
Fibroblast Heterogeneity in Inflammatory Bowel Disease

Abstract
:1. Introduction
2. Heterogeneity of Fibroblasts in Development and Homeostasis
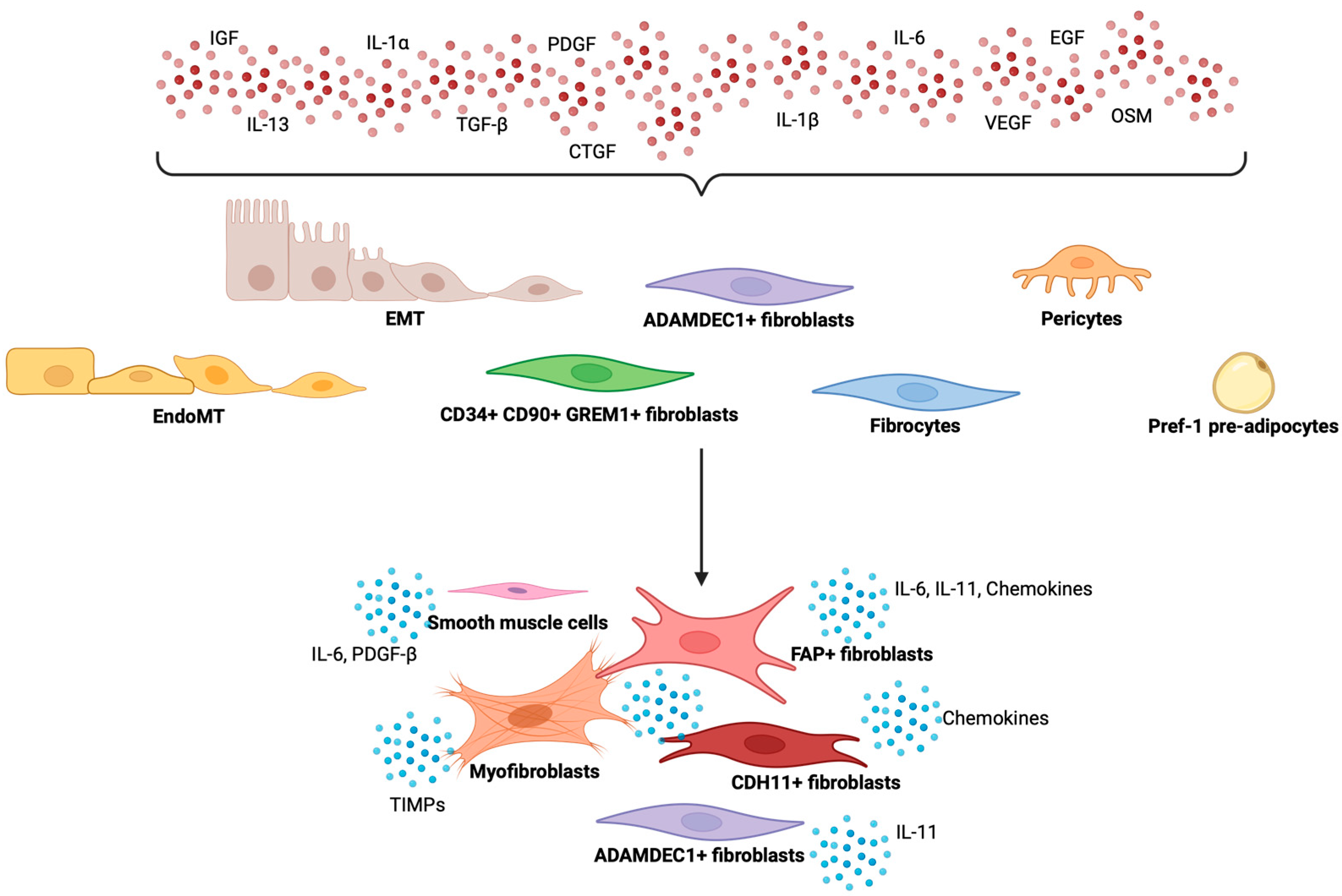
3. Heterogeneity of Fibroblasts in Chronic Inflammation and Intestinal Fibrosis
4. Immune-Stromal Cell Interaction in Intestinal Fibrosis
5. Strategies for Targeting Fibroblast Activity
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Sheng, G. The Developmental Basis of Mesenchymal Stem/Stromal Cells (MSCs). BMC Dev. Biol. 2015, 15, 44. [Google Scholar] [CrossRef] [PubMed]
- Lefebvre, V.; Bhattaram, P. Chapter Eight—Vertebrate Skeletogenesis. In Current Topics in Developmental Biology; Koopman, P., Ed.; Academic Press: Oxford, UK, 2010; Volume 90, pp. 291–317. ISBN 0070-2153. [Google Scholar]
- Zhang, G.; Eames, B.F.; Cohn, M.J. Chapter 2 Evolution of Vertebrate Cartilage Development. In Current Topics in Developmental Biology; Academic Press: Oxford, UK, 2009; Volume 86, pp. 15–42. ISBN 0070-2153. [Google Scholar]
- Sowa, Y.; Imura, T.; Numajiri, T.; Takeda, K.; Mabuchi, Y.; Matsuzaki, Y.; Nishino, K. Adipose Stromal Cells Contain Phenotypically Distinct Adipogenic Progenitors Derived from Neural Crest. PLoS ONE 2014, 8, e84206. [Google Scholar] [CrossRef] [PubMed]
- Damerell, V.; Pepper, M.S.; Prince, S. Molecular Mechanisms Underpinning Sarcomas and Implications for Current and Future Therapy. Signal Transduct. Target. Ther. 2021, 6, 246. [Google Scholar] [CrossRef] [PubMed]
- Dominici, M.; Le Blanc, K.; Mueller, I.; Slaper-Cortenbach, I.; Marini, F.C.; Krause, D.S.; Deans, R.J.; Keating, A.; Prockop, D.J.; Horwitz, E.M. Minimal Criteria for Defining Multipotent Mesenchymal Stromal Cells. The International Society for Cellular Therapy Position Statement. Cytotherapy 2006, 8, 315–317. [Google Scholar] [CrossRef]
- Jacob, J.-M.; Di Carlo, S.E.; Stzepourginski, I.; Lepelletier, A.; Ndiaye, P.D.; Varet, H.; Legendre, R.; Kornobis, E.; Benabid, A.; Nigro, G.; et al. PDGFRα-Induced Stromal Maturation Is Required to Restrain Postnatal Intestinal Epithelial Stemness and Promote Defense Mechanisms. Cell Stem Cell 2022, 29, 856–868.e5. [Google Scholar] [CrossRef]
- Farahani, R.M.; Xaymardan, M. Platelet-Derived Growth Factor Receptor Alpha as a Marker of Mesenchymal Stem Cells in Development and Stem Cell Biology. Stem Cells Int. 2015, 2015, 362753. [Google Scholar] [CrossRef]
- Worthley, D.L.; Churchill, M.; Compton, J.T.; Tailor, Y.; Rao, M.; Si, Y.; Levin, D.; Schwartz, M.G.; Uygur, A.; Hayakawa, Y.; et al. Gremlin 1 Identifies a Skeletal Stem Cell with Bone, Cartilage, and Reticular Stromal Potential. Cell 2015, 160, 269–284. [Google Scholar] [CrossRef]
- McCarthy, N.; Manieri, E.; Storm, E.E.; Saadatpour, A.; Luoma, A.M.; Kapoor, V.N.; Madha, S.; Gaynor, L.T.; Cox, C.; Keerthivasan, S.; et al. Distinct Mesenchymal Cell Populations Generate the Essential Intestinal BMP Signaling Gradient. Cell Stem Cell 2020, 26, 391–402.e5. [Google Scholar] [CrossRef]
