
Integrated Analysis of Transcriptome and Metabolome in the Brain After Cold Stress of Red Tilapia During Overwintering
 , and
, and
Abstract
1. Introduction
2. Results
2.1. Fish Serum Physiological Parameters
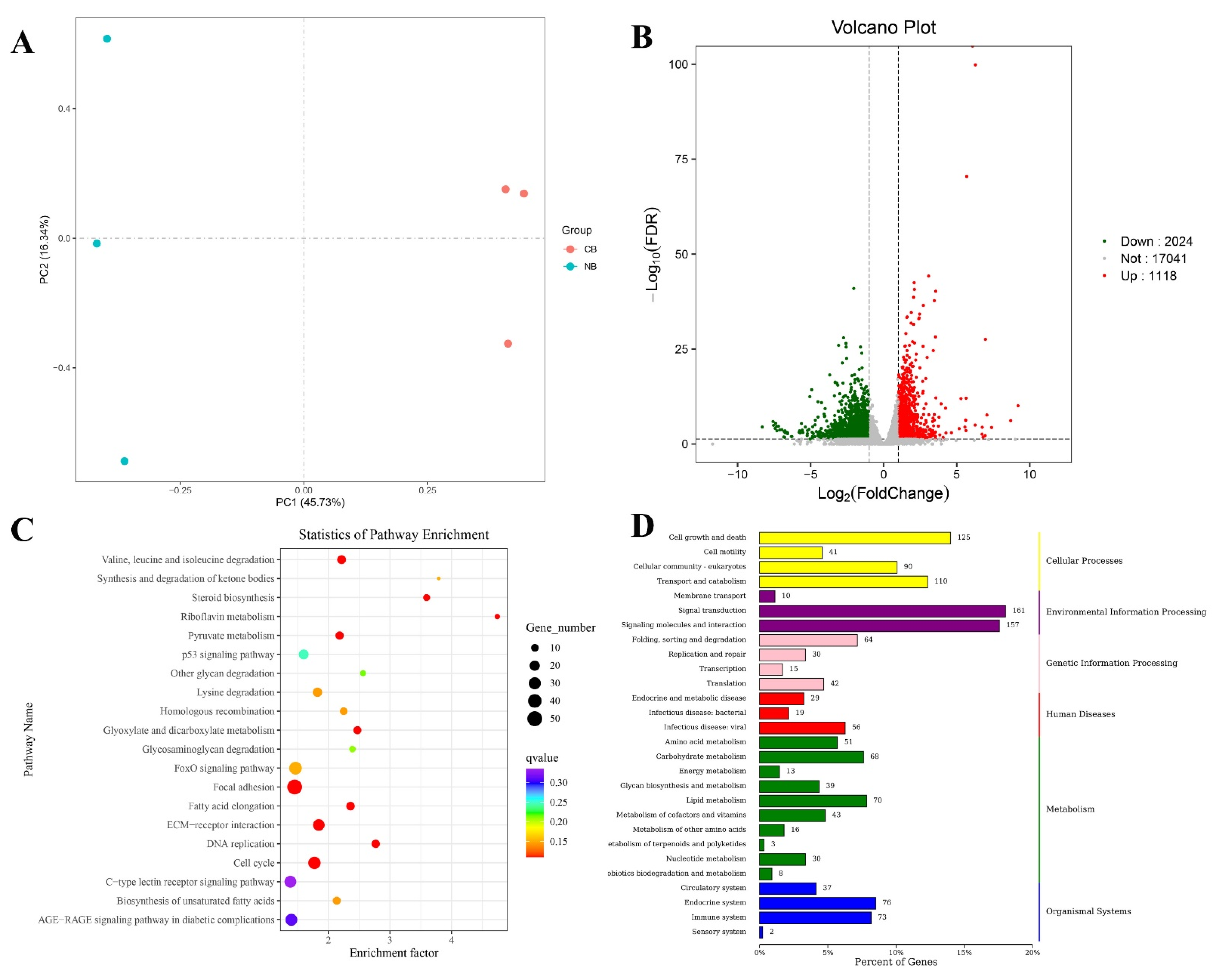
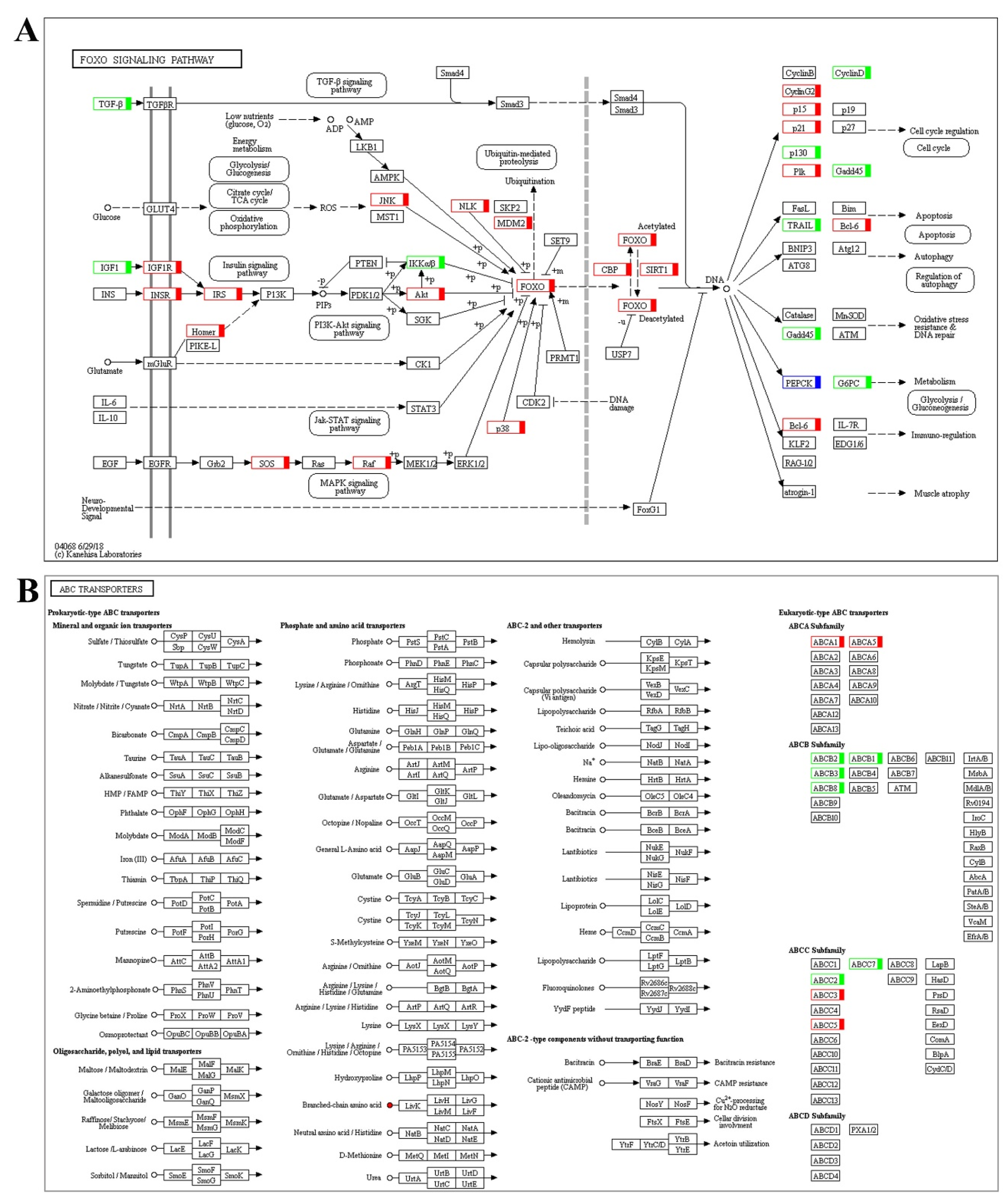
2.2. Differentially Expressed Genes (DEGs) Identification and Functional Enrichment
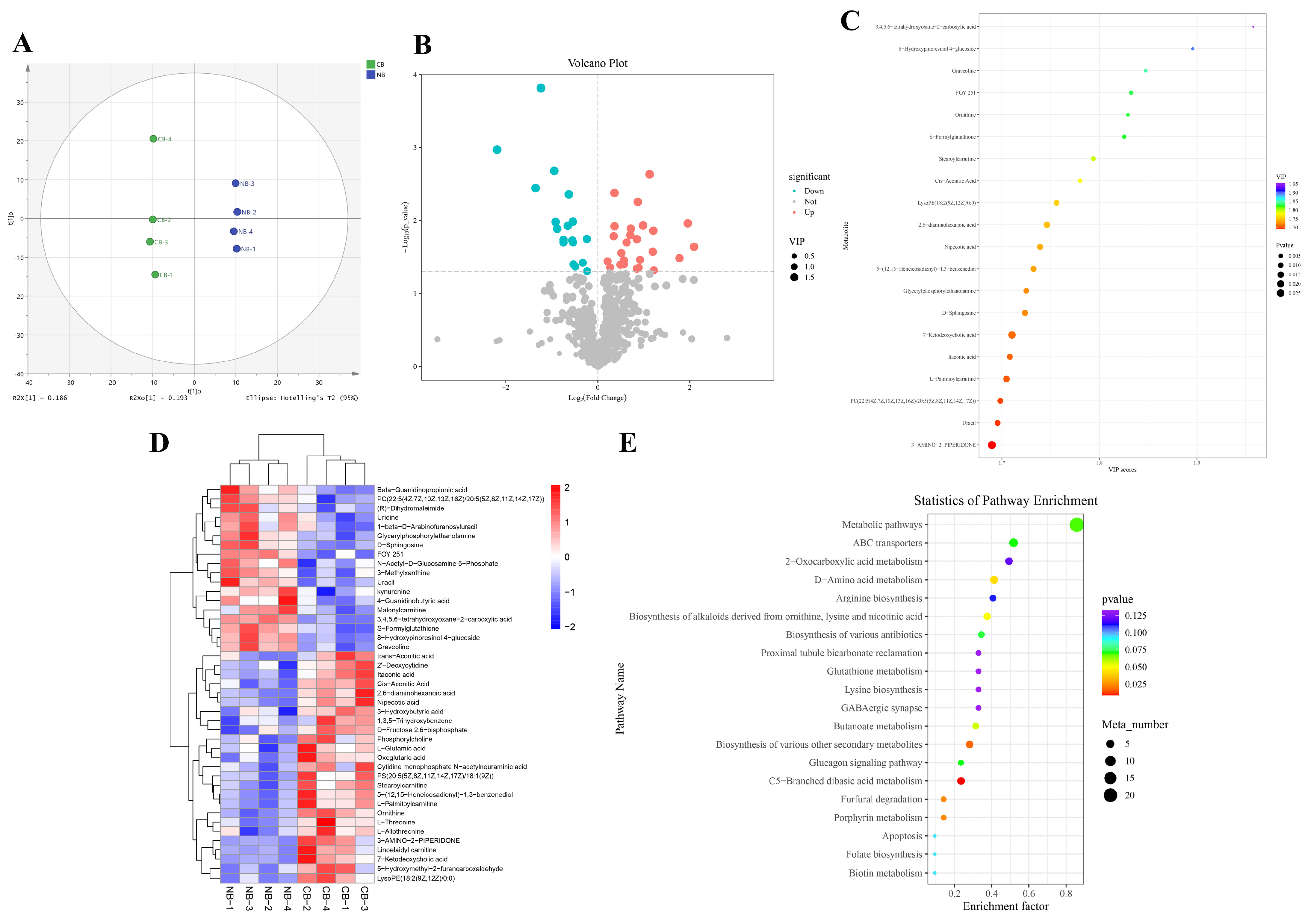
2.3. Differential Metabolites (DMs) Identification and Functional Enrichment
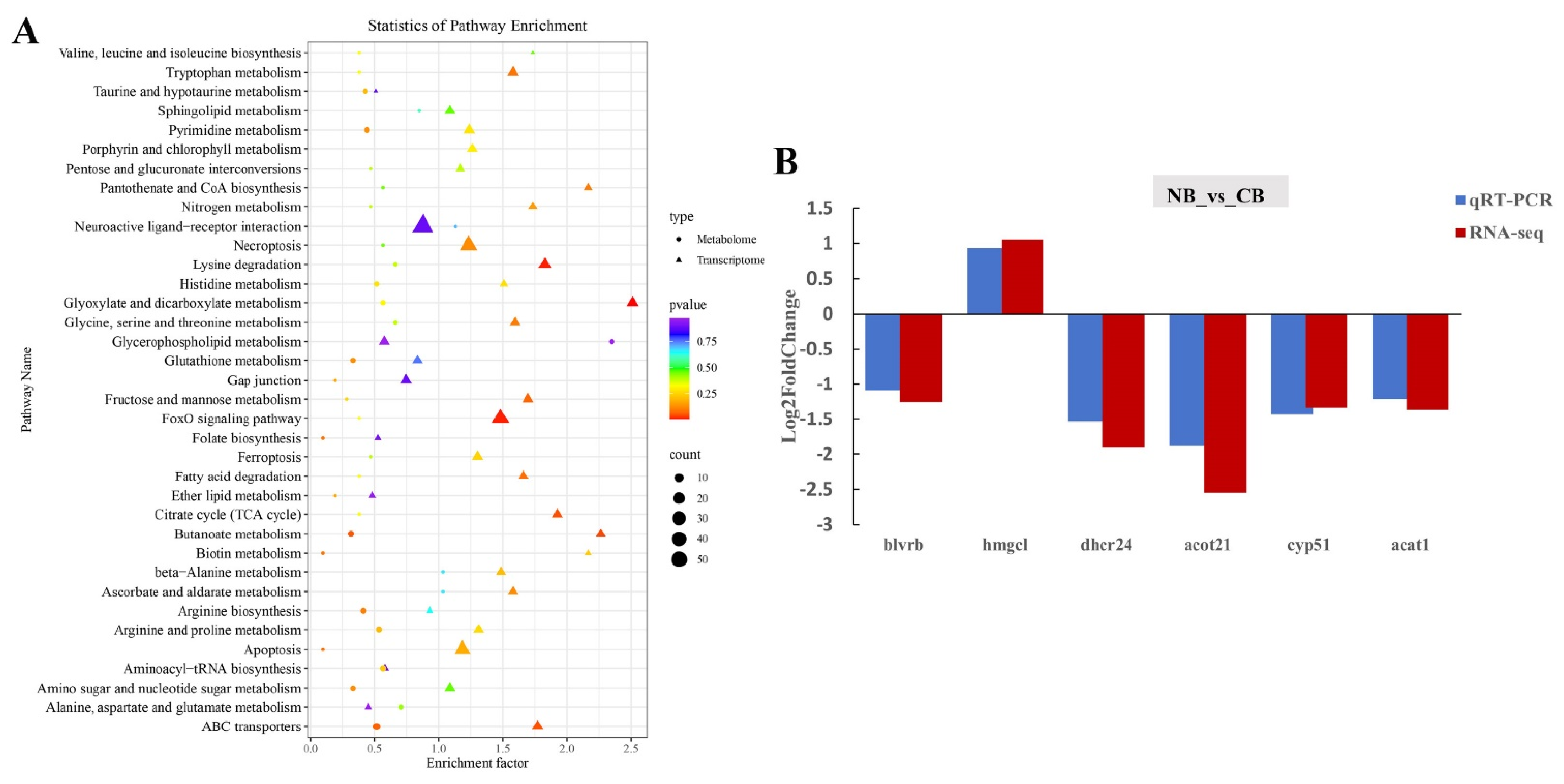
2.4. DEGs Validation and Co-Analysis of DEGs and DMs
3. Discussion
4. Materials and Methods
4.1. Ethics Statement
4.2. Fish and Experiment
4.3. Transcriptome and Metabolome Analysis
4.4. qRT-PCR Validation of DEGs
4.5. Conjoint Analysis of Transcriptome and Metabolome Data
4.6. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Abdullah, S.; Omar, N.; Yusoff, S.M.; Obukwho, E.B.; Nwunuji, T.P.; Hanan, L.; Samad, J. Clinicopathological features and immunohistochemical detection of antigens in acute experimental Streptococcus agalactiae infection in red tilapia (Oreochromis spp.). SpringerPlus 2013, 2, 286. [Google Scholar] [CrossRef]
- Zhu, W.; Wang, L.; Dong, Z.; Chen, X.; Song, F.; Liu, N.; Yang, H.; Fu, J. Comparative transcriptome analysis identifies candidate genes related to skin color differentiation in red tilapia. Sci. Rep. 2016, 6, 31347. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.M.; Zhu, W.B.; Fu, J.J.; Luo, M.K.; Dong, Z.J. Identification of differentially expressed miRNA involved in skin color variation in red tilapia during overwintering. J. Fish. China 2023, 47, 049602, (In Chinese with English abstract). [Google Scholar]
