

Variations in BDNF and Their Role in the Neurotrophic Antidepressant Mechanisms of Ketamine and Esketamine: A Review


Abstract
1. Introduction
2. BDNF: Functions, Distribution, and Implications
3. BDNF, Depression, and Antidepressants
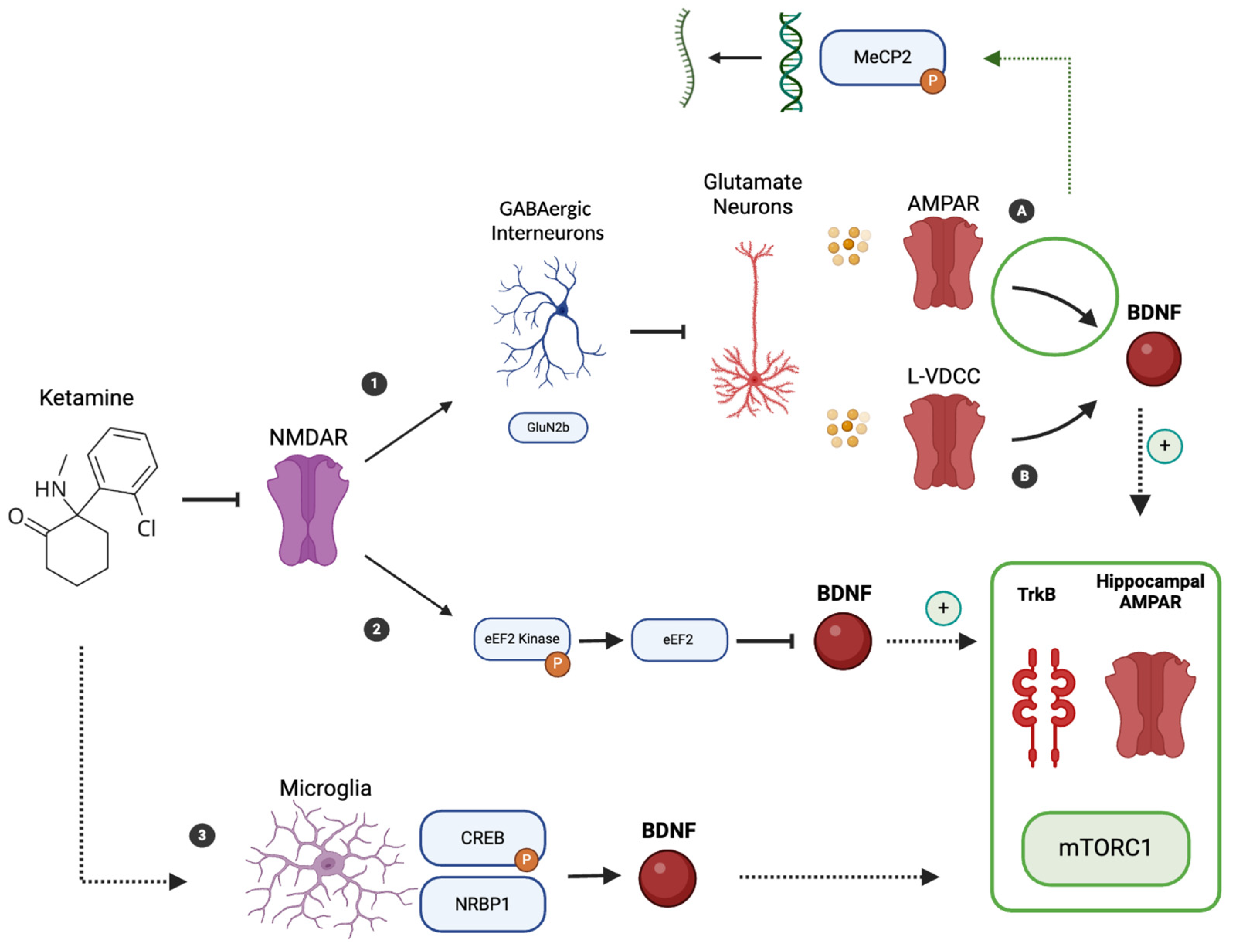
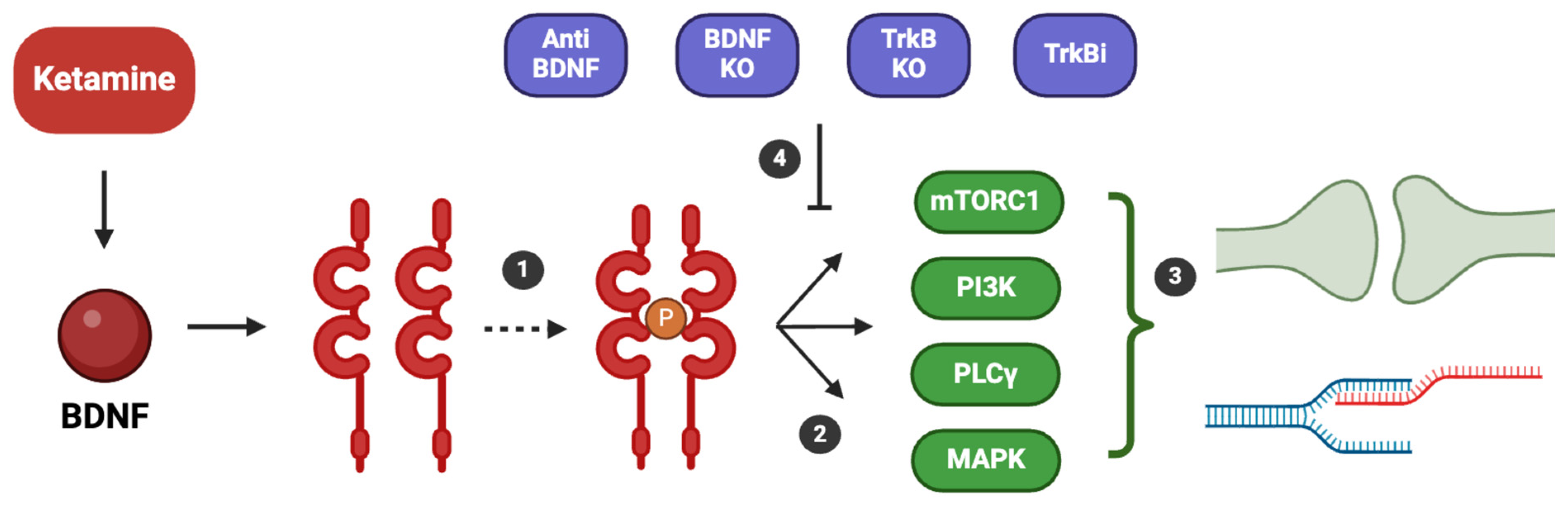
4. Ketamine Mechanism of Action and BDNF Involvement
5. Evidence for the Effects of Ketamine and Esketamine on BDNF Levels in Humans
5.1. Ketamine
5.2. Esketamine
6. Discussion
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Hasler, G. Pathophysiology of Depression: Do We Have Any Solid Evidence of Interest to Clinicians? World Psychiatry 2010, 9, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Duman, R.S.; Deyama, S.; Fogaça, M.V. Role of BDNF in the Pathophysiology and Treatment of Depression: Activity-Dependent Effects Distinguish Rapid-Acting Antidepressants. Eur. J. Neurosci. 2021, 53, 126–139. [Google Scholar] [CrossRef] [PubMed]
- Cavaleri, D.; Moretti, F.; Bartoccetti, A.; Mauro, S.; Crocamo, C.; Carrà, G.; Bartoli, F. The Role of BDNF in Major Depressive Disorder, Related Clinical Features, and Antidepressant Treatment: Insight from Meta-Analyses. Neurosci. Biobehav. Rev. 2023, 149, 105159. [Google Scholar] [CrossRef] [PubMed]
- Miyanishi, H.; Nitta, A. A Role of BDNF in the Depression Pathogenesis and a Potential Target as Antidepressant: The Modulator of Stress Sensitivity “Shati/Nat8l-BDNF System” in the Dorsal Striatum. Pharmaceuticals 2021, 14, 889. [Google Scholar] [CrossRef]
- Yu, H.; Chen, Z. The Role of BDNF in Depression on the Basis of Its Location in the Neural Circuitry. Acta Pharmacol. Sin. 2011, 32, 3–11. [Google Scholar] [CrossRef]
- Yang, T.; Nie, Z.; Shu, H.; Kuang, Y.; Chen, X.; Cheng, J.; Yu, S.; Liu, H. The Role of BDNF on Neural Plasticity in Depression. Front. Cell. Neurosci. 2020, 14, 82. [Google Scholar] [CrossRef]
