1. Introduction
As the global population continues to grow at an unprecedented rate, the demand for energy is rapidly increasing. This rising energy demand places significant pressure on nonrenewable resources, particularly fossil fuels, which are both limited and environmentally harmful [
1,
2]. To meet this escalating need, it is essential to explore and develop renewable energy sources that are not only efficient but also sustainable and environmentally friendly. Among these, harnessing solar energy to generate clean, renewable electricity presents a promising pathway toward reducing our reliance on fossil fuels and mitigating the environmental impacts of energy production [
3,
4,
5]. To achieve this vision, researchers are actively seeking a way to replicate the natural process of photosynthesis, in which sunlight is absorbed by chlorophyll molecules to drive the conversion of light energy into chemical energy. In photosynthesis, sunlight excites electrons in chlorophyll, which are then transferred to electron-deficient molecules, with electrons from water molecules filling the void left behind, initiating a series of chemical reactions that ultimately produce energy [
6,
7]. By mimicking this process, it may be possible to develop artificial systems for efficient solar energy conversion.
In our previous work with para-phenylene dimers, we demonstrated that the electronic properties of para-phenylene systems can be tuned by introducing specific substitutions and coupling them with π-acceptors [
8]. We found that the band gap of an unsubstituted para-phenylene (4.13 eV) could be reduced to 2.9 eV when linked to a diphenylamine-substituted para-phenylene and a π-acceptor. Furthermore, we observed significant fluorescence quenching when the para-phenylene dimer was mixed with reduced graphene oxide (RGO), suggesting a phenomenon of excited-state electron transfer. This finding prompted us to explore the effects of RGO on polypyridyl Ru(II)-based systems, specifically aiming to understand and control the interactions between these materials. To facilitate efficient charge transport and improve system mobility, we designed our Ru(II) complex with a conjugated backbone, which enhances π–π interactions. The primary goal of this study was to investigate the electron and energy transfer dynamics in novel Ru(II) polypyridyl oligomers, particularly in the presence of reduced graphene oxide (RGO). Polypyridyl ligands, particularly those with electron-donating groups like -NH
2 or -OCH
3, can donate electron density to the metal center via their nitrogen lone pairs. This can alter the oxidation state of the ruthenium center, often stabilizing a lower oxidation state (such as Ru
2+) and influencing the electronic structure of the complex. These ligands can absorb light, and their electronic structure allows them to participate in ligand-to-metal charge transfer (LMCT) transitions, where an electron is excited from the ligand (usually the nitrogen lone pair of the pyridine) to the metal center. In ruthenium complexes, these LMCT transitions can often occur in the visible-to-UV region and can significantly influence optical properties, contributing to the color and light absorption characteristics of the complex. Conversely, the ruthenium metal center can also participate in metal-to-ligand charge transfer (MLCT) transitions, where electrons are excited from the metal’s d-orbitals to the π* orbitals of the ligands. The presence of polypyridyl ligands enhances the possibility of these MLCT transitions, which are typically responsible for strong visible absorption bands in ruthenium-based complexes. Ruthenium polypyridyl complexes exhibit luminescence properties, often with long-lived excited states due to the strong spin–orbit coupling of the ruthenium metal center. The ligands can also modulate the emission wavelength and quantum yield by influencing the metal’s electronic structure and the rates of non-radiative decay. This is important for applications such as catalysis, electrochemical sensing, or energy storage. Therefore, the redox potentials of ruthenium-based complexes can be tuned by modifying the ligand structure. These interactions are crucial for the development of more-efficient energy conversion systems, such as light-driven catalytic water oxidation in photovoltaic cells. The nature of the interactions between the oligomers and RGO can vary from covalent bonding to electrostatic interactions and can occur over distances ranging from a short range to several nanometers [
9,
10,
11,
12]. Understanding these interactions will provide insights that can guide the design of more-effective devices for solar energy conversion. The study aims to evaluate the potential of graphene and graphene-based hybrid materials as electron-accepting components in energy systems. Given graphene’s excellent electrical conductivity, mechanical strength, and availability, it is an ideal material for creating efficient hybrid composites. These materials are expected to improve the solubility and processability of graphene, making it a versatile component in various applications, including energy storage and solar energy harvesting [
13,
14,
15]. In this context, we developed two graphene-hybrid composites to study electron/energy transfer interactions. Specifically, we modified Hummers’ and Offeman’s method to oxidize graphite and employed a reducing agent to obtain RGO. We propose that RGO can serve as an effective alternative to TiO
2 in future artificial light-harvesting systems, enhancing their efficiency and performance.
3. Materials and Methods
All reagents were purchased from Fisher Scientific (Raleigh, NC, USA) and used as received unless otherwise stated. Proton nuclear magnetic resonance spectra (1H NMR) were recorded on a Varian (500 MHz) spectrophotometer. Chemical shifts δ were reported in parts per million (ppm) units, with tetramethyl silane (TMS at 0.0 ppm) as an internal reference. All reagents were purchased from Fisher Scientific and used as received unless otherwise stated. C642 is commercially available and was purchased from Fisher Scientific. Proton nuclear magnetic resonance spectra (1H NMR) were recorded on a Varian (500 MHz) spectrophotometer. Chemical shifts δ were reported in parts per million (ppm) units with tetramethyl silane (TMS at 0.0 ppm) as an internal reference. All reagents were purchased from Fisher Scientific and used as received unless otherwise stated.
3.1. Synthesis of Graphene Oxide (GO)
A mixture of graphite (5 g), NaNO3 (5 g), and H2SO4 (230 mL) was stirred in an ice bath. KMnO4 (15 g) was slowly added while stirring. The rate of addition was controlled to prevent the reaction temperature from exceeding 20 °C. The mixture was then transferred to a 35 °C oil bath and stirred for about 30 min, forming a thick paste. Subsequently, deionized water (130 mL) was added gradually, causing an increase in temperature. After 15 min, the mixture was further treated with de-ionized water (700 mL) and 30% H2O2 solution (50 mL). The warm solution was then filtered and washed with deionized water until neutralized (pH 7) and dried under vacuum.
3.2. Synthesis of Reduced Graphene Oxide (RGO) by Sodium Borohydride
GO (1 g) was placed in a 500 mL round-bottom flask, which was followed by the addition of deionized water (250 mL). After stirring and ultra-sonication for 30 min, sodium borohydride (5 g) was gradually added to the solution. Sodium borohydride was chosen for its selective, efficient, and safe reduction of GO to rGO, which leads to significant improvements in electrical conductivity, while still allowing for tunable surface chemistry. Sodium borohydride reduction reduces many oxygen-containing groups and causes partial reduction where the surface of rGO can still undergo chemical functionalization, making rGO suitable for applications like sensors, batteries, and supercapacitors, where specific surface interactions are needed.
The mixture was stirred for 4 h at room temperature and then filtered. The filter cake was washed with deionized water until the filtrate was clear. The remaining black solid was dried under vacuum.
3.3. Synthesis of 4,7-Dibromobenzo[1,2,5]Thiadiazole (1)
A mixture of 1,2,5-benzothiadiazole (20.0 g) in aq. HBr (48%, 60 mL) was heated to reflux with stirring, while Br2 (22.6 mL) was added slowly over 1 h. Towards the end of the addition, the mixture became a suspension. To facilitate stirring, aq. HBr (48%, 40 mL) was added. The mixture was heated to reflux for an additional 2 h then filtered while hot, cooled, filtered again, and washed well with deionized water. The compound was dried over Na2SO4 and recrystallized with MeOH to give a white needle product (38.0 g, 88%). 1H NMR (500 MHz, chloroform-d) δ 7.75–7.73 (s, 3H).
3.4. Synthesis of 3,6-Dibromobenzene-1,2-Diamine (2)
To a suspension of 1 (5.0 g) in EtOH (170 mL), NaBH4 (11.4 g) was added portionwise at 0 °C. The mixture was then stirred for 20 h at room temperature. Afterwards, the solvent was removed under vacuum and deionized water (100 mL) was added. The mixture was extracted with Et2O. The organic phase was washed with saturated aq. NaCl solution and dried over Na2SO4. The compound was then dried under vacuum to give a white solid product (3.9 g, 87%). 1H NMR (500 MHz, chloroform-d) δ 6.97–6.76 (s, 2H), 3.94–3.80 (s, 4H).