- Azaza, M.S.; Dhraïef, M.N.; Kraïem, M.M. Effects of water temperature on growth and sex ratio of juvenile Nile tilapia Oreochromis niloticus (Linnaeus) reared in geothermal waters in southern Tunisia. J. Therm. Biol. 2008, 33, 98–105. [Google Scholar] [CrossRef]
- Ma, X.Y.; Qiang, J.; He, J.; Gabriel, N.N.; Xu, P. Changes in the physiological parameters, fatty acid metabolism, and SCD activity and expression in juvenile GIFT tilapia (Oreochromis niloticus) reared at three different temperatures. Fish Physiol. Biochem. 2015, 41, 937–950. [Google Scholar] [CrossRef] [PubMed]
- Charo-Karisa, H.; Rezk, M.A.; Bovenhuis, H.; Komen, H. Heritability of cold tolerance in Nile tilapia, Oreochromis niloticus, juveniles. Aquaculture 2005, 249, 115–123. [Google Scholar] [CrossRef]
- Zhu, W.B.; Liu, H.L.; Chen, Z.Z.; Liang, Z.Y.; Dong, Z.J. Effects of cooling temperature stress on serum biochemical indexes in Malaysian red tilapia (Oreochromis mossambicus × O. niloticus). Chin. J. Fish. 2013, 26, 16–20. [Google Scholar]
- Jeffries, K.M.; Connon, R.E.; Davis, B.E.; Komoroske, L.M.; Britton, M.T.; Sommer, T.; Todgham, A.E.; Fangue, N.A. Effects of high temperatures on threatened estuarine fishes during periods of extreme drought. J. Exp. Biol. 2016, 219, 1705–1716. [Google Scholar] [CrossRef] [PubMed]
- Terrazas, M.M.; Adams, J.R.; Sudheesh, P.S.; Cain, K.D. Effects of diel temperature fluctuation on growth, stress response, and immune function of burbot. Trans. Am. Fish. Soc. 2017, 146, 996–1007. [Google Scholar] [CrossRef]
