
Mitochondrial Transplantation Ameliorates Pulmonary Fibrosis by Suppressing Myofibroblast Activation
 , , , , and
, , , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
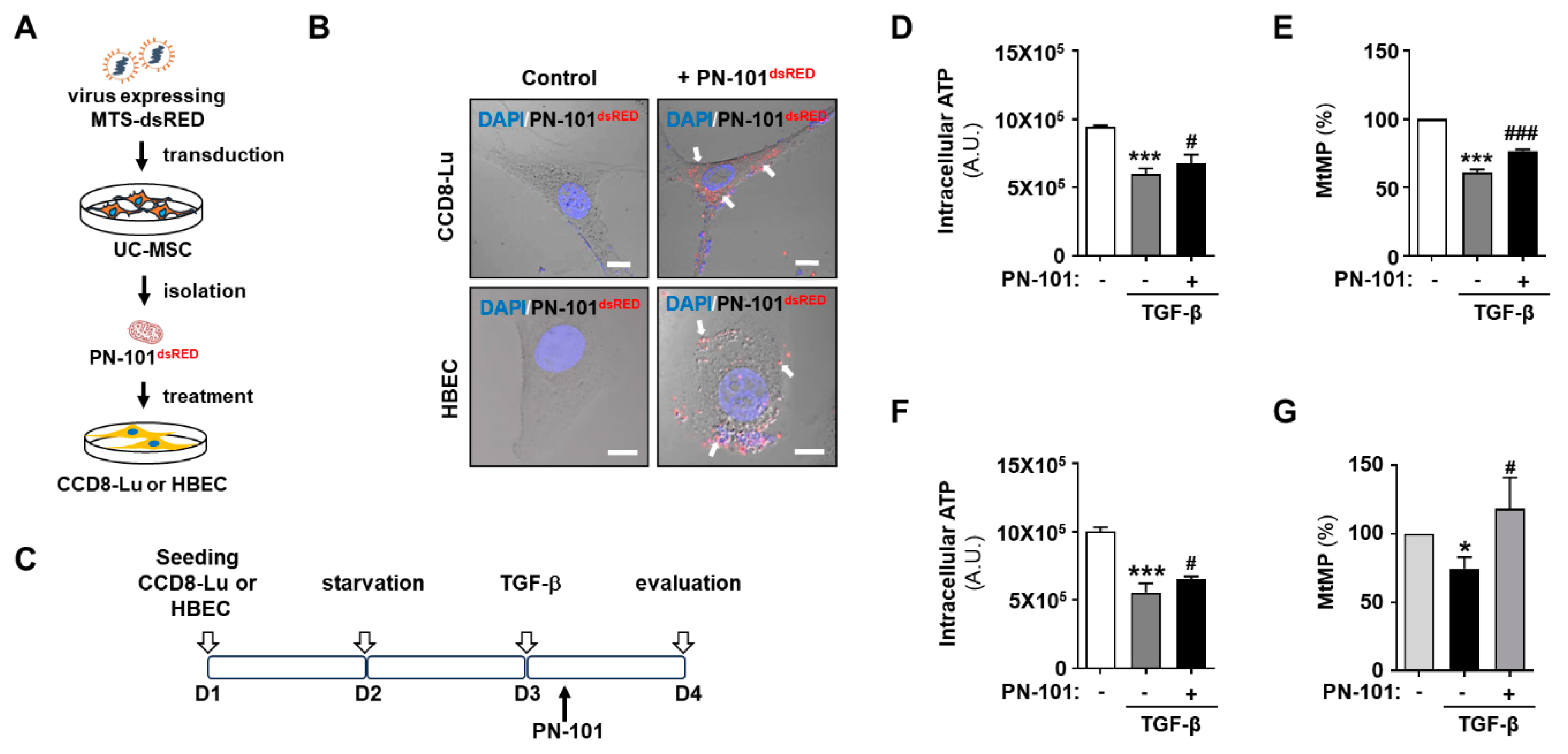
2.1. Mitochondrial Transplantation Ameliorated Mitochondrial Damage in TGF-β-Treated Pulmonary Cells
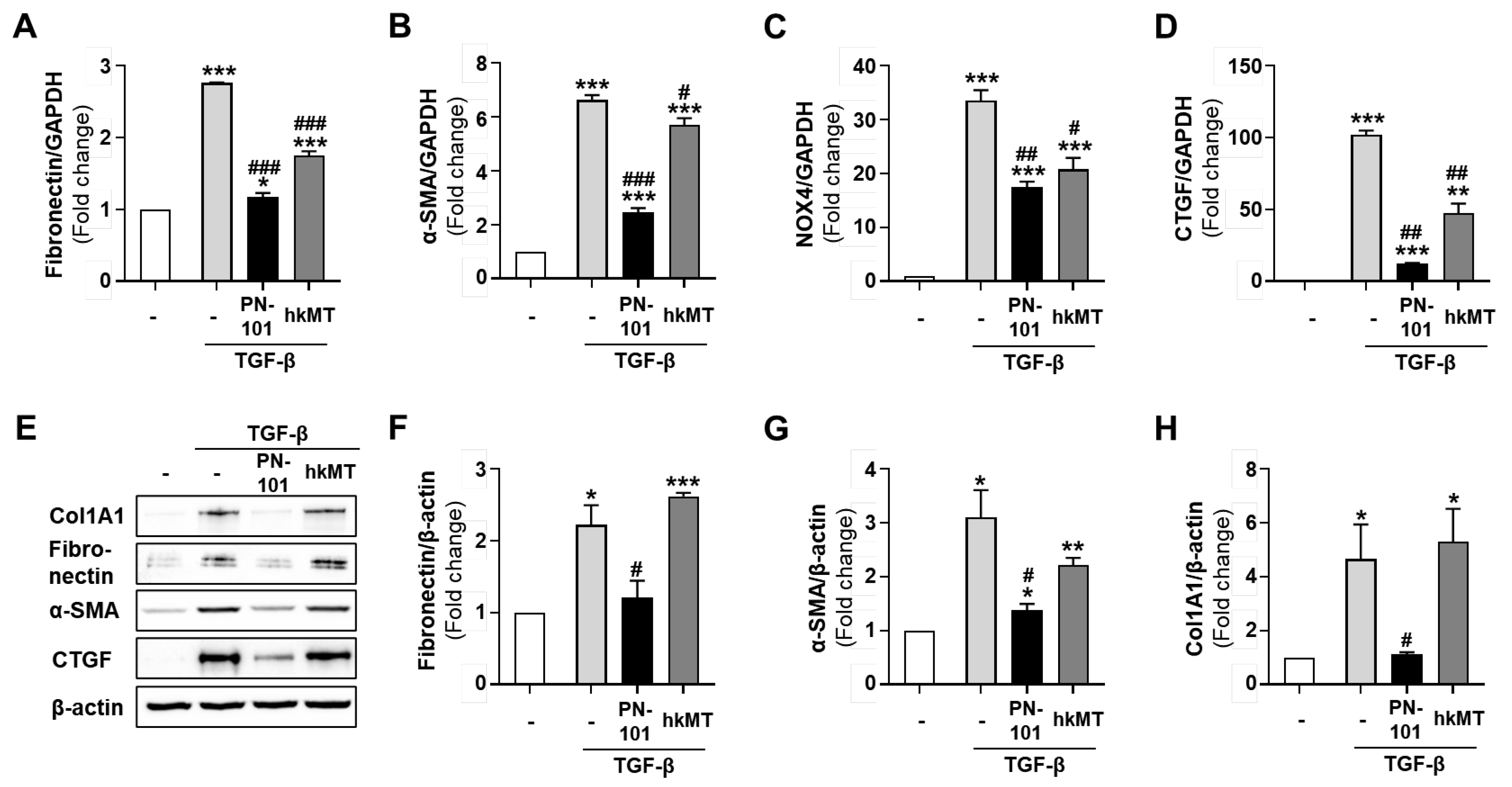
2.2. Mitochondrial Transplantation Reduced the Expression of Fibrotic Genes in TGF-β-Treated Lung Fibroblasts
2.3. Stem Cell-Derived Mitochondria Demonstrated an Excellent Anti-Fibrotic Effect