3.5. Synthesis of 1,10-Phenanthroline-5,6-Dione (3)
Concentrated H2SO4 (20 mL) and nitric acid (10 mL) were added dropwise to a mixture of 1,10-phenanthroline (1.00 g) and KBr (5.95 g) at 0 °C. The mixture was refluxed at 80 °C for 2 h and then cooled to room temperature. The contents of the reaction flask were diluted with deionized water (400 mL) and neutralized with sodium bicarbonate (NaHCO3). The product was extracted with methylene chloride and dried over anhydrous MgSO4. After all solvents were removed under vacuum, the compound was recrystallized with MeOH to give a yellow solid (1.1 g, 95%). 1H NMR (500 MHz, chloroform-d) δ 9.25–9.08 (s, 2H), 8.61–8.33 (m, 2H), 7.62–7.58 (s, 2H).
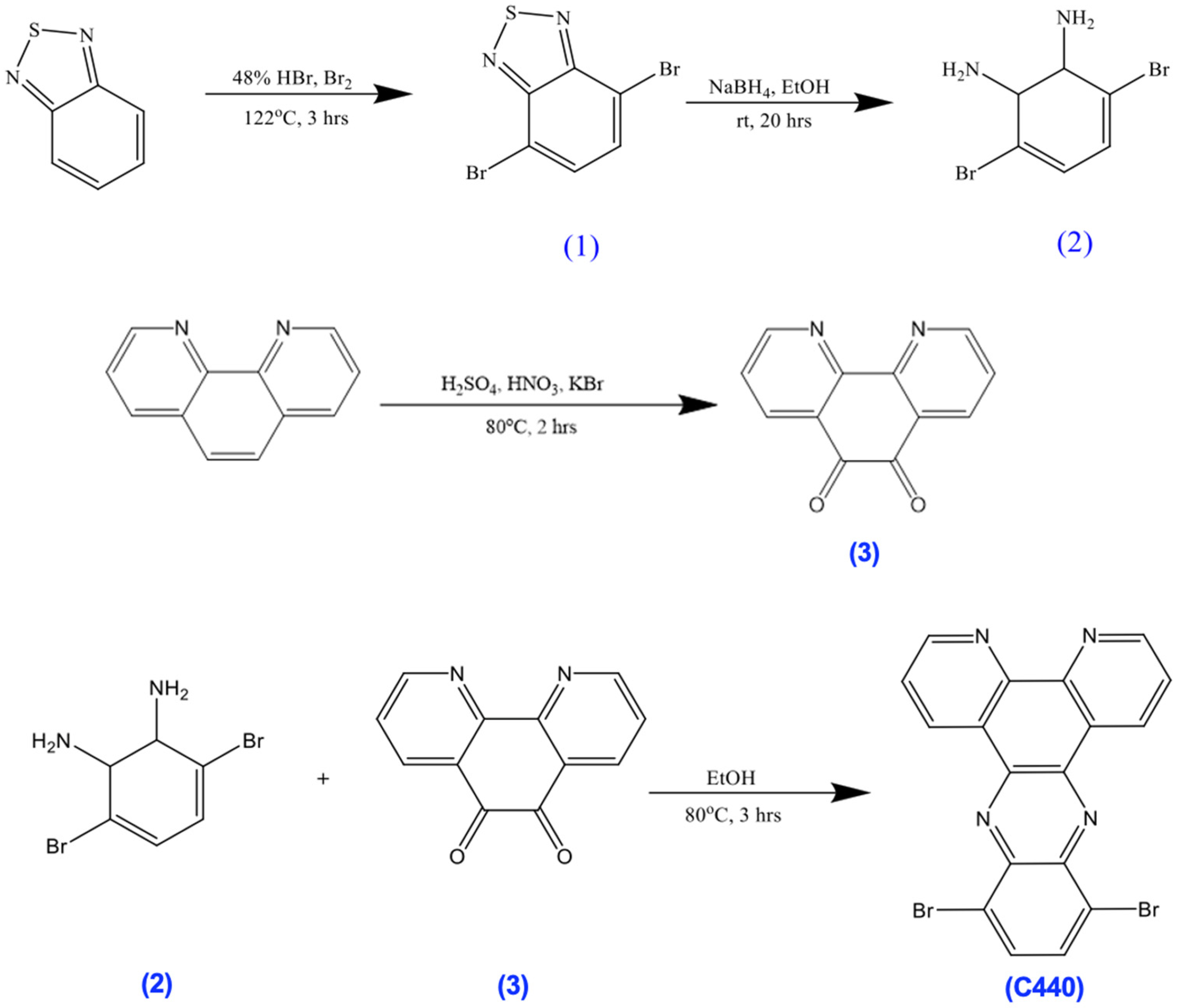
3.6. Synthesis of 10,13-Dibromodipyrido[3,2-a:2,3-c]Phenazine (C440)
A mixture of 2 (0.53 g) and 3 (0.42 g) in EtOH (25 mL) was refluxed and stirred for 3 h. The reaction mixture was allowed to cool to room temperature, vacuum filtered, and then washed with (CH3)2CO (20 mL) and Et2O (40 mL) to give a yellow solid (0.364 g, 91%). 1H NMR (500 MHz, chloroform-d) δ 9.79–9.67 (s, 2H), 9.37–9.27 (s, 2H), 8.16–8.05 (s, 2H), 7.89–7.79 (s, 2H).
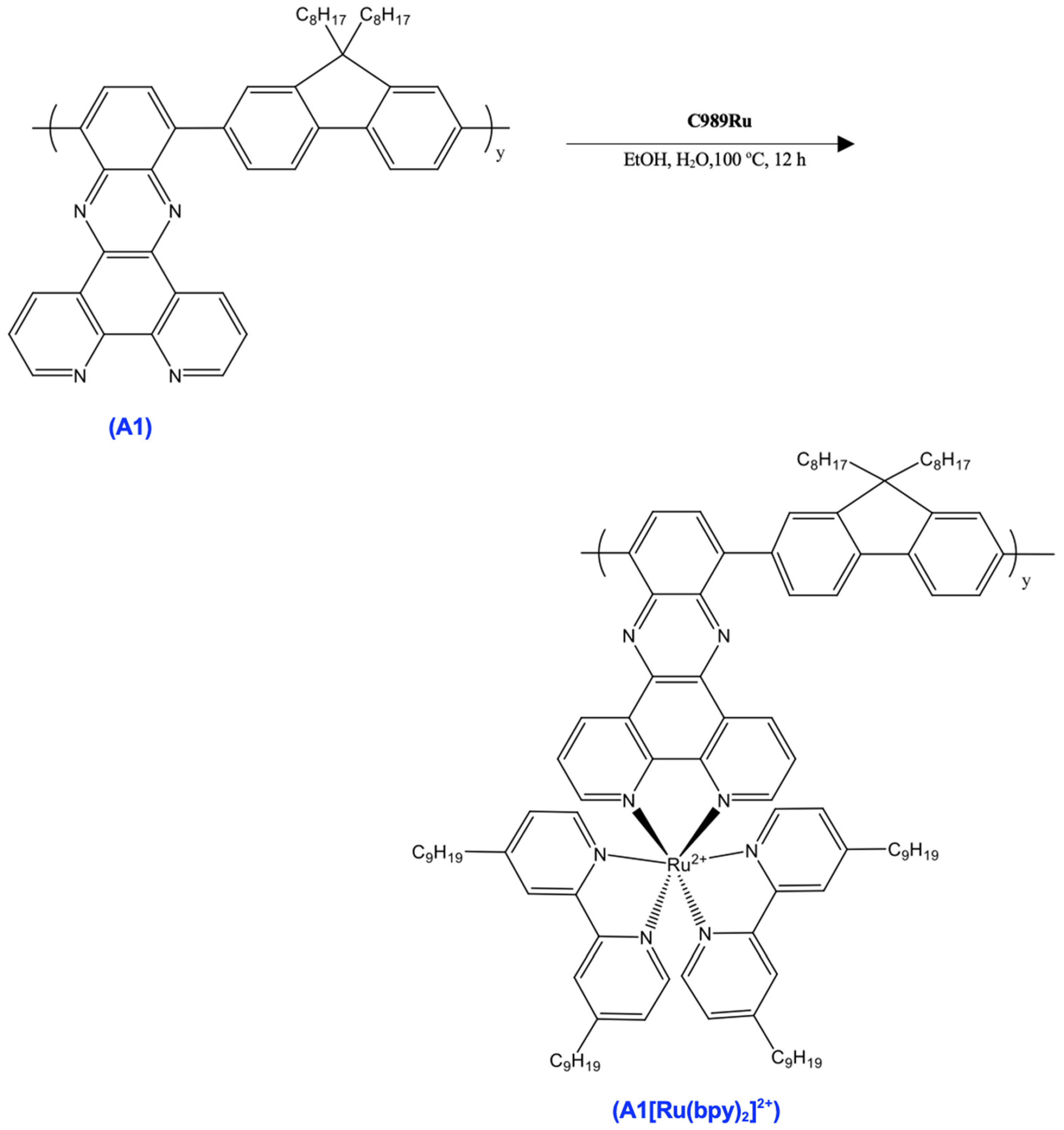
3.7. Synthesis of A1
A mixture of C440 (100 mg), C642 (100 mg), Pd(PPh3)4 (10 mg), and Cs2CO3 (276 mg) in DMF (4 mL) was stirred at 120 °C for 36 h under dry nitrogen. The reaction mixture was allowed to cool to room temperature then precipitated in methanol (MeOH). The collected solid was washed with MeOH and deionized water, then with MeOH again. The crude product was extracted with acetone (200 mL) for 3 days in a Soxhlet apparatus to remove impurities. The residual solid in the thimble was dried under vacuum to give an orange solid (132 mg). 1H NMR (500 MHz, Chloroform-d) δ 9.03–8.94 (ddd, 3H), 8.88–8.82 (dd, 1H), 8.30–8.24 (dd, 1H), 8.09–8.02 (m, 2H), 8.01–7.82 (m, 4H), 7.74–7.68 (t, 2H), 7.66–7.60 (dd, 1H), 7.58–7.50 (td, 1H), 7.50–7.43 (td, 1H), 2.19–2.12 (m, 2H), 1.98–1.91 (m, 2H), 1.53–1.44 (m, 4H), 1.38–1.21 (m, 21H), 0.90–0.82 (m, 6H).