- El-Sayed, A.F.M.; Khaled, A.A.; Hamdan, A.M.; Makled, S.O.; Hafez, E.E.; Saleh, A.A. The role of antifreeze genes in the tolerance of cold stress in the Nile tilapia (Oreochromis niloticus). BMC Genom. 2023, 24, 476. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Tan, X.; Liu, Q.; Ye, H.; Zou, C.; Xu, M.; Zhang, Y.; Ye, C. Physiological, immune responses and liver lipid metabolism of orange-spotted grouper (Epinephelus coioides) under cold stress. Aquaculture 2019, 498, 545–555. [Google Scholar] [CrossRef]
- Velmurugan, B.K.; Chan, C.R.; Weng, C.F. Innate-immune responses of tilapia (Oreochromis mossambicus) exposure to acute cold stress. J. Cell. Physiol. 2019, 234, 16125–16135. [Google Scholar] [CrossRef]
- Dellagostin, E.N.; Martins, A.W.; Blödorn, E.B.; Silveira, T.L.R.; Komninou, E.R.; Junior, A.S.V.; Corcini, C.D.; Nunes, L.S.; Remião, M.H.; Collares, G.L. Chronic cold exposure modulates genes related to feeding and immune system in Nile tilapia (Oreochromis niloticus). Fish Shellfish. Immunol. 2022, 128, 269–278. [Google Scholar] [CrossRef]
- Cheng, C.H.; Ye, C.X.; Guo, Z.X.; Wang, A.L. Immune and physiological responses of pufferfish (Takifugu obscurus) under cold stress. Fish Shellfish. Immunol. 2017, 64, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Shahjahan, M.; Islam, M.J.; Hossain, M.T.; Mishu, M.A.; Hasan, J.; Brown, C. Blood biomarkers as diagnostic tools: An overview of climate-driven stress. Sci. Total Environ. 2022, 843, 156910. [Google Scholar] [CrossRef]