- Mosiołek, A.; Mosiołek, J.; Jakima, S.; Pięta, A.; Szulc, A. Effects of Antidepressant Treatment on Neurotrophic Factors (BDNF and IGF-1) in Patients with Major Depressive Disorder (MDD). J. Clin. Med. 2021, 10, 3377. [Google Scholar] [CrossRef]
- McIntyre, R.S.; Alsuwaidan, M.; Baune, B.T.; Berk, M.; Demyttenaere, K.; Goldberg, J.F.; Gorwood, P.; Ho, R.; Kasper, S.; Kennedy, S.H.; et al. Treatment-Resistant Depression: Definition, Prevalence, Detection, Management, and Investigational Interventions. World Psychiatry 2023, 22, 394–412. [Google Scholar] [CrossRef]
- Gaynes, B.N.; Lux, L.; Gartlehner, G.; Asher, G.; Forman-Hoffman, V.; Green, J.; Boland, E.; Weber, R.P.; Randolph, C.; Bann, C.; et al. Defining Treatment-Resistant Depression. Depress. Anxiety 2020, 37, 134–145. [Google Scholar] [CrossRef]
- Demyttenaere, K.; Van Duppen, Z. The Impact of (the Concept of) Treatment-Resistant Depression: An Opinion Review. Int. J. Neuropsychopharmacol. 2019, 22, 85–92. [Google Scholar] [CrossRef]
- Pigott, H.E.; Kim, T.; Xu, C.; Kirsch, I.; Amsterdam, J. What Are the Treatment Remission, Response and Extent of Improvement Rates After up to Four Trials of Antidepressant Therapies in Real-World Depressed Patients? A Reanalysis of the STAR*D Study’s Patient-Level Data with Fidelity to the Original Research Protocol. BMJ Open 2023, 13, e063095. [Google Scholar] [CrossRef] [PubMed]
- Food and Drug Administration (FDA). FDA Approves New Nasal Spray Medication for Treatment-Resistant Depression, Available Only at a Certified Doctor’s Office or Clinic; Food and Drug Administration (FDA): Silver Spring, MD, USA, 2019. Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-new-nasal-spray-medication-treatment-resistant-depression-available-only-certified (accessed on 26 November 2024).