- Elmentaite, R.; Ross, A.D.B.; Roberts, K.; James, K.R.; Ortmann, D.; Gomes, T.; Nayak, K.; Tuck, L.; Pritchard, S.; Bayraktar, O.A.; et al. Single-Cell Sequencing of Developing Human Gut Reveals Transcriptional Links to Childhood Crohn’s Disease. Dev. Cell 2020, 55, 771–783.e5. [Google Scholar] [CrossRef]
- Davis, H.; Irshad, S.; Bansal, M.; Rafferty, H.; Boitsova, T.; Bardella, C.; Jaeger, E.; Lewis, A.; Freeman-Mills, L.; Giner, F.C.; et al. Aberrant Epithelial GREM1 Expression Initiates Colonic Tumorigenesis from Cells Outside the Stem Cell Niche. Nat. Med. 2015, 21, 62–70. [Google Scholar] [CrossRef]
- Greicius, G.; Kabiri, Z.; Sigmundsson, K.; Liang, C.; Bunte, R.; Singh, M.K.; Virshup, D.M. PDGFRα+ Pericryptal Stromal Cells Are the Critical Source of Wnts and RSPO3 for Murine Intestinal Stem Cells in Vivo. Proc. Natl. Acad. Sci. USA 2018, 115, E3173–E3181. [Google Scholar] [CrossRef] [PubMed]
- Bhanja, P.; Saha, S.; Kabarriti, R.; Liu, L.; Roy-Chowdhury, N.; Roy-Chowdhury, J.; Sellers, R.S.; Alfieri, A.A.; Guha, C. Protective Role of R-Spondin1, an Intestinal Stem Cell Growth Factor, against Radiation-Induced Gastrointestinal Syndrome in Mice. PLoS ONE 2009, 4, e8014. [Google Scholar] [CrossRef]
- Ugurlu, B.; Karaoz, E. Comparison of Similar Cells: Mesenchymal Stromal Cells and Fibroblasts. Acta Histochem. 2020, 122, 151634. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.H.; Shah, B.; Moioli, E.K.; Mao, J.J. CTGF Directs Fibroblast Differentiation from Human Mesenchymal Stem/Stromal Cells and Defines Connective Tissue Healing in a Rodent Injury Model. J. Clin. Investig. 2010, 120, 3340–3349. [Google Scholar] [CrossRef] [PubMed]
- Fawkner-Corbett, D.; Antanaviciute, A.; Parikh, K.; Jagielowicz, M.; Gerós, A.S.; Gupta, T.; Ashley, N.; Khamis, D.; Fowler, D.; Morrissey, E.; et al. Spatiotemporal Analysis of Human Intestinal Development at Single-Cell Resolution. Cell 2021, 184, 810–826.e23. [Google Scholar] [CrossRef]
- Holloway, E.M.; Czerwinski, M.; Tsai, Y.-H.; Wu, J.H.; Wu, A.; Childs, C.J.; Walton, K.D.; Sweet, C.W.; Yu, Q.; Glass, I.; et al. Mapping Development of the Human Intestinal Niche at Single-Cell Resolution. Cell Stem Cell 2021, 28, 568–580.e4. [Google Scholar] [CrossRef]
- Qian, J.; Olbrecht, S.; Boeckx, B.; Vos, H.; Laoui, D.; Etlioglu, E.; Wauters, E.; Pomella, V.; Verbandt, S.; Busschaert, P.; et al. A Pan-Cancer Blueprint of the Heterogeneous Tumor Microenvironment Revealed by Single-Cell Profiling. Cell Res. 2020, 30, 745–762. [Google Scholar] [CrossRef]
- Kinchen, J.; Chen, H.H.; Parikh, K.; Antanaviciute, A.; Jagielowicz, M.; Fawkner-Corbett, D.; Ashley, N.; Cubitt, L.; Mellado-Gomez, E.; Attar, M.; et al. Structural Remodeling of the Human Colonic Mesenchyme in Inflammatory Bowel Disease. Cell 2018, 175, 372–386.e17. [Google Scholar] [CrossRef]