- Luo, M.; Zhu, W.; Liang, Z.; Feng, B.; Xie, X.; Li, Y.; Liu, Y.; Shi, X.; Fu, J.; Miao, L.; et al. High-temperature stress response: Insights into the molecular regulation of American shad (Alosa sapidissima) using a multi-omics approach. Sci. Total Environ. 2024, 916, 170329. [Google Scholar] [CrossRef]
- Liu, L.; Zhang, R.; Wang, X.; Zhu, H.; Tian, Z. Transcriptome analysis reveals molecular mechanisms responsive to acute cold stress in the tropical stenothermal fish tiger barb (Puntius tetrazona). BMC Genom. 2020, 21, 1–14. [Google Scholar] [CrossRef]
- Qian, B.; Xue, L. Liver transcriptome sequencing and de novo annotation of the large yellow croaker (Larimichthy crocea) under heat and cold stress. Mar. Genom. 2016, 25, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Hu, J.; Zhu, J.; Wang, Y.; Zhang, Y.; Li, Y.; Xu, S.; Yan, X.; Zhang, D. Transcriptome, antioxidant enzymes and histological analysis reveal molecular mechanisms responsive to long-term cold stress in silver pomfret (Pampus argenteus). Fish Shellfish. Immunol. 2022, 121, 351–361. [Google Scholar] [CrossRef]
- Wen, B.; Jin, S.R.; Chen, Z.Z.; Gao, J.Z. Physiological responses to cold stress in the gills of discus fish (Symphysodon aequifasciatus) revealed by conventional biochemical assays and GC-TOF-MS metabolomics. Sci. Total Environ. 2018, 640, 1372–1381. [Google Scholar] [CrossRef]
- Jiao, S.; Nie, M.; Song, H.; Xu, D.; You, F. Physiological responses to cold and starvation stresses in the liver of yellow drum (Nibea albiflora) revealed by LC-MS metabolomics. Sci. Total Environ. 2020, 715, 136940. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Gao, J.; Cao, X.; Du, J.; Cao, L.; Nie, Z.; Xu, G.; Dong, Z. Integrated analysis of transcriptomics and metabolomics unveil the novel insight of one-year-old precocious mechanism in the chinese mitten crab, Eriocheir sinensis. Int. J. Mol. Sci. 2023, 24, 11171. [Google Scholar] [CrossRef]