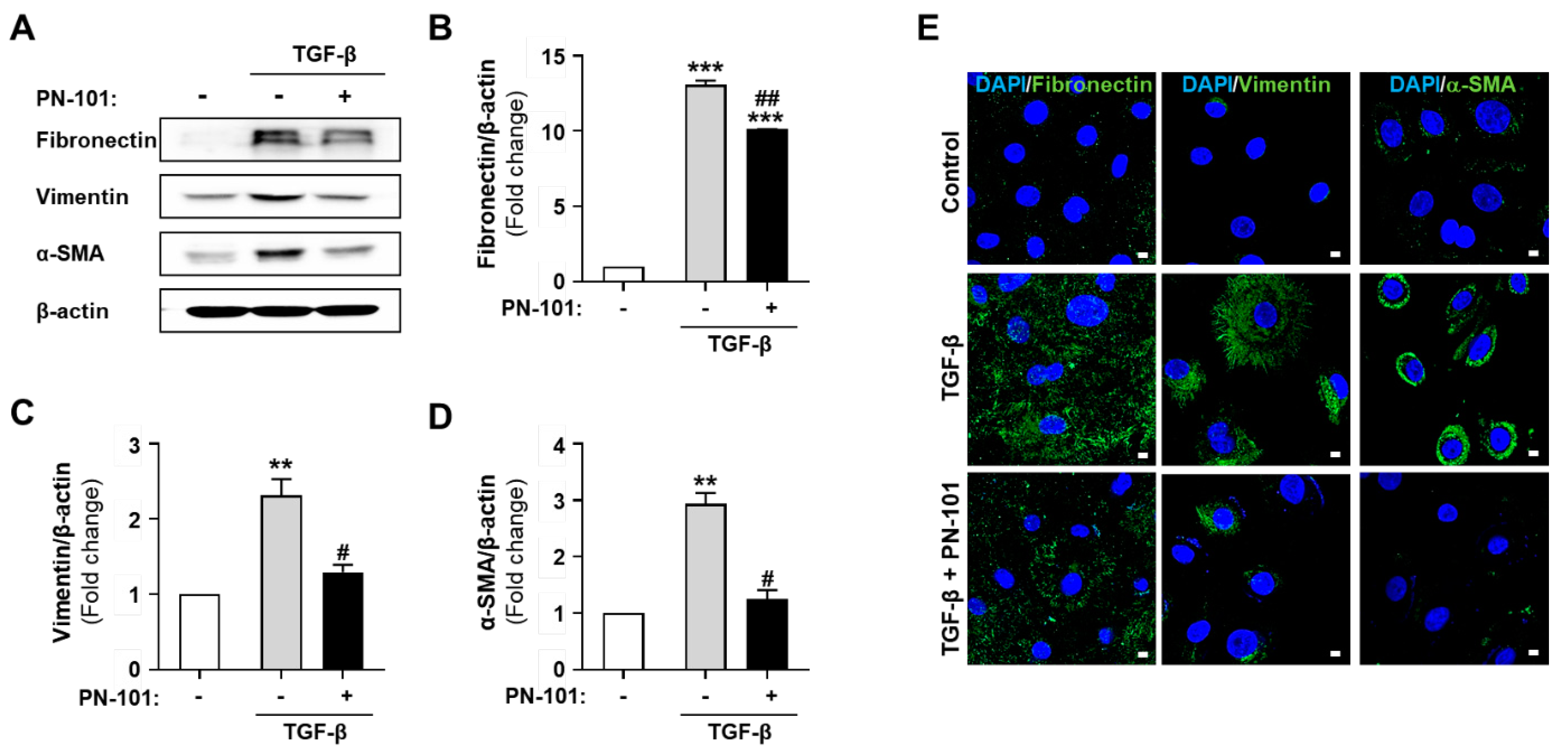
2.4. PN-101 Suppressed TGF-β-Induced Mesenchymal Phenotype in HBECs
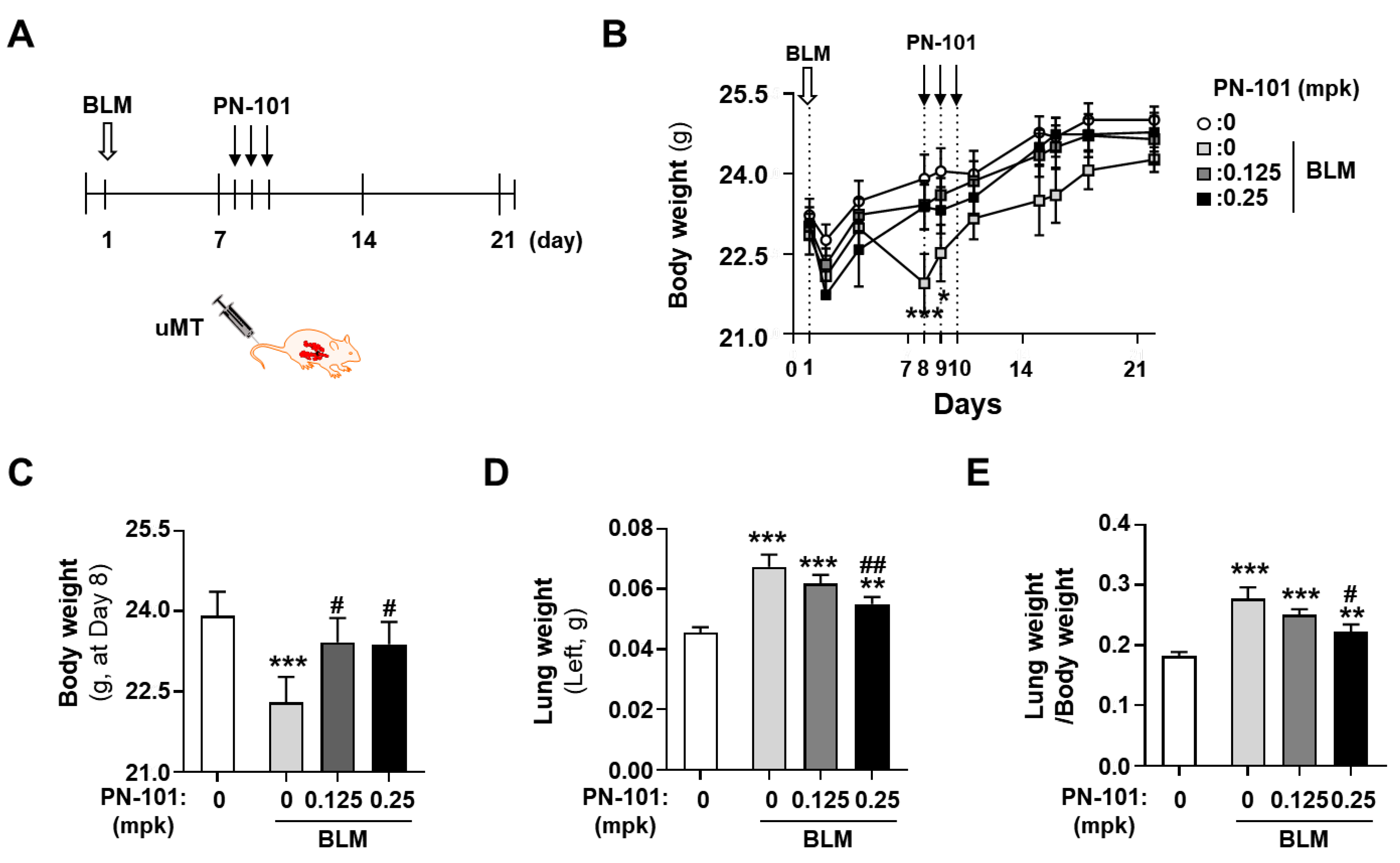
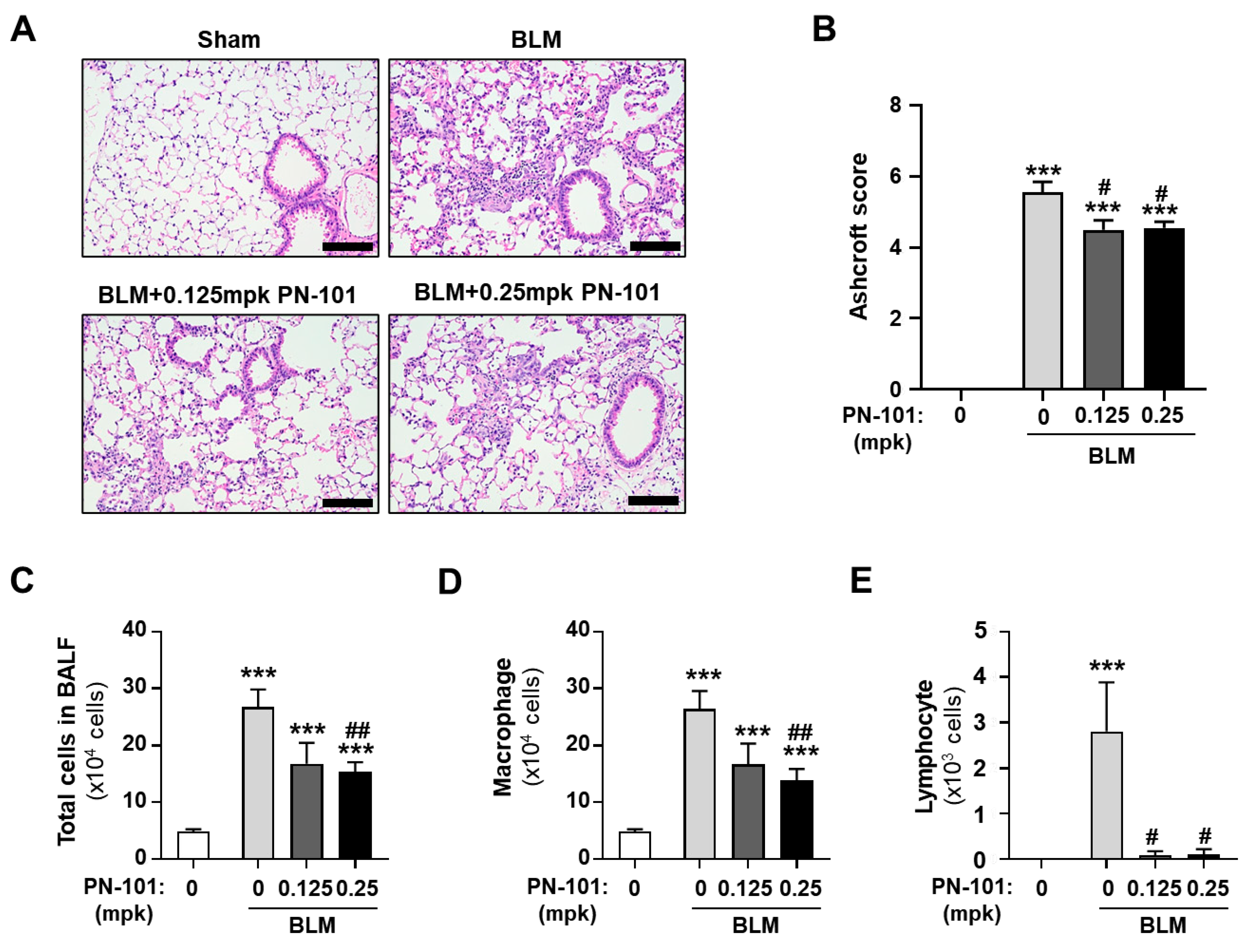
2.5. Effects of PN-101 on Fibrotic Processes in Bleomycin-Induced PF Mice
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Isolation of Mitochondria from PN-101 and hkMT
4.3. Labeling of the Mitochondria
4.4. ATP Assay
4.5. MtMP Assay
4.6. Quantitative Reverse Transcription-Polymerase Chain Reaction (RT-PCR) Assay