- Degirmenci, B.; Valenta, T.; Dimitrieva, S.; Hausmann, G.; Basler, K. GLI1-Expressing Mesenchymal Cells Form the Essential Wnt-Secreting Niche for Colon Stem Cells. Nature 2018, 558, 449–453. [Google Scholar] [CrossRef]
- Buechler, M.B.; Pradhan, R.N.; Krishnamurty, A.T.; Cox, C.; Calviello, A.K.; Wang, A.W.; Yang, Y.A.; Tam, L.; Caothien, R.; Roose-Girma, M.; et al. Cross-Tissue Organization of the Fibroblast Lineage. Nature 2021, 593, 575–579. [Google Scholar] [CrossRef]
- Jasso, G.J.; Jaiswal, A.; Varma, M.; Laszewski, T.; Grauel, A.; Omar, A.; Silva, N.; Dranoff, G.; Porter, J.A.; Mansfield, K.; et al. Colon Stroma Mediates an Inflammation-Driven Fibroblastic Response Controlling Matrix Remodeling and Healing. PLoS Biol. 2022, 20, e3001532. [Google Scholar] [CrossRef] [PubMed]
- Stzepourginski, I.; Nigro, G.; Jacob, J.-M.; Dulauroy, S.; Sansonetti, P.J.; Eberl, G.; Peduto, L. CD34+ Mesenchymal Cells Are a Major Component of the Intestinal Stem Cells Niche at Homeostasis and after Injury. Proc. Natl. Acad. Sci. USA 2017, 114, E506–E513. [Google Scholar] [CrossRef] [PubMed]
- Acharjee, A.; Shivaji, U.N.; Santacroce, G.; Jeffery, L.; Reynolds, G.; Majumder, S.; Akiror, S.; Zardo, D.; Gkoutos, G.V.; Iacucci, M.; et al. DOP61 Up-Regulated Gremlin 1 in Fibroblasts from Crohn’s Disease Fibrotic Strictures: A Potential Therapeutic Target. J. Crohns Colitis 2024, 18, i185–i186. [Google Scholar] [CrossRef]
- Yang, Y.; Zeng, Q.-S.; Zou, M.; Zeng, J.; Nie, J.; Chen, D.; Gan, H.-T. Targeting Gremlin 1 Prevents Intestinal Fibrosis Progression by Inhibiting the Fatty Acid Oxidation of Fibroblast Cells. Front. Pharmacol. 2021, 12, 663774. [Google Scholar] [CrossRef] [PubMed]
- Yao, L.; Rathnakar, B.H.; Kwon, H.R.; Sakashita, H.; Kim, J.H.; Rackley, A.; Tomasek, J.J.; Berry, W.L.; Olson, L.E. Temporal Control of PDGFRα Regulates the Fibroblast-to-Myofibroblast Transition in Wound Healing. Cell Rep. 2022, 40, 111192. [Google Scholar] [CrossRef]
- Brügger, M.D.; Valenta, T.; Fazilaty, H.; Hausmann, G.; Basler, K. Distinct Populations of Crypt-Associated Fibroblasts Act as Signaling Hubs to Control Colon Homeostasis. PLoS Biol. 2020, 18, e3001032. [Google Scholar] [CrossRef]
- Ke, B.-J.; Abdurahiman, S.; Biscu, F.; Zanella, G.; Dragoni, G.; Santhosh, S.; De Simone, V.; Zouzaf, A.; van Baarle, L.; Stakenborg, M.; et al. Intercellular Interaction between FAP+ Fibroblasts and CD150+ Inflammatory Monocytes Mediates Fibro-Stenosis in Crohn’s Disease. J. Clin. Investig. 2024, 134, 16. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, J.; Sun, H.; Xun, Z.; He, Z.; Zhao, Y.; Qi, J.; Sun, S.; Yang, Q.; Gu, Y.; et al. TWIST1+FAP+ Fibroblasts in the Pathogenesis of Intestinal Fibrosis in Crohn’s Disease. J. Clin. Investig. 2024, 134, 18. [Google Scholar] [CrossRef]
- Friedrich, M.; Pohin, M.; Jackson, M.A.; Korsunsky, I.; Bullers, S.J.; Rue-Albrecht, K.; Christoforidou, Z.; Sathananthan, D.; Thomas, T.; Ravindran, R.; et al. IL-1-Driven Stromal–Neutrophil Interactions Define a Subset of Patients with Inflammatory Bowel Disease That Does Not Respond to Therapies. Nat. Med. 2021, 27, 1970–1981. [Google Scholar] [CrossRef]