- Wang, J.; Hou, X.; Chen, X.; Zhang, K.; Wang, J.; Wang, C. Comprehensive analysis of metabolomics and transcriptomics provides insights into growth difference of juvenile Eriocheir sinensis during the molting cycle. Aquaculture 2021, 539, 736661. [Google Scholar] [CrossRef]
- Donaldson, M.; Cooke, S.; Patterson, D.; Macdonald, J. Cold shock and fish. J. Fish Biol. 2008, 73, 1491–1530. [Google Scholar] [CrossRef]
- Seibel, H.; Baßmann, B.; Rebl, A. Blood will tell: What hematological analyses can reveal about fish welfare. Front. Vet. Sci. 2021, 8, 616955. [Google Scholar] [CrossRef]
- Atli, G.; Ariyurek, S.Y.; Kanak, E.G.; Canli, M. Alterations in the serum biomarkers belonging to different metabolic systems of fish (Oreochromis niloticus) after Cd and Pb exposures. Environ. Toxicol. Phar. 2015, 40, 508–515. [Google Scholar] [CrossRef]
- Latif, M.; Faheem, M.; Asmatullah; Hoseinifar, S.H.; Van Doan, H. Dietary black seed effects on growth performance, proximate composition, antioxidant and histo-biochemical parameters of a culturable fish, rohu (Labeo rohita). Animals 2020, 11, 48. [Google Scholar] [CrossRef]
- Li, D.S.; Huang, Q.F.; Guan, L.H.; Zhang, H.Z.; Li, X.; Fu, K.L.; Chen, Y.-X.; Wan, J.-B.; Huang, M.; Bi, C.-H. Targeted bile acids and gut microbiome profiles reveal the hepato-protective effect of WZ tablet (Schisandra sphenanthera extract) against LCA-induced cholestasis. Chin. J. Nat. Med. 2020, 18, 211–218. [Google Scholar] [CrossRef]