- Vasiliu, O. Esketamine for Treatment-Resistant Depression: A Review of Clinical Evidence (Review). Exp. Ther. Med. 2023, 25, 111. [Google Scholar] [CrossRef] [PubMed]
- Swainson, J.; Thomas, R.K.; Archer, S.; Chrenek, C.; MacKay, M.-A.; Baker, G.; Dursun, S.; Klassen, L.J.; Chokka, P.; Demas, M.L. Esketamine for Treatment Resistant Depression. Expert Rev. Neurother. 2019, 19, 899–911. [Google Scholar] [CrossRef] [PubMed]
- Dean, R.L.; Hurducas, C.; Hawton, K.; Spyridi, S.; Cowen, P.J.; Hollingsworth, S.; Marquardt, T.; Barnes, A.; Smith, R.; McShane, R.; et al. Ketamine and Other Glutamate Receptor Modulators for Depression in Adults with Unipolar Major Depressive Disorder. Cochrane Database Syst. Rev. 2021, 9, CD011612. [Google Scholar] [CrossRef] [PubMed]
- Castro, M.; Wilkinson, S.T.; Al Jurdi, R.K.; Petrillo, M.P.; Zaki, N.; Borentain, S.; Fu, D.J.; Turkoz, I.; Sun, L.; Brown, B.; et al. Efficacy and Safety of Esketamine Nasal Spray in Patients with Treatment-Resistant Depression Who Completed a Second Induction Period: Analysis of the Ongoing SUSTAIN-3 Study. CNS Drugs 2023, 37, 715–723. [Google Scholar] [CrossRef] [PubMed]
- McIntyre, R.S.; Carvalho, I.P.; Lui, L.M.W.; Majeed, A.; Masand, P.S.; Gill, H.; Rodrigues, N.B.; Lipsitz, O.; Coles, A.C.; Lee, Y.; et al. The Effect of Intravenous, Intranasal, and Oral Ketamine in Mood Disorders: A Meta-Analysis. J. Affect. Disord. 2020, 276, 576–584. [Google Scholar] [CrossRef] [PubMed]
- Yavi, M.; Lee, H.; Henter, I.D.; Park, L.T.; Zarate, C.A. Ketamine Treatment for Depression: A Review. Discov. Ment. Health 2022, 2, 9. [Google Scholar] [CrossRef] [PubMed]
- Björkholm, C.; Monteggia, L.M. BDNF—A Key Transducer of Antidepressant Effects. Neuropharmacology 2016, 102, 72–79. [Google Scholar] [CrossRef] [PubMed]
- Caliman-Fontes, A.T.; Leal, G.; Correia-Melo, F.S.; Paixão, C.S.; Carvalho, M.S.; Jesus-Nunes, A.P.; Vieira, F.; Magnavita, G.; Bandeira, I.D.; Mello, R.P.; et al. Brain-Derived Neurotrophic Factor Serum Levels Following Ketamine and Esketamine Intervention for Treatment-Resistant Depression: Secondary Analysis from a Randomized Trial. Trends Psychiatry Psychother. 2023, 45, e20210298. [Google Scholar] [CrossRef] [PubMed]
- Binder, D.K.; Scharfman, H.E. Brain-Derived Neurotrophic Factor. Growth Factors 2004, 22, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Autry, A.E.; Monteggia, L.M. Brain-Derived Neurotrophic Factor and Neuropsychiatric Disorders. Pharmacol. Rev. 2012, 64, 238–258. [Google Scholar] [CrossRef] [PubMed]
- Roux, P. Neurotrophin Signaling Through the P75 Neurotrophin Receptor. Prog. Neurobiol. 2002, 67, 203–233. [Google Scholar] [CrossRef] [PubMed]
- Eggert, S.; Kins, S.; Endres, K.; Brigadski, T. Brothers in Arms: ProBDNF/BDNF and sAPPα/Aβ-Signaling and Their Common Interplay with ADAM10, TrkB, p75NTR, Sortilin, and sorLA in the Progression of Alzheimer’s Disease. Biol. Chem. 2022, 403, 43–71. [Google Scholar] [CrossRef]
- Levine, E.S.; Crozier, R.A.; Black, I.B.; Plummer, M.R. Brain-Derived Neurotrophic Factor Modulates Hippocampal Synaptic Transmission by Increasing N-Methyl-d-Aspartic Acid Receptor Activity. Proc. Natl. Acad. Sci. USA 1998, 95, 10235–10239. [Google Scholar] [CrossRef]
- Yoshii, A.; Constantine-Paton, M. BDNF Induces Transport of PSD-95 to Dendrites Through PI3K-AKT Signaling After NMDA Receptor Activation. Nat. Neurosci. 2007, 10, 702–711. [Google Scholar] [CrossRef] [PubMed]
- Miranda, M.; Morici, J.F.; Zanoni, M.B.; Bekinschtein, P. Brain-Derived Neurotrophic Factor: A Key Molecule for Memory in the Healthy and the Pathological Brain. Front. Cell. Neurosci. 2019, 13, 363. [Google Scholar] [CrossRef]
- Bathina, S.; Das, U.N. Brain-Derived Neurotrophic Factor and Its Clinical Implications. Arch. Med. Sci. 2015, 6, 1164–1178. [Google Scholar] [CrossRef]
- Karpova, N.N. Role of BDNF Epigenetics in Activity-Dependent Neuronal Plasticity. Neuropharmacology 2014, 76, 709–718. [Google Scholar] [CrossRef]
- Hashimoto, K.; Shimizu, E.; Iyo, M. Critical Role of Brain-Derived Neurotrophic Factor in Mood Disorders. Brain Res. Rev. 2004, 45, 104–114. [Google Scholar] [CrossRef]
- Gao, L.; Zhang, Y.; Sterling, K.; Song, W. Brain-Derived Neurotrophic Factor in Alzheimer’s Disease and Its Pharmaceutical Potential. Transl. Neurodegener. 2022, 11, 4. [Google Scholar] [CrossRef]
- Nieto, R.R.; Carrasco, A.; Corral, S.; Castillo, R.; Gaspar, P.A.; Bustamante, M.L.; Silva, H. BDNF as a Biomarker of Cognition in Schizophrenia/Psychosis: An Updated Review. Front. Psychiatry 2021, 12, 662407. [Google Scholar] [CrossRef] [PubMed]