3.8. Synthesis of C989Ru
A mixture of RuCl3·3H2O (84 mg), LiCl (92 mg), and 4,4′-Dinonyl-2,2′-dipyridyl (262 mg) in DMF (5 mL) was heated to 120 °C for 8 h under dry nitrogen. After the removal of the solvent in vacuum, the resulting purple solid was dissolved in DCM and washed with deionized water (2 × 50 mL). After the evaporation of the DCM, the resulting solid was washed with hexane (4 × 40 mL) upon sonication. Subsequent removal of the hexane resulted in a purple solid (200 mg). 1H NMR (500 MHz, Chloroform-d) δ 8.67–8.63 (d, 1H), 8.63–8.57 (d, 1H), 7.22–7.16 (dd, 1H), 3.43–3.37 (t, 2H), 1.65–1.57 (q, 2H), 1.30–1.22 (m, 12H), 0.90–0.82 (m, 3H).
3.9. Synthesis of A1[Ru(bpy)2]2+
A mixture of A1 and C989Ru in EtOH (5 mL) and deionized water (5 mL) was refluxed overnight under dry nitrogen. The reaction mixture was cooled to room temperature and the solvent was evaporated. The residue was then suspended in EtOH and filtered. The filtrate was precipitated in MeOH, and the solid was collected and dried under vacuum (384 mg). 1H NMR (500 MHz, chloroform-d) δ 8.67–8.63 (d, 1H), 8.63–8.57 (d, 1H), 7.22–7.16 (dd, 1H), 3.43–3.37 (t, 2H), 1.86–1.80 (t, 1H), 1.65–1.57 (q, 2H), 1.35–1.20 (m, 17H), 0.90–0.82 (m, 4H).
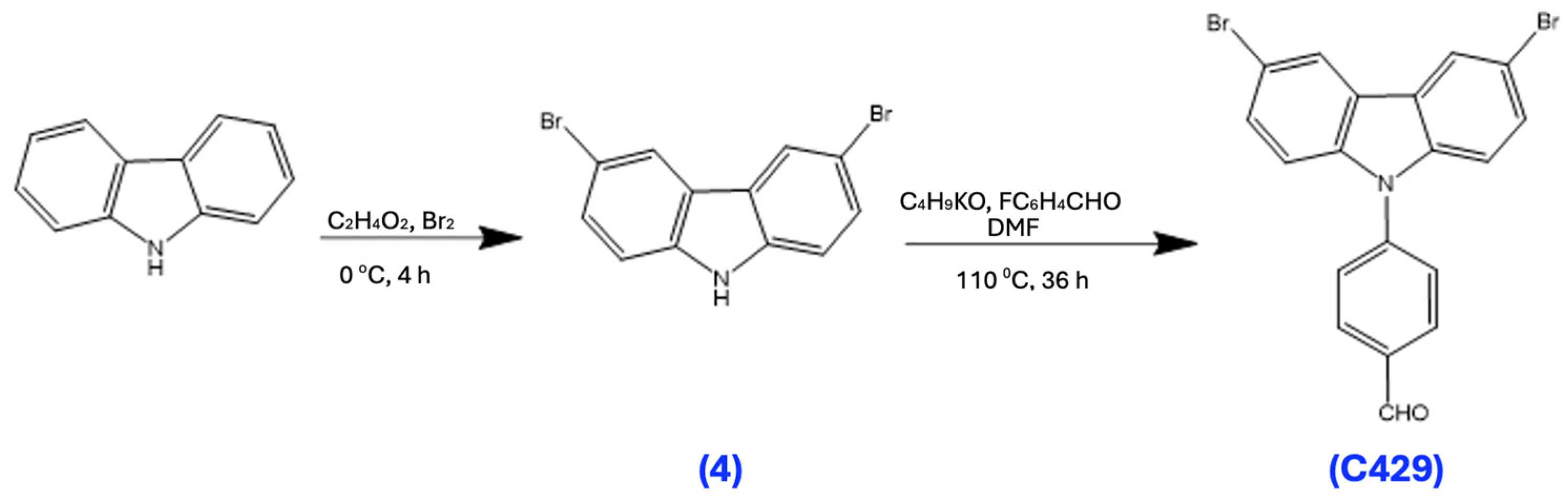
3.10. Synthesis of 3,6-Dibromocarbazole (4)
To a solution of carbazole (10 g) in acetic acid and sodium acetate buffer solution, bromine (7.5 mL) was added dropwise at 0 °C and stirred for 4 h. Afterward, a 2M NaOH aqueous solution was poured into the reaction mixture. The crude product was filtered and washed with deionized water and then recrystallized from ethanol, resulting in a white solid (15.4 g, 79%). 1H NMR (500 MHz, Chloroform-d) δ 8.12–8.08 (d, 2H), 7.39–7.29 (m, 3H), 7.28–7.24 (s, 1H).
3.11. Synthesis of C429
A mixture of compound 4 (8.4 g) and potassium tert-butoxide (5.5 g) in anhydrous DMF (200 mL) was heated at 110 °C for 30 min, then 4-fluorobenzaldehyde (7 mL) was added dropwise to the solution, followed by heating and stirring for an additional 36 h. The mixture was then cooled to room temperature and poured into ice water, then filtered and dried. The purified yellow product was obtained after recrystallization from acetone/water. 1H NMR (500 MHz, Chloroform-d) δ 10.20–10.09 (m, 1H), 8.26–8.00 (m, 4H), 7.80–7.67 (dd, 2H), 7.59–7.50 (dq, 2H), 7.39–7.29 (m, 2H).