- Kurahara, L.H.; Hiraishi, K.; Hu, Y.; Koga, K.; Onitsuka, M.; Doi, M.; Aoyagi, K.; Takedatsu, H.; Kojima, D.; Fujihara, Y.; et al. Activation of Myofibroblast TRPA1 by Steroids and Pirfenidone Ameliorates Fibrosis in Experimental Crohn’s Disease. Cell Mol. Gastroenterol. Hepatol. 2018, 5, 299–318. [Google Scholar] [CrossRef]
- Biel, C.; Faber, K.N.; Bank, R.A.; Olinga, P. Matrix Metalloproteinases in Intestinal Fibrosis. J. Crohns Colitis 2024, 18, 462–478. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Deng, Y.; Liu, A.; Shen, N.; Wang, W.; Du, X.; Tang, Q.; Li, S.; Odeh, Z.; Wu, T.; et al. Novel Mechanism of the Pericyte-Myofibroblast Transition in Renal Interstitial Fibrosis: Core Fucosylation Regulation. Sci. Rep. 2017, 7, 16914. [Google Scholar] [CrossRef] [PubMed]
- Gu, W.; Hong, X.; Le Bras, A.; Nowak, W.N.; Issa Bhaloo, S.; Deng, J.; Xie, Y.; Hu, Y.; Ruan, X.Z.; Xu, Q. Smooth Muscle Cells Differentiated from Mesenchymal Stem Cells Are Regulated by MicroRNAs and Suitable for Vascular Tissue Grafts. J. Biol. Chem. 2018, 293, 8089–8102. [Google Scholar] [CrossRef]
- Severi, C.; Sferra, R.; Scirocco, A.; Vetuschi, A.; Pallotta, N.; Pronio, A.; Caronna, R.; Di Rocco, G.; Gaudio, E.; Corazziari, E.; et al. Contribution of Intestinal Smooth Muscle to Crohn’s Disease Fibrogenesis. Eur. J. Histochem. 2014, 58, 2457. [Google Scholar] [CrossRef]
- Graham, M.F.; Bryson, G.R.; Diegelmann, R.F. Transforming Growth Factor β1 Selectively Augments Collagen Synthesis by Human Intestinal Smooth Muscle Cells. Gastroenterology 1990, 99, 447–453. [Google Scholar] [CrossRef]
- Geng, Z.; Li, J.; Zuo, L.; Zhang, X.; Wang, L.; Xia, Y.; Yang, J.; Yin, L.; Song, X.; Wang, Y.; et al. Intestinal Adipocytes Transdifferentiate into Myofibroblast-like Cells and Contribute to Fibrosis in Crohn’s Disease. J. Crohns Colitis 2024, 18, jjae036. [Google Scholar] [CrossRef]
- Hwang, N.; Kang, D.; Shin, S.-J.; Yoon, B.K.; Chun, J.; Kim, J.; Fang, S. Creeping Fat Exhibits Distinct Inflammation-Specific Adipogenic Preadipocytes in Crohn’s Disease. Front. Immunol. 2023, 14, 1198905. [Google Scholar] [CrossRef]
- Dragoni, G.; Ke, B.-J.; Picariello, L.; Abdurahiman, S.; Ceni, E.; Biscu, F.; Mello, T.; Polvani, S.; Innocenti, T.; Spalart, V.; et al. The Impact of PAD4-Dependent Neutrophil Extracellular Trap Formation on the Early Development of Intestinal Fibrosis in Crohn’s Disease. J. Crohns Colitis 2024, jjae121. [Google Scholar] [CrossRef]
- Stawski, L.; Trojanowska, M. Oncostatin M and Its Role in Fibrosis. Connect. Tissue Res. 2019, 60, 40–49. [Google Scholar] [CrossRef]
- Wei, K.; Nguyen, H.N.; Brenner, M.B. Fibroblast Pathology in Inflammatory Diseases. J. Clin. Investig. 2021, 131, e149538. [Google Scholar] [CrossRef]