- Melis, R.; Sanna, R.; Braca, A.; Bonaglini, E.; Cappuccinelli, R.; Slawski, H.; Roggio, T.; Uzzau, S.; Anedda, R. Molecular details on gilthead sea bream (Sparus aurata) sensitivity to low water temperatures from 1H NMR metabolomics. Comp. Biochem. Physiol. Part A Mol. Integr. Physiol. 2017, 204, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Zhao, H.; Wang, G.; Sun, Y.; Wang, L. Energy consumption and intestinal microbiome disorders of yellow catfish (Pelteobagrus fulvidraco) under cold stress. Front. Physiol. 2022, 13, 985046. [Google Scholar] [CrossRef]
- Chang, C.C.; Tsai, W.L.; Jiang, J.R.; Cheng, W. The acute modulation of norepinephrine on immune responses and genes expressions via adrenergic receptors in the giant freshwater prawn, Macrobrachium rosenbergii. Fish Shellfish. Immunol. 2015, 46, 459–467. [Google Scholar] [CrossRef]
- Accili, D.; Arden, K.C. FoxOs at the crossroads of cellular metabolism, differentiation, and transformation. Cell 2004, 117, 421–426. [Google Scholar] [CrossRef] [PubMed]
- Eijkelenboom, A.; Burgering, B.M. FOXOs: Signalling integrators for homeostasis maintenance. Nat. Rev. Mol. Cell Biol. 2013, 14, 83–97. [Google Scholar] [CrossRef]
- Liu, R.; Liu, R.; Song, G.; Li, Q.; Cui, Z.; Long, Y. Mitochondria dysfunction and cell apoptosis limit resistance of nile tilapia (Oreochromis niloticus) to lethal cold stress. Animals 2022, 12, 2382. [Google Scholar] [CrossRef]