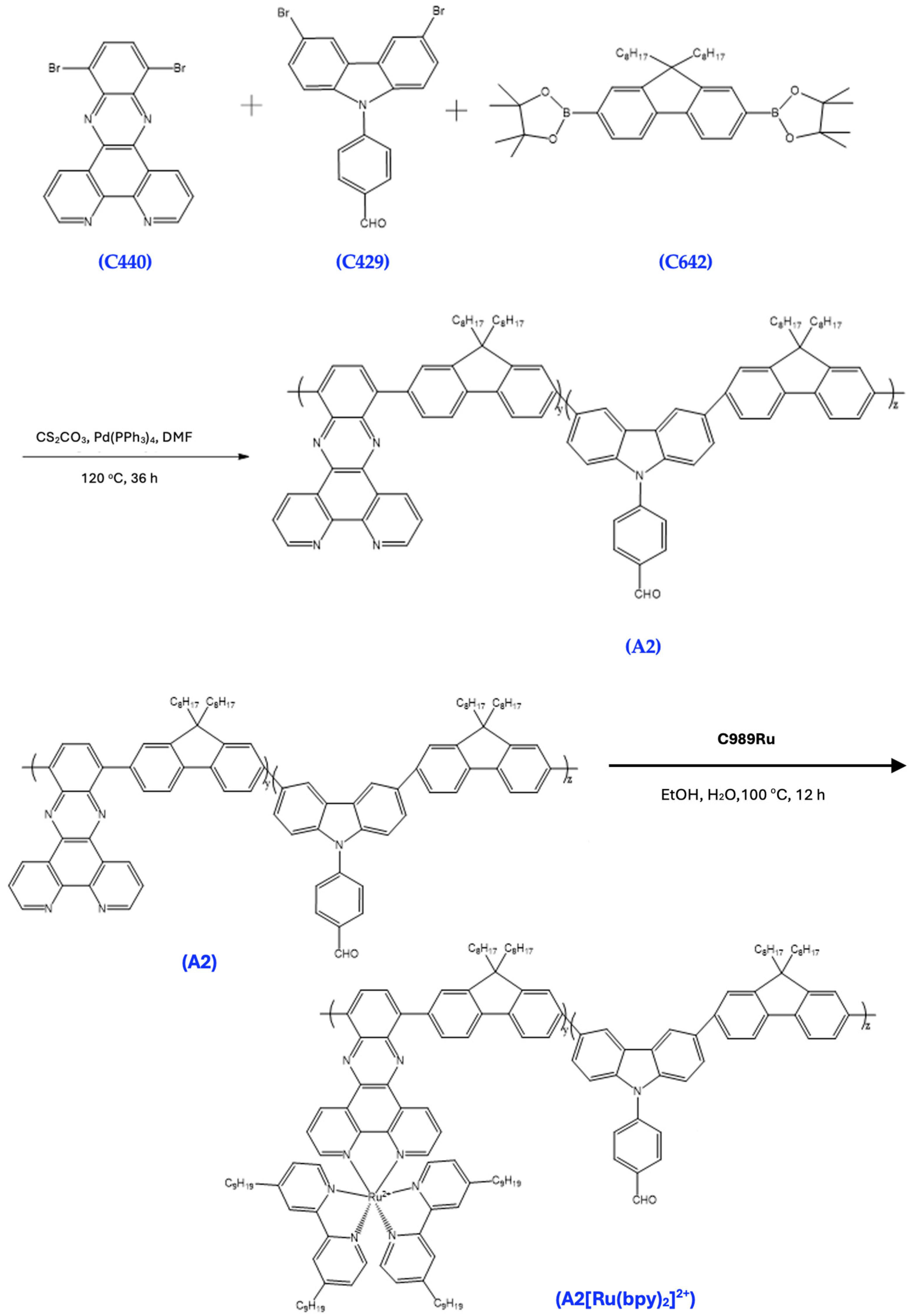
3.12. Synthesis of A2
A mixture of C440 (100 mg), C642 (100 mg), C429 (97 mg), Pd(PPh3)4 (10 mg), and Cs2CO3 (276 mg) in 4 mL DMF was stirred at 120 °C for 36 h under dry nitrogen. The reaction mixture was allowed to cool to room temperature and then precipitated in MeOH. The collected solid was washed with MeOH and deionized water, then with MeOH again. The collected crude product was extracted with acetone (200 mL) for 3 days in a Soxhlet apparatus to remove impurities. The residual was dried under vacuum to give an orange solid (143 mg). 1H NMR (500 MHz, Chloroform-d) δ 9.00–8.94 (dd, 1H), 8.26–8.17 (m, 2H), 8.15–8.07 (m, 2H), 8.05–7.95 (m, 2H), 7.94–7.83 (m, 2H), 7.80–7.68 (m, 2H), 3.82–3.75 (t, 1H), 3.26–3.16 (p, 2H), 2.95–2.88 (t, 1H), 2.27–2.20 (m, 1H), 2.11–2.04 (m, 1H), 1.65–1.53 (m, 3H), 1.44–1.34 (m, 2H), 1.31–1.21 (m, 17H), 0.90–0.82 (m, 6H).
3.13. Synthesis of A2[Ru(bpy)2]2+
A mixture of A2 and C989Ru in EtOH (5 mL) and H2O (5 mL) was refluxed overnight under dry nitrogen. The reaction mixture was cooled to room temperature and the solvent was evaporated. The residue was then suspended in EtOH and filtered. The filtrate was then precipitated in MeOH, and the solid was collected and dried under vacuum (150 mg). 1H NMR (500 MHz, chloroform-d) δ 8.67–8.63 (d, 1H), 8.63–8.57 (d, 1H), 8.10–8.04 (d, 1H), 7.91–7.81 (m, 2H), 7.80–7.73 (ddd, 1H), 7.22–7.16 (dd, 1H), 3.43–3.37 (t, 2H), 1.86–1.80 (t, 2H), 1.65–1.57 (q, 2H), 1.36–1.20 (m, 24H), 0.90–0.82 (m, 6H).
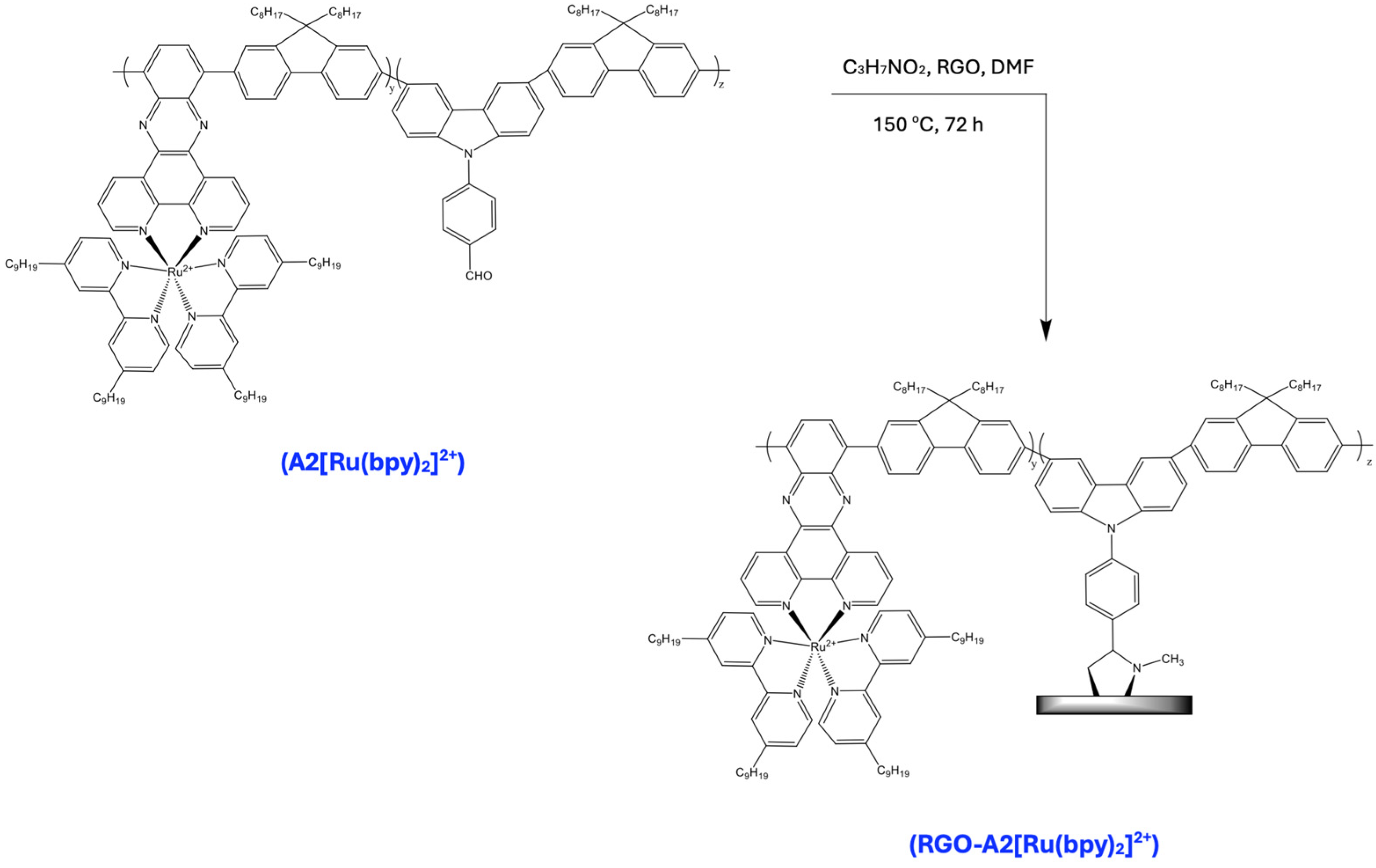
3.14. Synthesis of RGO-A2[Ru(bpy)2]2+
A mixture of A2[Ru(bpy)2]2+ (100 mg), RGO (5 mg), and sarcosine (5 mg) in anhydrous DMF (50 mL) was refluxed for 72 h. Afterward, the mixture was cooled to room temperature and 100 mL of methanol was added. The crude product was collected by filtration using a polycarbonate film (0.45 μm), washed with ethanol and dichloromethane, and then dissolved in a small amount of DMF, before undergoing precipitation from methanol. The procedure was repeated at least three times. The gray-black solid obtained was dried under vacuum overnight.