- Garrido-Trigo, A.; Corraliza, A.M.; Veny, M.; Dotti, I.; Melón-Ardanaz, E.; Rill, A.; Crowell, H.L.; Corbí, Á.; Gudiño, V.; Esteller, M.; et al. Macrophage and Neutrophil Heterogeneity at Single-Cell Spatial Resolution in Human Inflammatory Bowel Disease. Nat. Commun. 2023, 14, 4506. [Google Scholar] [CrossRef] [PubMed]
- Salvador, P.; Macías-Ceja, D.C.; Gisbert-Ferrándiz, L.; Hernández, C.; Bernardo, D.; Alós, R.; Navarro-Vicente, F.; Esplugues, J.V.; Ortiz-Masiá, D.; Barrachina, M.D.; et al. CD16+ Macrophages Mediate Fibrosis in Inflammatory Bowel Disease. J. Crohns Colitis 2018, 12, 589–599. [Google Scholar] [CrossRef] [PubMed]
- Rivera-Ortega, P.; Hayton, C.; Blaikley, J.; Leonard, C.; Chaudhuri, N. Nintedanib in the Management of Idiopathic Pulmonary Fibrosis: Clinical Trial Evidence and Real-World Experience. Ther. Adv. Respir. Dis. 2018, 12, 1753466618800618. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Li, J.; Xiao, T.; Hu, Y.; Yang, Y.; Gu, X.; Jin, G.; Cao, H.; Zhou, H.; Yang, C. Nintedanib Alleviates Experimental Colitis by Inhibiting CEBPB/PCK1 and CEBPB/EFNA1 Pathways. Front. Pharmacol. 2022, 13, 904420. [Google Scholar] [CrossRef]
- Kataria, J.; Kerr, J.; Lourenssen, S.R.; Blennerhassett, M.G. Nintedanib Regulates Intestinal Smooth Muscle Hyperplasia and Phenotype in Vitro and in TNBS Colitis in Vivo. Sci. Rep. 2022, 12, 10275. [Google Scholar] [CrossRef]
- Antoniu, S.A. Pirfenidone for the Treatment of Idiopathic Pulmonary Fibrosis. Expert. Opin. Investig. Drugs 2006, 15, 823–828. [Google Scholar] [CrossRef]
- Li, G.; Ren, J.; Hu, Q.; Deng, Y.; Chen, G.; Guo, K.; Li, R.; Li, Y.; Wu, L.; Wang, G.; et al. Oral Pirfenidone Protects against Fibrosis by Inhibiting Fibroblast Proliferation and TGF-β Signaling in a Murine Colitis Model. Biochem. Pharmacol. 2016, 117, 57–67. [Google Scholar] [CrossRef]
- Sun, Y.-W.; Zhang, Y.-Y.; Ke, X.-J.; Wu, X.; Chen, Z.-F.; Chi, P. Pirfenidone Prevents Radiation-Induced Intestinal Fibrosis in Rats by Inhibiting Fibroblast Proliferation and Differentiation and Suppressing the TGF-Β1/Smad/CTGF Signaling Pathway. Eur. J. Pharmacol. 2018, 822, 199–206. [Google Scholar] [CrossRef]
- Ali, M.; Barash, M.; Mohammed, A.; Ramalingam, V. Severe Pancolitis: A Rare Adverse Effect of Nintedanib. Chest 2018, 154, 446A. [Google Scholar] [CrossRef]
- Kato, M.; Sasaki, S.; Nakamura, T.; Kurokawa, K.; Yamada, T.; Ochi, Y.; Ihara, H.; Takahashi, F.; Takahashi, K. Gastrointestinal Adverse Effects of Nintedanib and the Associated Risk Factors in Patients with Idiopathic Pulmonary Fibrosis. Sci. Rep. 2019, 9, 12062. [Google Scholar] [CrossRef]
- Lipson, K.; Moustafa, M.; Akbarpour, M.; Fouse, S.; Kriegsmann, M.; Zhou, C.; Liu, K.; Lasitschka, F.; Weichert, W.; Seeley, T.; et al. Therapeutic Pamrevlumab (FG-3019) Is More Effective than Pirfenidone or Nintedanib in a Mouse Radiation-Induced Lung Fibrosis Model. Eur. Respir. J. 2017, 50, PA908. [Google Scholar] [CrossRef]
- di Mola, F.F.; Di Sebastiano, P.; Gardini, A.; Innocenti, P.; Zimmermann, A.; Büchler, M.W.; Friess, H. Differential Expression of Connective Tissue Growth Factor in Inflammatory Bowel Disease. Digestion 2004, 69, 245–253. [Google Scholar] [CrossRef] [PubMed]