- Bremner, J.D.; Narayan, M.; Anderson, E.R.; Staib, L.H.; Miller, H.L.; Charney, D.S. Hippocampal Volume Reduction in Major Depression. Am. J. Psychiatry 2000, 157, 115–118. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, V.; Han, M.-H.; Graham, D.L.; Berton, O.; Renthal, W.; Russo, S.J.; LaPlant, Q.; Graham, A.; Lutter, M.; Lagace, D.C.; et al. Molecular Adaptations Underlying Susceptibility and Resistance to Social Defeat in Brain Reward Regions. Cell 2007, 131, 391–404. [Google Scholar] [CrossRef] [PubMed]
- Tebartz Van Elst, L.; Woermann, F.; Lemieux, L.; Trimble, M.R. Increased Amygdala Volumes in Female and Depressed Humans. A Quantitative Magnetic Resonance Imaging Study. Neurosci. Lett. 2000, 281, 103–106. [Google Scholar] [CrossRef] [PubMed]
- Frodl, T.; Meisenzahl, E.; Zetzsche, T.; Bottlender, R.; Born, C.; Groll, C.; Jäger, M.; Leinsinger, G.; Hahn, K.; Möller, H.-J. Enlargement of the Amygdala in Patients with a First Episode of Major Depression. Biol. Psychiatry 2002, 51, 708–714. [Google Scholar] [CrossRef]
- Castrén, E.; Kojima, M. Brain-Derived Neurotrophic Factor in Mood Disorders and Antidepressant Treatments. Neurobiol. Dis. 2017, 97, 119–126. [Google Scholar] [CrossRef]
- Molendijk, M.L.; Spinhoven, P.; Polak, M.; Bus, B.A.A.; Penninx, B.W.J.H.; Elzinga, B.M. Serum BDNF Concentrations as Peripheral Manifestations of Depression: Evidence from a Systematic Review and Meta-Analyses on 179 Associations (N=9484). Mol. Psychiatry 2014, 19, 791–800. [Google Scholar] [CrossRef]
- Liu, X.; Li, P.; Ma, X.; Zhang, J.; Sun, X.; Luo, X.; Zhang, Y. Association Between Plasma Levels of BDNF and GDNF and the Diagnosis, Treatment Response in First-Episode MDD. J. Affect. Disord. 2022, 315, 190–197. [Google Scholar] [CrossRef]
- Zelada, M.I.; Garrido, V.; Liberona, A.; Jones, N.; Zúñiga, K.; Silva, H.; Nieto, R.R. Brain-Derived Neurotrophic Factor (BDNF) as a Predictor of Treatment Response in Major Depressive Disorder (MDD): A Systematic Review. Int. J. Mol. Sci. 2023, 24, 14810. [Google Scholar] [CrossRef]
- Cubillos, S.; Engmann, O.; Brancato, A. BDNF as a Mediator of Antidepressant Response: Recent Advances and Lifestyle Interactions. Int. J. Mol. Sci. 2022, 23, 14445. [Google Scholar] [CrossRef]
- Nibuya, M.; Nestler, E.; Duman, R. Chronic Antidepressant Administration Increases the Expression of cAMP Response Element Binding Protein (CREB) in Rat Hippocampus. J. Neurosci. 1996, 16, 2365–2372. [Google Scholar] [CrossRef]
- Nibuya, M.; Morinobu, S.; Duman, R. Regulation of BDNF and trkB mRNA in Rat Brain by Chronic Electroconvulsive Seizure and Antidepressant Drug Treatments. J. Neurosci. 1995, 15, 7539–7547. [Google Scholar] [CrossRef]
- Deyama, S.; Duman, R.S. Neurotrophic Mechanisms Underlying the Rapid and Sustained Antidepressant Actions of Ketamine. Pharmacol. Biochem. Behav. 2020, 188, 172837. [Google Scholar] [CrossRef]
- Shirayama, Y.; Chen, A.C.-H.; Nakagawa, S.; Russell, D.S.; Duman, R.S. Brain-Derived Neurotrophic Factor Produces Antidepressant Effects in Behavioral Models of Depression. J. Neurosci. 2002, 22, 3251–3261. [Google Scholar] [CrossRef]
- Castrén, E.; Monteggia, L.M. Brain-Derived Neurotrophic Factor Signaling in Depression and Antidepressant Action. Biol. Psychiatry 2021, 90, 128–136. [Google Scholar] [CrossRef]
- Duclot, F.; Kabbaj, M. Epigenetic Mechanisms Underlying the Role of Brain-Derived Neurotrophic Factor in Depression and Response to Antidepressants. J. Exp. Biol. 2015, 218, 21–31. [Google Scholar] [CrossRef]
- Boulle, F.; Van Den Hove, D.L.A.; Jakob, S.B.; Rutten, B.P.; Hamon, M.; Van Os, J.; Lesch, K.-P.; Lanfumey, L.; Steinbusch, H.W.; Kenis, G. Epigenetic Regulation of the BDNF Gene: Implications for Psychiatric Disorders. Mol. Psychiatry 2012, 17, 584–596. [Google Scholar] [CrossRef]
- Chen, K.-W.; Chen, L. Epigenetic Regulation of BDNF Gene During Development and Diseases. Int. J. Mol. Sci. 2017, 18, 571. [Google Scholar] [CrossRef]
- Hing, B.; Davidson, S.; Lear, M.; Breen, G.; Quinn, J.; McGuffin, P.; MacKenzie, A. A Polymorphism Associated with Depressive Disorders Differentially Regulates Brain Derived Neurotrophic Factor Promoter IV Activity. Biol. Psychiatry 2012, 71, 618–626. [Google Scholar] [CrossRef]
- Peña, C.J.; Nestler, E.J. Progress in Epigenetics of Depression. In Progress in Molecular Biology and Translational Science; Elsevier: Amsterdam, The Netherlands, 2018; Volume 157, pp. 41–66. ISBN 978-0-12-813565-5. [Google Scholar]
- Chen, Z.-Y.; Bath, K.; McEwen, B.; Hempstead, B.; Lee, F. Impact of Genetic Variant BDNF (Val66Met) on Brain Structure and Function. Novartis Found Symp. 2008, 289, 180–188, discussion 188–195. [Google Scholar] [CrossRef]
- Colle, R.; Gressier, F.; Verstuyft, C.; Deflesselle, E.; Lépine, J.-P.; Ferreri, F.; Hardy, P.; Guilloux, J.-P.; Petit, A.-C.; Fève, B.; et al. Brain-Derived Neurotrophic Factor Val66Met Polymorphism and 6-Month Antidepressant Remission in Depressed Caucasian Patients. J. Affect. Disord. 2015, 175, 233–240. [Google Scholar] [CrossRef]