3.15. Computational Methods
The calculations were performed using FHI-aims via Duke University’s computer cluster. FHI-aims uses a numeric atomic orbital (NAO) basis set, which is efficient and well-suited for large systems and periodic calculations. The NAO basis sets in FHI-aims are highly efficient because they are optimized for plane-wave expansion and do not require the explicit definition of Gaussian functions. They allow for a more systematic improvement in accuracy, with fewer computational resources for large systems. These studies were usually calculated under light relaxation settings with bfg of E-2, using either PBE or HF functions. PBE is a generalized gradient approximation (GGA) functional, widely used in DFT calculations for many materials and molecular systems. It is relatively efficient and provides a good balance between accuracy and computational cost. HF was chosen for its ability to provide a more rigorous treatment of the exchange interaction and is useful for validating the results obtained using PBE and PBE0. HF allowed the researchers to cross-check the results and ensure that the functional dependence did not significantly alter the outcome of the calculations. It is particularly suitable for systems with weak intermolecular interactions, such as van der Waals interactions or when researchers want to perform large-scale computations without sacrificing too much accuracy. PBE tends to underestimate the bandgap of semiconductors and systems with significant electron correlation, but it is still a widely used functional due to its efficiency. Geometric optimization was run first, followed by molecular orbital calculations on the relaxed molecule for both Gaussian and FHI-aims calculations. PBE functions were followed by single-point conformation conducted via PBE0. PBE0 was chosen for single-point energy calculations because of its improved accuracy for electronic structure properties (such as HOMO–LUMO gaps, ionization potentials, or electron affinities). In particular, PBE0 is known to give more-accurate electronic band gaps and is often used for systems where electronic properties are crucial. The HF functions were followed by single-point conformation conducted via HF again. Gaussian 09 is a more widely used software compared to the relatively new FHI-aims software (
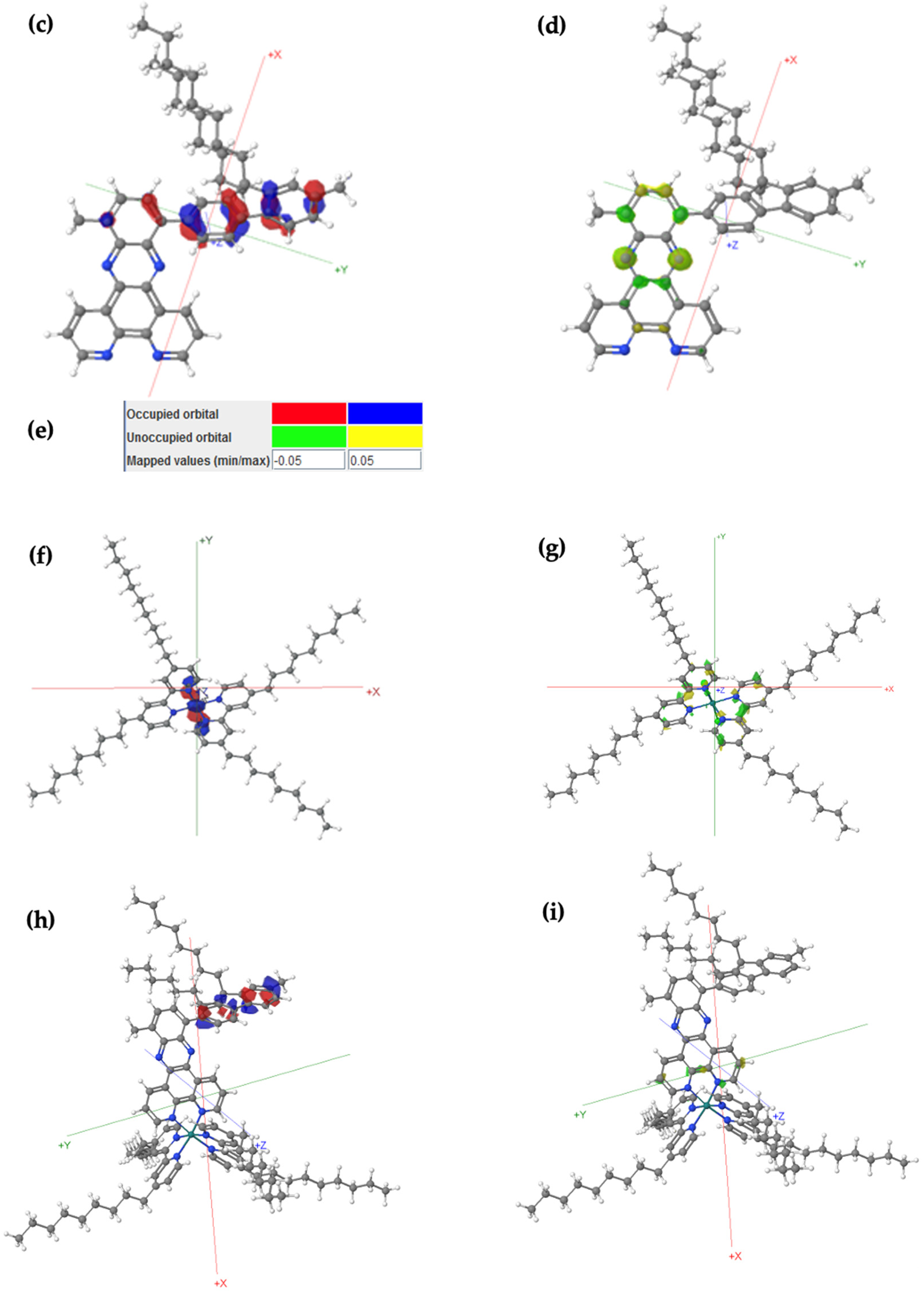
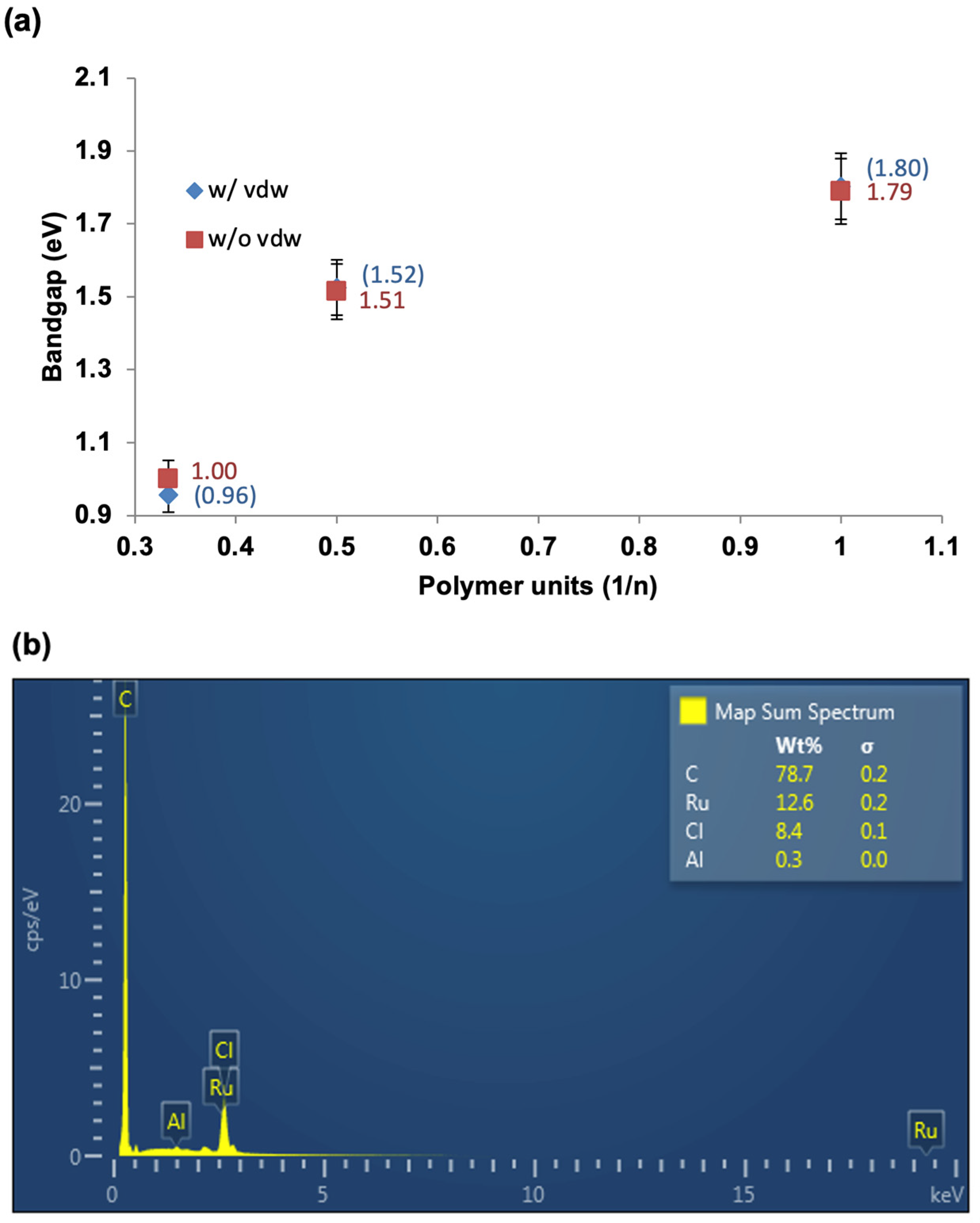
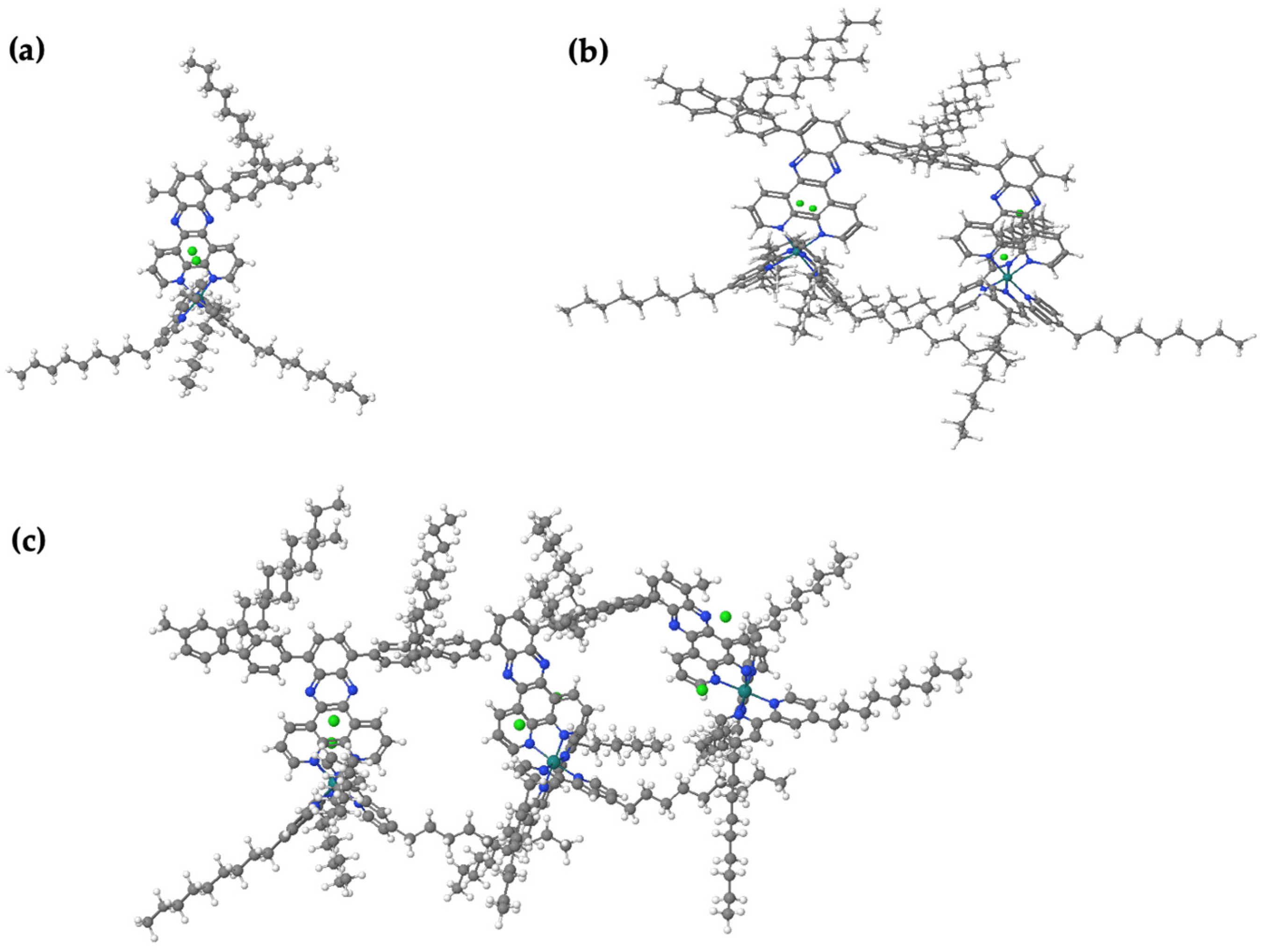
https://fhi-aims.org accessed on 12 May 2016), so HF and PBE calculations are run on both packages for comparison purposes. Gaussian typically uses Gaussian-type orbitals (GTOs), which are popular and well-established. GTOs are more compact in real space, making them more efficient for localized systems (especially small-to-medium-sized molecules) when compared to plane-wave-based approaches. The polymer properties for bandgaps were calculated by plotting results for oligomers with increasing chain length against 1/n, where n represents the number of monomer repeat units. Note that the minimum number of repeat units is three for a molecule to be considered a polymer. The data were extrapolated using second-order polynomial fits, and the extrapolating energy levels were calculated for monomers through trimers for A1[Ru].