- Solitano, V.; Dal Buono, A.; Gabbiadini, R.; Wozny, M.; Repici, A.; Spinelli, A.; Vetrano, S.; Armuzzi, A. Fibro-Stenosing Crohn’s Disease: What Is New and What Is Next? J. Clin. Med. 2023, 12, 3052. [Google Scholar] [CrossRef] [PubMed]
- Solitano, V.; Jairath, V.; Ungaro, F.; Peyrin-Biroulet, L.; Danese, S. TL1A Inhibition for Inflammatory Bowel Disease Treatment: From Inflammation to Fibrosis. Med 2024, 5, 386–400. [Google Scholar] [CrossRef]
- Loomba, R.; Sanyal, A.J.; Nakajima, A.; Neuschwander-Tetri, B.A.; Goodman, Z.D.; Harrison, S.A.; Lawitz, E.J.; Gunn, N.; Imajo, K.; Ravendhran, N.; et al. Pegbelfermin in Patients With Nonalcoholic Steatohepatitis and Stage 3 Fibrosis (FALCON 1): A Randomized Phase 2b Study. Clin. Gastroenterol. Hepatol. 2024, 22, 102–112.e9. [Google Scholar] [CrossRef]
- Sanyal, A.; Charles, E.D.; Neuschwander-Tetri, B.A.; Loomba, R.; Harrison, S.A.; Abdelmalek, M.F.; Lawitz, E.J.; Halegoua-DeMarzio, D.; Kundu, S.; Noviello, S.; et al. Pegbelfermin (BMS-986036), a PEGylated Fibroblast Growth Factor 21 Analogue, in Patients with Non-Alcoholic Steatohepatitis: A Randomised, Double-Blind, Placebo-Controlled, Phase 2a Trial. Lancet 2018, 392, 2705–2717. [Google Scholar] [CrossRef]
- Harrison, S.A.; Neff, G.; Guy, C.D.; Bashir, M.R.; Paredes, A.H.; Frias, J.P.; Younes, Z.; Trotter, J.F.; Gunn, N.T.; Moussa, S.E.; et al. Efficacy and Safety of Aldafermin, an Engineered FGF19 Analog, in a Randomized, Double-Blind, Placebo-Controlled Trial of Patients With Nonalcoholic Steatohepatitis. Gastroenterology 2021, 160, 219–231.e1. [Google Scholar] [CrossRef]
- Feagan, B.G.; Sandborn, W.J.; Gasink, C.; Jacobstein, D.; Lang, Y.; Friedman, J.R.; Blank, M.A.; Johanns, J.; Gao, L.L.; Miao, Y.; et al. Ustekinumab as Induction and Maintenance Therapy for Crohn’s Disease. N. Engl. J. Med. 2024, 375, 1946–1960. [Google Scholar] [CrossRef]
- Hirschfield, G.M.; Gershwin, M.E.; Strauss, R.; Mayo, M.J.; Levy, C.; Zou, B.; Johanns, J.; Nnane, I.P.; Dasgupta, B.; Li, K.; et al. Ustekinumab for Patients with Primary Biliary Cholangitis Who Have an Inadequate Response to Ursodeoxycholic Acid: A Proof-of-Concept Study. Hepatology 2016, 64, 189–199. [Google Scholar] [CrossRef]
- Holvoet, T.; Devriese, S.; Castermans, K.; Boland, S.; Leysen, D.; Vandewynckel, Y.-P.; Devisscher, L.; Van den Bossche, L.; Van Welden, S.; Dullaers, M.; et al. Treatment of Intestinal Fibrosis in Experimental Inflammatory Bowel Disease by the Pleiotropic Actions of a Local Rho Kinase Inhibitor. Gastroenterology 2017, 153, 1054–1067. [Google Scholar] [CrossRef]
- Gambardella, A.; Guisot, N.E.S.; Bunyard, P.R.; Offer, E. Effects of RXC007, a Highly Potent and Selective ROCK2 Inhibitor, in Ex-Vivo and in Vivo Models of Pulmonary Fibrosis. Eur. Respir. J. 2022, 60, 3033. [Google Scholar] [CrossRef]