- Smit, A.J.T.; Wu, G.W.Y.; Rampersaud, R.; Reus, V.I.; Wolkowitz, O.M.; Mellon, S.H. Serum Brain-Derived Neurotrophic Factor, Val66Met Polymorphism and Open-Label SSRI Treatment Response in Major Depressive Disorder. Psychoneuroendocrinology 2024, 165, 107045. [Google Scholar] [CrossRef]
- Yan, T.; Wang, L.; Kuang, W.; Xu, J.; Li, S.; Chen, J.; Yang, Y. Brain-Derived Neurotrophic Factor Val66Met Polymorphism Association with Antidepressant Efficacy: A Systematic Review and Meta-Analysis: BDNF Val66Met and Antidepressant Efficacy. Asia-Pac. Psychiatry 2014, 6, 241–251. [Google Scholar] [CrossRef]
- Hirota, K.; Lambert, D.G. Ketamine; History and Role in Anesthetic Pharmacology. Neuropharmacology 2022, 216, 109171. [Google Scholar] [CrossRef]
- Mion, G.; Villevieille, T. Ketamine Pharmacology: An Update (Pharmacodynamics and Molecular Aspects, Recent Findings). CNS Neurosci. Ther. 2013, 19, 370–380. [Google Scholar] [CrossRef]
- Li, N.; Lee, B.; Liu, R.-J.; Banasr, M.; Dwyer, J.M.; Iwata, M.; Li, X.-Y.; Aghajanian, G.; Duman, R.S. mTOR-Dependent Synapse Formation Underlies the Rapid Antidepressant Effects of NMDA Antagonists. Science 2010, 329, 959–964. [Google Scholar] [CrossRef]
- Zanos, P.; Gould, T.D. Mechanisms of Ketamine Action as an Antidepressant. Mol. Psychiatry 2018, 23, 801–811. [Google Scholar] [CrossRef]
- Autry, A.E.; Adachi, M.; Nosyreva, E.; Na, E.S.; Los, M.F.; Cheng, P.; Kavalali, E.T.; Monteggia, L.M. NMDA Receptor Blockade at Rest Triggers Rapid Behavioural Antidepressant Responses. Nature 2011, 475, 91–95. [Google Scholar] [CrossRef]
- Moda-Sava, R.N.; Murdock, M.H.; Parekh, P.K.; Fetcho, R.N.; Huang, B.S.; Huynh, T.N.; Witztum, J.; Shaver, D.C.; Rosenthal, D.L.; Alway, E.J.; et al. Sustained Rescue of Prefrontal Circuit Dysfunction by Antidepressant-Induced Spine Formation. Science 2019, 364, eaat8078. [Google Scholar] [CrossRef]
- Krystal, J.H.; Kavalali, E.T.; Monteggia, L.M. Ketamine and Rapid Antidepressant Action: New Treatments and Novel Synaptic Signaling Mechanisms. Neuropsychopharmacology 2024, 49, 41–50. [Google Scholar] [CrossRef]
- Kopelman, J.; Keller, T.A.; Panny, B.; Griffo, A.; Degutis, M.; Spotts, C.; Cruz, N.; Bell, E.; Do-Nguyen, K.; Wallace, M.L.; et al. Rapid Neuroplasticity Changes and Response to Intravenous Ketamine: A Randomized Controlled Trial in Treatment-Resistant Depression. Transl. Psychiatry 2023, 13, 159. [Google Scholar] [CrossRef]
- Inserra, A.; Campanale, A.; Rezai, T.; Romualdi, P.; Rubino, T. Epigenetic Mechanisms of Rapid-Acting Antidepressants. Transl. Psychiatry 2024, 14, 359. [Google Scholar] [CrossRef]
- Duman, R.S.; Aghajanian, G.K.; Sanacora, G.; Krystal, J.H. Synaptic Plasticity and Depression: New Insights from Stress and Rapid-Acting Antidepressants. Nat. Med. 2016, 22, 238–249. [Google Scholar] [CrossRef]
- Maeng, S.; Zarate, C.A.; Du, J.; Schloesser, R.J.; McCammon, J.; Chen, G.; Manji, H.K. Cellular Mechanisms Underlying the Antidepressant Effects of Ketamine: Role of α-Amino-3-Hydroxy-5-Methylisoxazole-4-Propionic Acid Receptors. Biol. Psychiatry 2008, 63, 349–352. [Google Scholar] [CrossRef]
- Moghaddam, B.; Adams, B.; Verma, A.; Daly, D. Activation of Glutamatergic Neurotransmission by Ketamine: A Novel Step in the Pathway from NMDA Receptor Blockade to Dopaminergic and Cognitive Disruptions Associated with the Prefrontal Cortex. J. Neurosci. 1997, 17, 2921–2927. [Google Scholar] [CrossRef]
- Lepack, A.E.; Fuchikami, M.; Dwyer, J.M.; Banasr, M.; Duman, R.S. BDNF Release Is Required for the Behavioral Actions of Ketamine. Int. J. Neuropsychopharmacol. 2015, 18, pyu033. [Google Scholar] [CrossRef]
- Jourdi, H.; Hsu, Y.-T.; Zhou, M.; Qin, Q.; Bi, X.; Baudry, M. Positive AMPA Receptor Modulation Rapidly Stimulates BDNF Release and Increases Dendritic mRNA Translation. J. Neurosci. 2009, 29, 8688–8697. [Google Scholar] [CrossRef]
- Miller, O.H.; Moran, J.T.; Hall, B.J. Two Cellular Hypotheses Explaining the Initiation of Ketamine’s Antidepressant Actions: Direct Inhibition and Disinhibition. Neuropharmacology 2016, 100, 17–26. [Google Scholar] [CrossRef]
- Nosyreva, E.; Szabla, K.; Autry, A.E.; Ryazanov, A.G.; Monteggia, L.M.; Kavalali, E.T. Acute Suppression of Spontaneous Neurotransmission Drives Synaptic Potentiation. J. Neurosci. 2013, 33, 6990–7002. [Google Scholar] [CrossRef]
- Lin, P.-Y.; Ma, Z.Z.; Mahgoub, M.; Kavalali, E.T.; Monteggia, L.M. A Synaptic Locus for TrkB Signaling Underlying Ketamine Rapid Antidepressant Action. Cell Rep. 2021, 36, 109513. [Google Scholar] [CrossRef]
- Lepack, A.E.; Bang, E.; Lee, B.; Dwyer, J.M.; Duman, R.S. Fast-Acting Antidepressants Rapidly Stimulate ERK Signaling and BDNF Release in Primary Neuronal Cultures. Neuropharmacology 2016, 111, 242–252. [Google Scholar] [CrossRef]