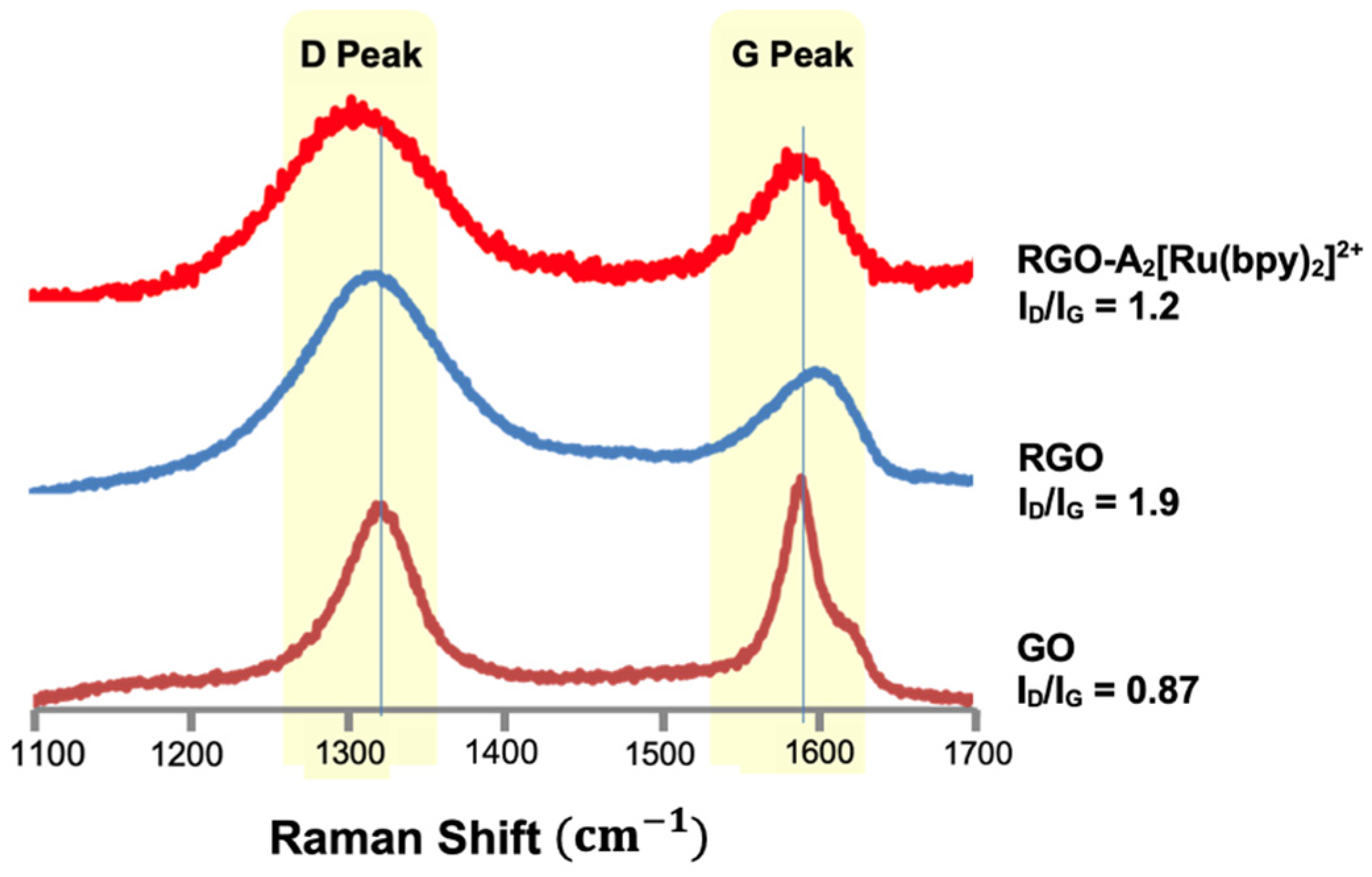
3.16. Raman Analysis
A sample was prepared from a stock solution of GO or RGO in ethanol (EtOH) (3 mg/10 mL) and was drop cast onto a silicon wafer. The wafer was allowed to dry for approximately 30 min. Afterwards, a 785 nm excitation laser attached to a Renishaw Ramascope instrument with an Olympus BH2-UMA microscope was used to obtain the spectra. The typical Raman spectrum of graphene commonly exhibits two major bands defined as the D and G bands. The D band contributes to transformation of sp2 carbons to sp3 carbons due to covalent functionalization [
32,
33,
34,
35]. The average ratio of the D-to-G peak (ID/IG) was compared to determine the degree of functionalization on the surface.
3.17. Atomic Force Microscopy Studies
A topographic image of RGO was captured using tapping mode atomic force microscopy (AFM) on the model Nanoscope III, from Digital Instruments, Inc., Santa Barbara, CA, USA. The sample was prepared using a diluted stock solution of RGO in EtOH (1 mg/10 mL) and was drop cast onto a mica wafer. The wafer was allowed to dry for approximately 30 min.
3.18. XPS Studies
XPS analysis was carried out using a monochromatic microspot X-ray beam originating from the A1 Kα source with a spot diameter of ~600 μm from a Surface Science Instrument 100-06 spectrometer (Kratos Analytical Inc., Nanuet, NY, USA). The XPS data were analyzed with a least-square fitting routine and into individual Gaussian peaks, assuming a linear background over the energy range of the fit. Deconvolution was performed with the constraint of equal full width at half maximum (FWHM) value for the prominent lines of different spectra.
3.19. Scanning Electron Microscopy Studies
SEM was performed on a Nova NanoSEM 600 instrument, (Schaumburg, IL, USA) and the sample was prepared from a dilute solution of RGO (1 mg/10 mL) deposited onto a silicon wafer and examined.
4. Conclusions
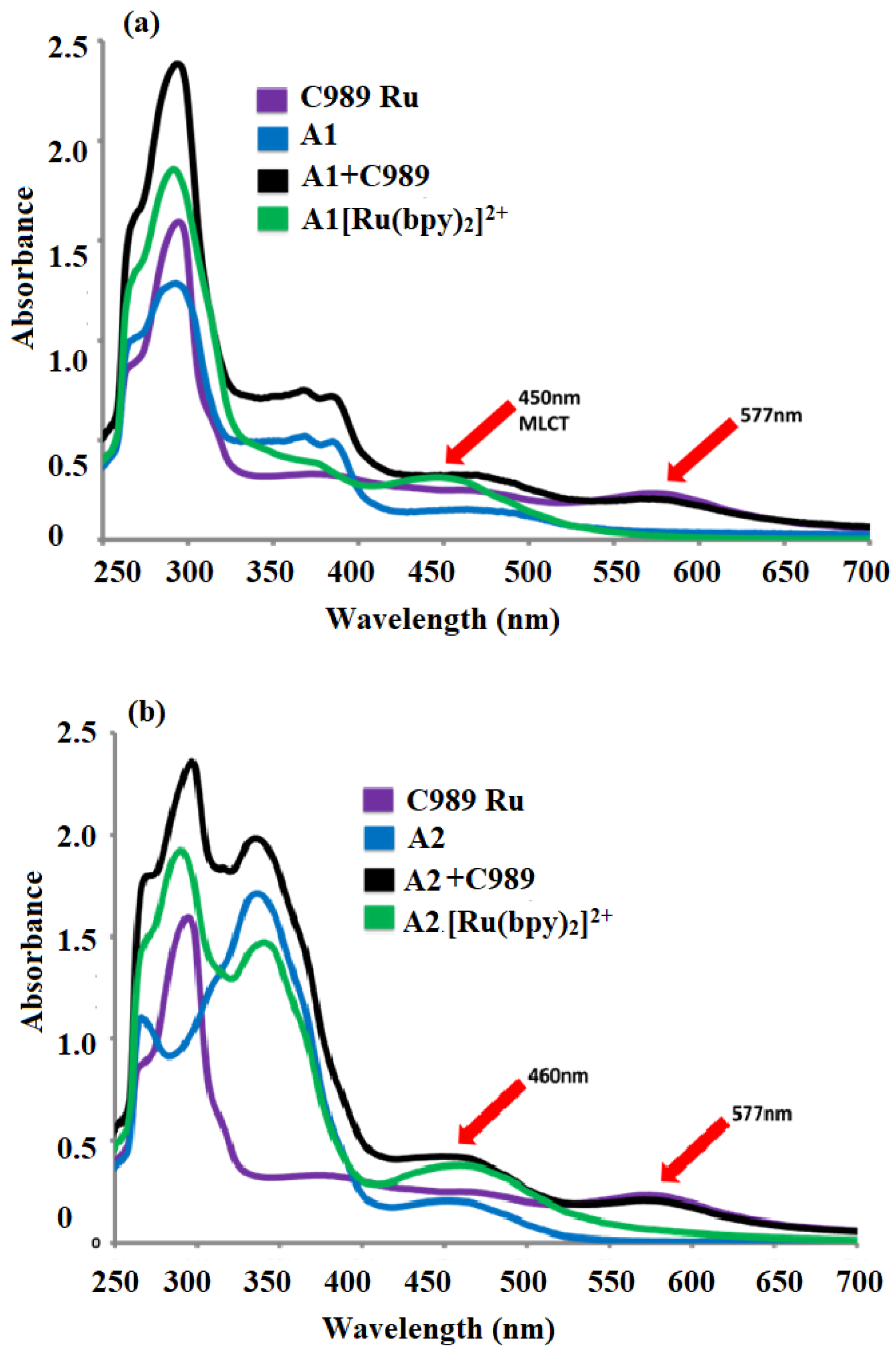
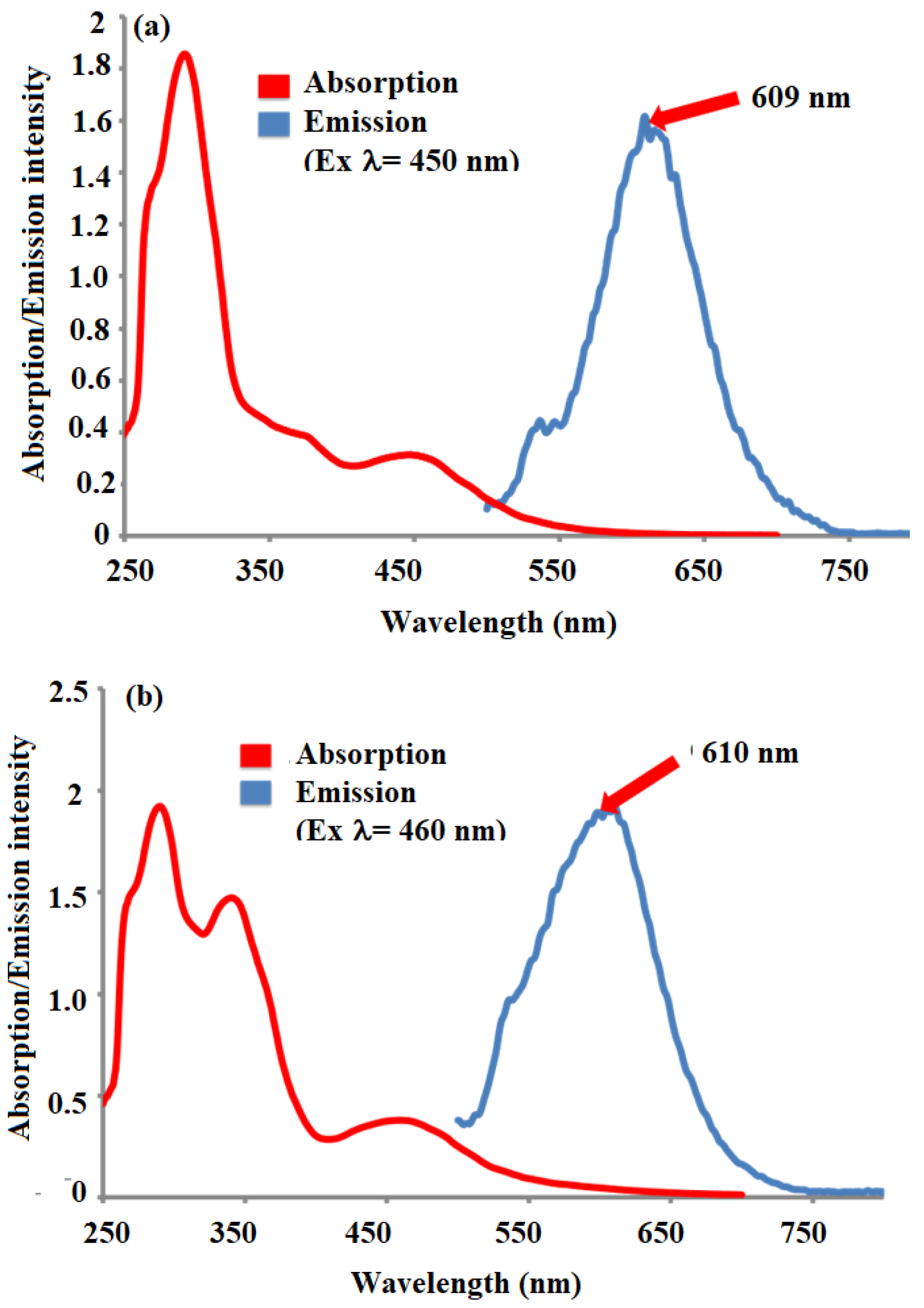
We have successfully developed and outlined the synthetic strategies employed to obtain the novel conjugated oligomer A2[Ru(bpy)2]2+, a complex incorporating a Ru(II) polypyridyl coordination with 2,2′-bipyridine (bpy) ligands. The synthesis of this oligomer involved a series of steps, culminating in a carefully controlled reaction to ensure the desired structural integrity and electronic properties of the final product. A2[[Ru(bpy)2]2+ was systematically studied using a variety of spectroscopic and electrochemical techniques to evaluate its physical, electronic, and optoelectronic properties. The fluorescence quenching studies revealed strong evidence of electron transfer through the conjugated oligomer backbone. These findings suggest that the oligomer possesses good charge transport characteristics, which