- Wang, W.; Bhattacharyya, S.; Marangoni, R.G.; Carns, M.; Dennis-Aren, K.; Yeldandi, A.; Wei, J.; Varga, J. The JAK/STAT Pathway Is Activated in Systemic Sclerosis and Is Effectively Targeted by Tofacitinib. J. Scleroderma Relat. Disord. 2019, 5, 40–50. [Google Scholar] [CrossRef] [PubMed]
- Sandborn, W.J.; Ghosh, S.; Panes, J.; Vranic, I.; Wang, W.; Niezychowski, W.; Vermeire, S.A.R.A.; Dewit, O.; Peeters, H.; Stehlik, J.; et al. A Phase 2 Study of Tofacitinib, an Oral Janus Kinase Inhibitor, in Patients with Crohn’s Disease. Clin. Gastroenterol. Hepatol. 2014, 12, 1485–1493.e2. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Kavanaugh, A.; Wicklund, J.; McInnes, I.B. Filgotinib, a Novel JAK1-Preferential Inhibitor for the Treatment of Rheumatoid Arthritis: An Overview from Clinical Trials. Mod. Rheumatol. 2022, 32, 1–11. [Google Scholar] [CrossRef]
- Fanizza, J.; D’Amico, F.; Lauri, G.; Martinez-Dominguez, S.J.; Allocca, M.; Furfaro, F.; Zilli, A.; Fiorino, G.; Parigi, T.L.; Radice, S.; et al. The Role of Filgotinib in Ulcerative Colitis and Crohn’s Disease. Immunotherapy 2023, 16, 59–74. [Google Scholar] [CrossRef]
- Abou Zahr, A.; Salama, M.E.; Carreau, N.; Tremblay, D.; Verstovsek, S.; Mesa, R.; Hoffman, R.; Mascarenhas, J. Bone Marrow Fibrosis in Myelofibrosis: Pathogenesis, Prognosis and Targeted Strategies. Haematologica 2016, 101, 660–671. [Google Scholar] [CrossRef]
- Cervantes, F.; Ross, D.M.; Radinoff, A.; Palandri, F.; Myasnikov, A.; Vannucchi, A.M.; Zachee, P.; Gisslinger, H.; Komatsu, N.; Foltz, L.; et al. Efficacy and Safety of a Novel Dosing Strategy for Ruxolitinib in the Treatment of Patients with Myelofibrosis and Anemia: The REALISE Phase 2 Study. Leukemia 2021, 35, 3455–3465. [Google Scholar] [CrossRef]
- Traboulsi, C.; Ayoub, F.; Silfen, A.; Rodriguez, T.G.; Rubin, D.T. Upadacitinib Is Safe and Effective for Crohn’s Disease: Real-World Data from a Tertiary Center. Dig. Dis. Sci. 2023, 68, 385–388. [Google Scholar] [CrossRef]
- Richeldi, L.; Du Bois, R.M.; Raghu, G.; Azuma, A.; Brown, K.K.; Costabel, U.; Cottin, V.; Flaherty, K.R.; Hansell, D.M.; Inoue, Y.; et al. Efficacy and Safety of Nintedanib in Idiopathic Pulmonary Fibrosis. N. Engl. J. Med. 2024, 370, 2071–2082. [Google Scholar] [CrossRef]
- King, T.E., Jr.; Bradford, W.Z.; Castro-Bernardini, S.; Fagan, E.A.; Glaspole, I.; Glassberg, M.K.; Gorina, E.; Hopkins, P.M.; Kardatzke, D.; Lancaster, L.; et al. A Phase 3 Trial of Pirfenidone in Patients with Idiopathic Pulmonary Fibrosis. N. Engl. J. Med. 2024, 370, 2083–2092. [Google Scholar] [CrossRef]
- Noble, P.W.; Albera, C.; Bradford, W.Z.; Costabel, U.; Glassberg, M.K.; Kardatzke, D.; King, T.E., Jr.; Lancaster, L.; Sahn, S.A.; Szwarcberg, J.; et al. Pirfenidone in Patients with Idiopathic Pulmonary Fibrosis (CAPACITY): Two Randomised Trials. Lancet 2011, 377, 1760–1769. [Google Scholar] [CrossRef] [PubMed]