- Liu, R.-J.; Lee, F.S.; Li, X.-Y.; Bambico, F.; Duman, R.S.; Aghajanian, G.K. Brain-Derived Neurotrophic Factor Val66Met Allele Impairs Basal and Ketamine-Stimulated Synaptogenesis in Prefrontal Cortex. Biol. Psychiatry 2012, 71, 996–1005. [Google Scholar] [CrossRef]
- Deyama, S.; Bang, E.; Kato, T.; Li, X.-Y.; Duman, R.S. Neurotrophic and Antidepressant Actions of Brain-Derived Neurotrophic Factor Require Vascular Endothelial Growth Factor. Biol. Psychiatry 2019, 86, 143–152. [Google Scholar] [CrossRef]
- Kato, T.; Fogaça, M.V.; Deyama, S.; Li, X.-Y.; Fukumoto, K.; Duman, R.S. BDNF Release and Signaling Are Required for the Antidepressant Actions of GLYX-13. Mol. Psychiatry 2018, 23, 2007–2017. [Google Scholar] [CrossRef]
- Kim, J.-W.; Suzuki, K.; Kavalali, E.T.; Monteggia, L.M. Bridging Rapid and Sustained Antidepressant Effects of Ketamine. Trends Mol. Med. 2023, 29, 364–375. [Google Scholar] [CrossRef]
- Kim, J.-W.; Autry, A.E.; Na, E.S.; Adachi, M.; Björkholm, C.; Kavalali, E.T.; Monteggia, L.M. Sustained Effects of Rapidly Acting Antidepressants Require BDNF-Dependent MeCP2 Phosphorylation. Nat. Neurosci. 2021, 24, 1100–1109. [Google Scholar] [CrossRef]
- Cohen, S.; Gabel, H.W.; Hemberg, M.; Hutchinson, A.N.; Sadacca, L.A.; Ebert, D.H.; Harmin, D.A.; Greenberg, R.S.; Verdine, V.K.; Zhou, Z.; et al. Genome-Wide Activity-Dependent MeCP2 Phosphorylation Regulates Nervous System Development and Function. Neuron 2011, 72, 72–85. [Google Scholar] [CrossRef]
- Hutchinson, A.N.; Deng, J.V.; Cohen, S.; West, A.E. Phosphorylation of MeCP2 at Ser421 Contributes to Chronic Antidepressant Action. J. Neurosci. 2012, 32, 14355–14363. [Google Scholar] [CrossRef]
- Yao, W.; Cao, Q.; Luo, S.; He, L.; Yang, C.; Chen, J.; Qi, Q.; Hashimoto, K.; Zhang, J. Microglial ERK-NRBP1-CREB-BDNF Signaling in Sustained Antidepressant Actions of (R)-Ketamine. Mol. Psychiatry 2022, 27, 1618–1629. [Google Scholar] [CrossRef]
- Wang, Y.-L.; Han, Q.-Q.; Gong, W.-Q.; Pan, D.-H.; Wang, L.-Z.; Hu, W.; Yang, M.; Li, B.; Yu, J.; Liu, Q. Microglial Activation Mediates Chronic Mild Stress-Induced Depressive- and Anxiety-like Behavior in Adult Rats. J. Neuroinflamm. 2018, 15, 21. [Google Scholar] [CrossRef]
- Mandal, G.; Kirkpatrick, M.; Alboni, S.; Mariani, N.; Pariante, C.M.; Borsini, A. Ketamine Prevents Inflammation-Induced Reduction of Human Hippocampal Neurogenesis via Inhibiting the Production of Neurotoxic Metabolites of the Kynurenine Pathway. Int. J. Neuropsychopharmacol. 2024, 27, pyae041. [Google Scholar] [CrossRef]
- Onodera, J.; Nagata, H.; Nakashima, A.; Ikegaya, Y.; Koyama, R. Neuronal Brain-derived Neurotrophic Factor Manipulates Microglial Dynamics. Glia 2021, 69, 890–904. [Google Scholar] [CrossRef]
- Djalali, S.; Höltje, M.; Große, G.; Rothe, T.; Stroh, T.; Große, J.; Deng, D.R.; Hellweg, R.; Grantyn, R.; Hörtnagl, H.; et al. Effects of Brain-Derived Neurotrophic Factor (BDNF) on Glial Cells and Serotonergic Neurones During Development. J. Neurochem. 2005, 92, 616–627. [Google Scholar] [CrossRef]
- Abdallah, C.G.; Averill, L.A.; Gueorguieva, R.; Goktas, S.; Purohit, P.; Ranganathan, M.; Sherif, M.; Ahn, K.-H.; D’Souza, D.C.; Formica, R.; et al. Modulation of the Antidepressant Effects of Ketamine by the mTORC1 Inhibitor Rapamycin. Neuropsychopharmacology 2020, 45, 990–997. [Google Scholar] [CrossRef]
- Averill, L.A.; Averill, C.L.; Gueorguieva, R.; Fouda, S.; Sherif, M.; Ahn, K.-H.; Ranganathan, M.; D’Souza, D.C.; Southwick, S.M.; Sanacora, G.; et al. mTORC1 Inhibitor Effects on Rapid Ketamine-Induced Reductions in Suicidal Ideation in Patients with Treatment-Resistant Depression. J. Affect. Disord. 2022, 303, 91–97. [Google Scholar] [CrossRef]
- Tsokas, P.; Blitzer, R.D. mTOR and the Regulation of Translational Capacity in Late Forms of Synaptic Plasticity. In Synaptic Tagging and Capture; Sajikumar, S., Ed.; Springer New York: New York, NY, USA, 2015; pp. 99–132. ISBN 978-1-4939-1760-0. [Google Scholar]
- Izumi, Y.; Zorumski, C.F. Metaplastic Effects of Subanesthetic Ketamine on CA1 Hippocampal Function. Neuropharmacology 2014, 86, 273–281. [Google Scholar] [CrossRef]
- Cavalleri, L.; Merlo Pich, E.; Millan, M.J.; Chiamulera, C.; Kunath, T.; Spano, P.F.; Collo, G. Ketamine Enhances Structural Plasticity in Mouse Mesencephalic and Human iPSC-Derived Dopaminergic Neurons via AMPAR-Driven BDNF and mTOR Signaling. Mol. Psychiatry 2018, 23, 812–823. [Google Scholar] [CrossRef]
- Maiworm, M. The Relevance of BDNF for Neuroprotection and Neuroplasticity in Multiple Sclerosis. Front. Neurol. 2024, 15, 1385042. [Google Scholar] [CrossRef]