are critical for potential applications in fields such as organic photovoltaics, sensors, and electronic devices. UV–Visible (UV-Vis) spectroscopy was employed to assess the optical properties of A2[Ru(bpy)2]2+, and it was observed to exhibit well-defined absorption bands typical of Ru(II) polypyridyl complexes. Additionally, the calculated optical band gap of A2[Ru(bpy)2]2+ was found to be 1.98 eV, a value consistent with similar structures in the literature and indicative of its potential for efficient light absorption and charge separation in various optoelectronic applications. Cyclic voltammetry (CV) measurements provided further insight into the electrochemical behavior of Ru(bpy)2]2+ revealing key redox processes associated with the Ru(II/III) and bipyridine ligand. These data not only reinforce the stability of the complex but also suggest its suitability for electron transfer applications in hybrid materials. Additionally, transient absorption experiments were conducted, providing complementary data on the excited-state dynamics and kinetics of electron transfer in the conjugated structure. In an effort to enhance the properties of Ru(bpy)2]2+ for potential hybrid applications, a 1,3-dipolar cycloaddition reaction was performed between sarcosine, reduced graphene oxide (RGO), and A2[Ru(bpy)2]2+. This reaction resulted in the formation of a hybrid material, RGO-A2[Ru(bpy)2]2+, combining the electron-rich properties of the conjugated oligomer with the high surface area and conductivity of graphene. This hybrid material is expected to exhibit improved electronic properties, potentially useful for applications in energy storage, catalysis, and flexible electronics. To confirm the successful covalent attachment of A2[Ru(bpy)2]2+ to RGO, several characterization techniques were employed. Raman spectroscopy showed a characteristic decrease in the ID/IG ratio, suggesting the functionalization of RGO with the A2[Ru(bpy)2]2+ complex. Scanning electron microscopy (SEM) revealed significant morphological changes in the RGO-A2[Ru(bpy)2]2+ hybrid compared to pure RGO, with the formation of rough surface textures indicative of the polymer’s presence. X-ray photoelectron spectroscopy (XPS) further confirmed the covalent attachment of the Ru complex, providing evidence of elemental ruthenium and nitrogen on the surface of the material, with the Ru 3d and N 1s peaks matching those expected for the A2[Ru(bpy)2]2+ complex. These combined spectroscopic, electrochemical, and morphological analyses provide strong indirect evidence of the successful synthesis and covalent integration of A2[Ru(bpy)2]2+ onto RGO, forming a hybrid material with promising properties for future applications in electronic and energy-related devices.















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}