- Richeldi, L.; Fernández Pérez, E.R.; Costabel, U.; Albera, C.; Lederer, D.J.; Flaherty, K.R.; Ettinger, N.; Perez, R.; Scholand, M.B.; Goldin, J.; et al. Pamrevlumab, an Anti-Connective Tissue Growth Factor Therapy, for Idiopathic Pulmonary Fibrosis (PRAISE): A Phase 2, Randomised, Double-Blind, Placebo-Controlled Trial. Lancet Respir. Med. 2020, 8, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Verstovsek, S.; Mesa, R.A.; Gotlib, J.; Levy, R.S.; Gupta, V.; DiPersio, J.F.; Catalano, J.V.; Deininger, M.; Miller, C.; Silver, R.T.; et al. A Double-Blind, Placebo-Controlled Trial of Ruxolitinib for Myelofibrosis. N. Engl. J. Med. 2024, 366, 799–807. [Google Scholar] [CrossRef] [PubMed]
- Rinella, M.E.; Lieu, H.D.; Kowdley, K.V.; Goodman, Z.D.; Alkhouri, N.; Lawitz, E.; Ratziu, V.; Abdelmalek, M.F.; Wong, V.W.-S.; Younes, Z.H.; et al. A Randomized, Double-Blind, Placebo-Controlled Trial of Aldafermin in Patients with NASH and Compensated Cirrhosis. Hepatology 2024, 79, 674–689. [Google Scholar] [CrossRef]

{kind=link}
| Medication | Target | Status | Tissue | Outcomes | Main Side Effects |
|---|---|---|---|---|---|
| Nintedanib [71] | RTKs | Approved | IPF | Reduction of FVC decline | Diarrhoea, nausea |
| Pirfenidone [72,73] | TGF-βs | Approved | IPF | Reduction of FVC decline | Nausea, rash, headache, dyspepsia |
| Pamrevlumab [74] | CTGF | Phase III ongoing (NCT03955146) | IPF | Reduction of FVC decline * | Fatigue, urinary tract infections * |
| Ruxolitinib [75] | JAK1/JAK2 | Approved | Myelofibrosis | Reduction of spleen size and debilitating symptoms, Increase of OS | Anaemia, thrombocytopenia |
| AGMB-129 | TGFβR1 | Phase II ongoing (NCT05843578) | Fibro-stenotic CD | Unknown | Unknown |
| Pegbelfermin [57] | FGF21 | Phase II (NCT03486899) | Liver fibrosis | Primary endpoint not met | Unknown |
| Aldafermin [76] | FGF19 | Phase II (NCT03912532) | Liver fibrosis | Improvement of liver stiffness and aminotransferases | Diarrhoea |
| RXC008 | ROCK | Phase II ongoing (RD783.35406) | Fibro-stenotic CD | Unknown | Unknown |
| RXC007 | ROCK2 | Phase II ongoing (NCT05570058) | IPF | Unknown | Unknown |
| TEV-48574 [56] | TL1A | Phase II (NCT05499130) | IBD | Unknown | Unknown |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ke, B.-J.; Dragoni, G.; Matteoli, G. Fibroblast Heterogeneity in Inflammatory Bowel Disease. Int. J. Mol. Sci. 2024, 25, 13008. https://doi.org/10.3390/ijms252313008
Ke B-J, Dragoni G, Matteoli G. Fibroblast Heterogeneity in Inflammatory Bowel Disease. International Journal of Molecular Sciences. 2024; 25(23):13008. https://doi.org/10.3390/ijms252313008
Chicago/Turabian StyleKe, Bo-Jun, Gabriele Dragoni, and Gianluca Matteoli. 2024. "Fibroblast Heterogeneity in Inflammatory Bowel Disease" International Journal of Molecular Sciences 25, no. 23: 13008. https://doi.org/10.3390/ijms252313008
APA StyleKe, B.-J., Dragoni, G., & Matteoli, G. (2024). Fibroblast Heterogeneity in Inflammatory Bowel Disease. International Journal of Molecular Sciences, 25(23), 13008. https://doi.org/10.3390/ijms252313008