- Kang, M.J.Y.; Hawken, E.; Vazquez, G.H. The Mechanisms Behind Rapid Antidepressant Effects of Ketamine: A Systematic Review with a Focus on Molecular Neuroplasticity. Front. Psychiatry 2022, 13, 860882. [Google Scholar] [CrossRef]
- Mohammad Shehata, I.; Masood, W.; Nemr, N.; Anderson, A.; Bhusal, K.; Edinoff, A.N.; Cornett, E.M.; Kaye, A.M.; Kaye, A.D. The Possible Application of Ketamine in the Treatment of Depression in Alzheimer’s Disease. Neurol. Int. 2022, 14, 310–321. [Google Scholar] [CrossRef]
- Haile, C.N.; Murrough, J.W.; Iosifescu, D.V.; Chang, L.C.; Al Jurdi, R.K.; Foulkes, A.; Iqbal, S.; Mahoney, J.J.; De La Garza, R.; Charney, D.S.; et al. Plasma Brain Derived Neurotrophic Factor (BDNF) and Response to Ketamine in Treatment-Resistant Depression. Int. J. Neuropsychopharm. 2014, 17, 331–336. [Google Scholar] [CrossRef]
- Duncan, W.C.; Sarasso, S.; Ferrarelli, F.; Selter, J.; Riedner, B.A.; Hejazi, N.S.; Yuan, P.; Brutsche, N.; Manji, H.K.; Tononi, G.; et al. Concomitant BDNF and Sleep Slow Wave Changes Indicate Ketamine-Induced Plasticity in Major Depressive Disorder. Int. J. Neuropsychopharmacol. 2013, 16, 301–311. [Google Scholar] [CrossRef]
- Woelfer, M.; Li, M.; Colic, L.; Liebe, T.; Di, X.; Biswal, B.; Murrough, J.; Lessmann, V.; Brigadski, T.; Walter, M. Ketamine-Induced Changes in Plasma Brain-Derived Neurotrophic Factor (BDNF) Levels Are Associated with the Resting-State Functional Connectivity of the Prefrontal Cortex. World J. Biol. Psychiatry 2020, 21, 696–710. [Google Scholar] [CrossRef]
- Machado-Vieira, R.; Yuan, P.; Brutsche, N.; DiazGranados, N.; Luckenbaugh, D.; Manji, H.K.; Zarate, C.A. Brain-Derived Neurotrophic Factor and Initial Antidepressant Response to an N-Methyl-D-Aspartate Antagonist. J. Clin. Psychiatry 2009, 70, 1662–1666. [Google Scholar] [CrossRef]
- Medeiros, G.C.; Greenstein, D.; Kadriu, B.; Yuan, P.; Park, L.T.; Gould, T.D.; Zarate, C.A. Treatment of Depression with Ketamine Does Not Change Plasma Levels of Brain-Derived Neurotrophic Factor or Vascular Endothelial Growth Factor. J. Affect. Disord. 2021, 280, 136–139. [Google Scholar] [CrossRef]
- Wang, J.; Wang, Y.; Xu, X.; Peng, S.; Xu, F.; Liu, P. Use of Various Doses of S-Ketamine in Treatment of Depression and Pain in Cervical Carcinoma Patients with Mild/Moderate Depression After Laparoscopic Total Hysterectomy. Med. Sci. Monit. 2020, 26, e922028. [Google Scholar] [CrossRef]
- Zheng, W.; Gu, L.; Zhou, Y.; Wang, C.; Lan, X.; Zhang, B.; Li, Z.; Ning, Y. Baseline Plasma BDNF Levelsare Associated with Antianhedonic Effects ofRepeated-Dose Intravenous Ketamine in Major Depressive Disorder. Curr. Neuropharmacol. 2023, 21, 1013–1021. [Google Scholar] [CrossRef]
- Wang, M.; Zhang, B.; Zhou, Y.; Wang, C.; Zheng, W.; Liu, W.; Zhan, Y.; Lan, X.; Ning, Y. Sleep Improvement Is Associated with the Antidepressant Efficacy of Repeated-Dose Ketamine and Serum BDNF Levels: A Post-Hoc Analysis. Pharmacol. Rep. 2021, 73, 594–603. [Google Scholar] [CrossRef]
- Grunebaum, M.F.; Ellis, S.P.; Keilp, J.G.; Moitra, V.K.; Cooper, T.B.; Marver, J.E.; Burke, A.K.; Milak, M.S.; Sublette, M.E.; Oquendo, M.A.; et al. Ketamine Versus Midazolam in Bipolar Depression with Suicidal Thoughts: A Pilot Midazolam-Controlled Randomized Clinical Trial. Bipolar Disord. 2017, 19, 176–183. [Google Scholar] [CrossRef]
- Li, C.; Cai, Q.; Su, Z.; Chen, Z.; Cao, J.; Xu, F. Could Peripheral 5-HT Level Be Used as a Biomarker for Depression Diagnosis and Treatment? A Narrative Minireview. Front. Pharmacol. 2023, 14, 1149511. [Google Scholar] [CrossRef]
- Jiang, Q.; Qi, Y.; Zhou, M.; Dong, Y.; Zheng, W.; Zhu, L.; Li, Y.; Zhou, H.; Wang, L. Effect of Esketamine on Serum Neurotransmitters in Patients with Postpartum Depression: A Randomized Controlled Trial. BMC Anesthesiol. 2024, 24, 293. [Google Scholar] [CrossRef] [PubMed]
- Luo, T.; Deng, Z.; Ren, Q.; Mu, F.; Zhang, Y.; Wang, H. Effects of Esketamine on Postoperative Negative Emotions and Early Cognitive Disorders in Patients Undergoing Non-Cardiac Thoracic Surgery: A Randomized Controlled Trial. J. Clin. Anesth. 2024, 95, 111447. [Google Scholar] [CrossRef] [PubMed]
- Klein, A.B.; Williamson, R.; Santini, M.A.; Clemmensen, C.; Ettrup, A.; Rios, M.; Knudsen, G.M.; Aznar, S. Blood BDNF Concentrations Reflect Brain-Tissue BDNF Levels across Species. Int. J. Neuropsychopharm. 2011, 14, 347–353. [Google Scholar] [CrossRef] [PubMed]
- Karege, F.; Schwald, M.; Cisse, M. Postnatal Developmental Profile of Brain-Derived Neurotrophic Factor in Rat Brain and Platelets. Neurosci. Lett. 2002, 328, 261–264. [Google Scholar] [CrossRef]
- Sartorius, A.; Hellweg, R.; Litzke, J.; Vogt, M.; Dormann, C.; Vollmayr, B.; Danker-Hopfe, H.; Gass, P. Correlations and Discrepancies Between Serum and Brain Tissue Levels of Neurotrophins after Electroconvulsive Treatment in Rats. Pharmacopsychiatry 2009, 42, 270–276. [Google Scholar] [CrossRef]
