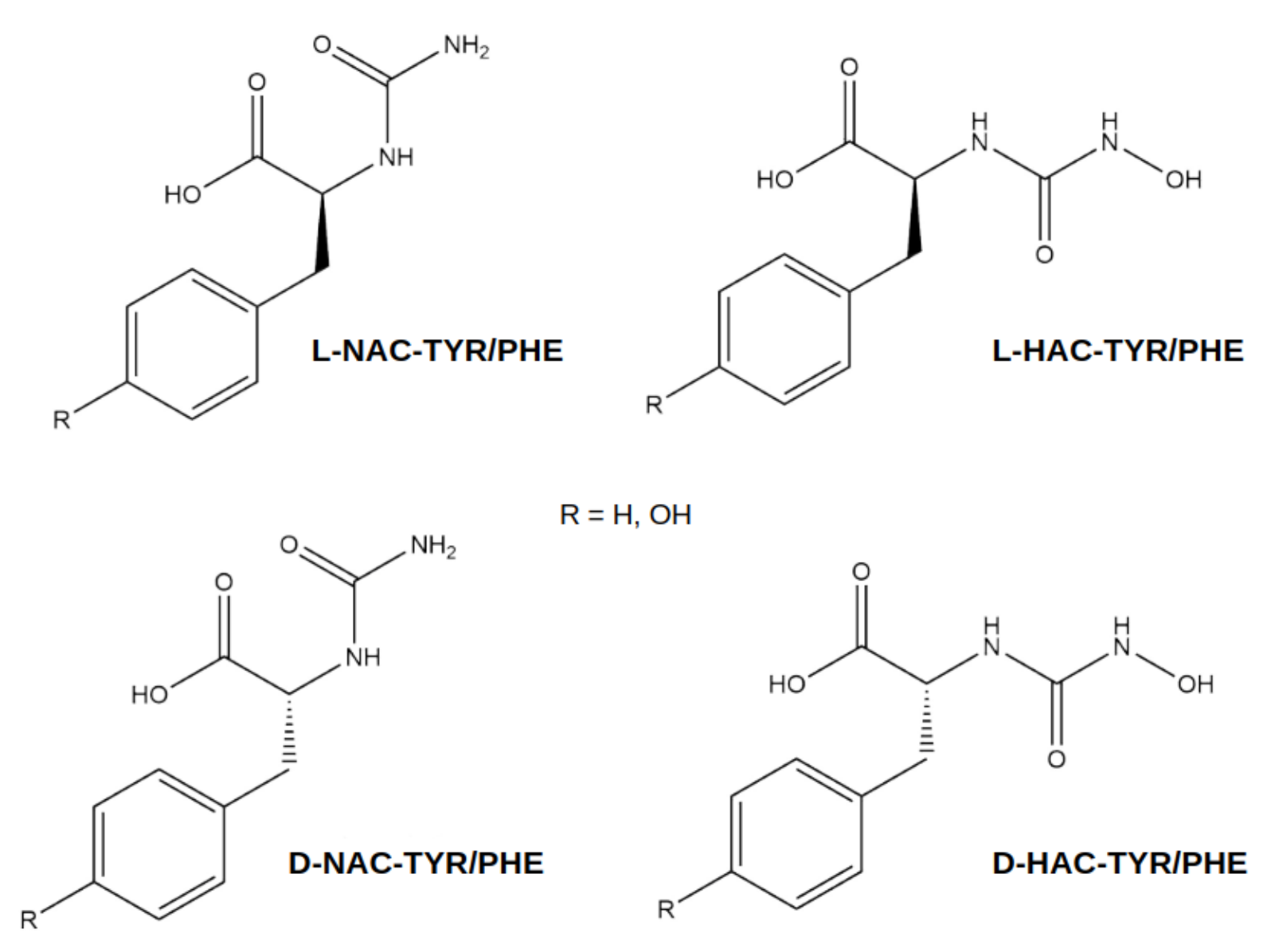
| N-Terminal Part | Ligand | Ligand | PDB |
|---|---|---|---|
| Charged glycine | GLY-TYR | GLY-PHE | 3CPA |
| Neutral glycine | NGY-TYR | NGY-PHE | 3CPA |
| (N-hydroxyamino)carbonyl | D-HAC-TYR | D-HAC-PHE | 1HDQ |
| (N-hydroxyamino)carbonyl | L-HAC-TYR | L-HAC-PHE | 1HEE |
| (amino)carbonyl | D-NAC-TYR | D-NAC-PHE | 1HDU |
| (amino)carbonyl | L-NAC-TYR | L-NAC-PHE | 1HDU |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Amador Balderas, J.A.; Beierlein, F.; Horn, A.H.C.; Volkenandt, S.; Völcker, L.; Mokhtari, N.; Ndongue, J.C.E.; Imhof, P. Mode of Metal Ligation Governs Inhibition of Carboxypeptidase A. Int. J. Mol. Sci. 2024, 25, 13725. https://doi.org/10.3390/ijms252413725
Amador Balderas JA, Beierlein F, Horn AHC, Volkenandt S, Völcker L, Mokhtari N, Ndongue JCE, Imhof P. Mode of Metal Ligation Governs Inhibition of Carboxypeptidase A. International Journal of Molecular Sciences. 2024; 25(24):13725. https://doi.org/10.3390/ijms252413725
Chicago/Turabian StyleAmador Balderas, Jorge Antonio, Frank Beierlein, Anselm H. C. Horn, Senta Volkenandt, Leon Völcker, Nikoo Mokhtari, Jules Cesar Epee Ndongue, and Petra Imhof. 2024. "Mode of Metal Ligation Governs Inhibition of Carboxypeptidase A" International Journal of Molecular Sciences 25, no. 24: 13725. https://doi.org/10.3390/ijms252413725
APA StyleAmador Balderas, J. A., Beierlein, F., Horn, A. H. C., Volkenandt, S., Völcker, L., Mokhtari, N., Ndongue, J. C. E., & Imhof, P. (2024). Mode of Metal Ligation Governs Inhibition of Carboxypeptidase A. International Journal of Molecular Sciences, 25(24), 13725. https://doi.org/10.3390/ijms252413725