- Hu, P.; Liu, M.; Liu, Y.; Wang, J.; Zhang, D.; Niu, H.; Jiang, S.; Wang, J.; Zhang, D.; Han, B. Transcriptome comparison reveals a genetic network regulating the lower temperature limit in fish. Sci. Rep. 2016, 6, 28952. [Google Scholar] [CrossRef]
- Xu, D.; Wang, Z.; Xia, Y.; Shao, F.; Xia, W.; Wei, Y.; Li, X.; Qian, X.; Lee, J.-H.; Du, L. The gluconeogenic enzyme PCK1 phosphorylates INSIG1/2 for lipogenesis. Nature 2020, 580, 530–535. [Google Scholar] [CrossRef] [PubMed]
- Bieczynski, F.; Painefilú, J.C.; Venturino, A.; Luquet, C.M. Expression and function of ABC proteins in fish intestine. Front. Physiol. 2021, 12, 791834. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Shen, M.; Xu, G.; Yu, H.; Jia, C.; Zhu, F.; Meng, Q.; Xu, D.; Du, S.; Zhang, D. Comprehensive analysis of histophysiology, transcriptome and metabolome tolerance mechanisms in black porgy (Acanthopagrus schlegelii) under low temperature stress. Sci. Total Environ. 2024, 927, 172318. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.M.; Yang, H.R.; Munyaradzia, H.B.; Zhu, W.B.; Dong, Z.J. The mRNA and protein datasets after cold stress of red tilapia. Sci. Data 2024, 11, 1177. [Google Scholar] [CrossRef]
- Wang, L.; Song, F.; Zhu, W.; Fu, J.; Dong, Z.; Xu, P. The stage-specific long non-coding RNAs and mRNAs identification and analysis during early development of common carp, Cyprinus carpio. Genomics 2021, 113, 20–28. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Items | Cold | Control | Significance |
|---|---|---|---|
| AST 1 | 648.43 ± 18.88 | 554.13 ± 53.69 | |
| ALP | 14.60 ± 0.12 | 27.45 ± 0.66 | ** |
| T-BIL | 6.63 ± 0.63 | 11.83 ± 0.64 | ** |
| TP | 34.64 ± 1.94 | 32.56 ± 0.54 | |