- Elfving, B.; Plougmann, P.H.; Müller, H.K.; Mathé, A.A.; Rosenberg, R.; Wegener, G. Inverse Correlation of Brain and Blood BDNF Levels in a Genetic Rat Model of Depression. Int. J. Neuropsychopharm. 2010, 13, 563–572. [Google Scholar] [CrossRef]
- Pan, Z.; Park, C.; Brietzke, E.; Zuckerman, H.; Rong, C.; Mansur, R.B.; Fus, D.; Subramaniapillai, M.; Lee, Y.; McIntyre, R.S. Cognitive Impairment in Major Depressive Disorder. CNS Spectr. 2019, 24, 22–29. [Google Scholar] [CrossRef]
- Eisch, A.J.; Bolaños, C.A.; De Wit, J.; Simonak, R.D.; Pudiak, C.M.; Barrot, M.; Verhaagen, J.; Nestler, E.J. Brain-Derived Neurotrophic Factor in the Ventral Midbrain–Nucleus Accumbens Pathway: A Role in Depression. Biol. Psychiatry 2003, 54, 994–1005. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| Authors | Study Design | Time of Measurement | Outcome—Ketamine |
|---|---|---|---|
| Haile, et al. [94] | 44 patients with TRD treated with IV Ketamine (0.5 mg/kg) or IV Midazolam (0.045 mg/kg) | 240 min post-infusion | Ketamine significantly increased plasma BDNF in responders compared to non-responders |
| Duncan Jr, et al. [95] | 30 patients with TRD treated with IV Ketamine (0.5 mg/kg) | 230 min post-infusion | Ketamine significantly increased plasma BDNF |
| Woelfer, et al. [96] | 80 healthy volunteers treated with IV Ketamine (0.5 mg/kg) or IV NaCl 0.9% | 120 min and 24 h post-infusion | Ketamine significantly increased plasma BDNF at both time points compared to placebo |
| Machado-Vieira, et al. [97] | 23 patients with TRD treated with IV Ketamine (0.5 mg/kg) | 40, 80, 120, and 230 min post-infusion | No significant increase in plasma BDNF at any time point |
| Medeiros, et al. [98] | 39 patients with major depressive disorder (MDD) treated with IV Ketamine (0.5 mg/kg) or IV saline | 230 min, 24 h, and 72 h post-infusion | No significant increase in plasma BDNF at any time point compared to placebo |
| Wang, et al. [99] | 417 cervical carcinoma patients receiving 0.25 mg/kg IV esketamine or 0.5 mg/kg IV esketamine or 0.5 mg/kg IV ketamine or IV saline | 24, 48, 72, 120 and 168 h after the surgery | Ketamine significantly increased plasma BDNF at 24, 48 and 72 h compared to placebo |
| Caliman-Fontes, et al. [20] | 53 patients with TRD treated with IV Ketamine (0.5 mg/kg) or IV Esketamine (0.25 mg/kg) | 24 h and 168 h (1 week) post-infusion | No significant increase in plasma BDNF at any time point |
| Authors | Study Design | Time of Measurement | Outcome—Esketamine |
|---|---|---|---|
| Jiang, et al. [104] | 315 patients with postpartum depression treated with IV esketamine (0.25 mg/kg) or IV saline | 72 h post-infusion | Esketamine significantly increased plasma BDNF compared to placebo |
| Luo, et al. [105] | 129 adult patients that underwent elective non-cardiac thoracic surgery under general anesthesia treated with 0.2 mg/kg IV esketamine or 0.5 mg/kg IV esketamine or with IV saline | End of the surgery and three days after the surgery | 0.5 mg/kg esketamine significantly increased plasma BDNF compared to placebo |
| Wang, et al. [99] | 417 cervical carcinoma patients receiving 0.25 mg/kg IV esketamine or 0.5 mg/kg IV esketamine or 0.5 mg/kg IV ketamine or IV saline | 24, 48, 72, 120 and 168 h after the surgery | Both 0.25 mg/kg esketamine and 0.5 mg/kg esketamine significantly increased plasma BDNF at 24, 48 and 72 h compared to placebo |
| Caliman-Fontes, et al. [20] | 53 patients with TRD treated with IV Ketamine (0.5 mg/kg) or IV Esketamine (0.25 mg/kg) | 24 h and 168 h (1 week) post-infusion | No significant increase in plasma BDNF at any time point |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pardossi, S.; Fagiolini, A.; Cuomo, A. Variations in BDNF and Their Role in the Neurotrophic Antidepressant Mechanisms of Ketamine and Esketamine: A Review. Int. J. Mol. Sci. 2024, 25, 13098. https://doi.org/10.3390/ijms252313098
Pardossi S, Fagiolini A, Cuomo A. Variations in BDNF and Their Role in the Neurotrophic Antidepressant Mechanisms of Ketamine and Esketamine: A Review. International Journal of Molecular Sciences. 2024; 25(23):13098. https://doi.org/10.3390/ijms252313098
Chicago/Turabian StylePardossi, Simone, Andrea Fagiolini, and Alessandro Cuomo. 2024. "Variations in BDNF and Their Role in the Neurotrophic Antidepressant Mechanisms of Ketamine and Esketamine: A Review" International Journal of Molecular Sciences 25, no. 23: 13098. https://doi.org/10.3390/ijms252313098
APA StylePardossi, S., Fagiolini, A., & Cuomo, A. (2024). Variations in BDNF and Their Role in the Neurotrophic Antidepressant Mechanisms of Ketamine and Esketamine: A Review. International Journal of Molecular Sciences, 25(23), 13098. https://doi.org/10.3390/ijms252313098