| ALB | 14.02 ± 0.88 | 13.36 ± 0.23 | |
| UREA | 0.96 ± 0.08 | 0.59 ± 0.08 | * |
| TC | 4.54 ± 0.05 | 5.34 ± 0.30 | * |
| TG | 1.95 ± 0.09 | 1.68 ± 0.16 | |
| CREA | 13.93 ± 1.38 | 16.93 ± 0.82 | |
| GLU | 10.47 ± 0.22 | 8.23 ± 0.41 | ** |
| T4 | 13.07 ± 0.38 | 13.27 ± 0.61 | |
| Cortisol | 94.75 ± 1.86 | 94.45 ± 2.95 | |
| EPI | 9.47 ± 0.52 | 8.61 ± 0.26 | |
| NE | 74.33 ± 1.15 | 64.95 ± 4.10 | * |
| CA | 98.65 ± 5.35 | 92.08 ± 5.57 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, C.; Yang, H.; Zhu, W.; Jiang, Q.; Dong, Z.; Wang, L. Integrated Analysis of Transcriptome and Metabolome in the Brain After Cold Stress of Red Tilapia During Overwintering. Int. J. Mol. Sci. 2024, 25, 13372. https://doi.org/10.3390/ijms252413372
Zhu C, Yang H, Zhu W, Jiang Q, Dong Z, Wang L. Integrated Analysis of Transcriptome and Metabolome in the Brain After Cold Stress of Red Tilapia During Overwintering. International Journal of Molecular Sciences. 2024; 25(24):13372. https://doi.org/10.3390/ijms252413372
Chicago/Turabian StyleZhu, Chenxi, Haoran Yang, Wenbin Zhu, Qichen Jiang, Zaijie Dong, and Lanmei Wang. 2024. "Integrated Analysis of Transcriptome and Metabolome in the Brain After Cold Stress of Red Tilapia During Overwintering" International Journal of Molecular Sciences 25, no. 24: 13372. https://doi.org/10.3390/ijms252413372
APA StyleZhu, C., Yang, H., Zhu, W., Jiang, Q., Dong, Z., & Wang, L. (2024). Integrated Analysis of Transcriptome and Metabolome in the Brain After Cold Stress of Red Tilapia During Overwintering. International Journal of Molecular Sciences, 25(24), 13372. https://doi.org/10.3390/ijms252413372