4.7. Western Blotting Assay
4.8. Immunocytochemistry
4.9. Animals and Mitochondrial Treatment
4.10. Body Weight and Lung Weight
4.11. Bronchoalveolar Lavage Fluid (BALF) Analysis
4.12. Histopathological Examination
4.13. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Martinez, F.J.; Collard, H.R.; Pardo, A.; Raghu, G.; Richeldi, L.; Selman, M.; Swigris, J.J.; Taniguchi, H.; Wells, A.U. Idiopathic pulmonary fibrosis. Nat. Rev. Dis. Primers 2017, 3, 17074. [Google Scholar] [CrossRef] [PubMed]
- Richeldi, L.; du Bois, R.M.; Raghu, G.; Azuma, A.; Brown, K.K.; Costabel, U.; Cottin, V.; Flaherty, K.R.; Hansell, D.M.; Inoue, Y.; et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N. Engl. J. Med. 2014, 370, 2071–2082. [Google Scholar] [CrossRef]
- Shihab, F.S.; Bennett, W.M.; Yi, H.; Andoh, T.F. Pirfenidone treatment decreases transforming growth factor-beta1 and matrix proteins and ameliorates fibrosis in chronic cyclosporine nephrotoxicity. Am. J. Transplant. 2002, 2, 111–119. [Google Scholar] [CrossRef]
- Khor, Y.H.; Ng, Y.; Barnes, H.; Goh, N.S.L.; McDonald, C.F.; Holland, A.E. Prognosis of idiopathic pulmonary fibrosis without anti-fibrotic therapy: A systematic review. Eur. Respir. Rev. 2020, 29, 157. [Google Scholar] [CrossRef] [PubMed]
- Nunnari, J.; Suomalainen, A. Mitochondria: In sickness and in health. Cell 2012, 148, 1145–1159. [Google Scholar] [CrossRef] [PubMed]
- Suomalainen, A.; Battersby, B.J. Mitochondrial diseases: The contribution of organelle stress responses to pathology. Nat. Rev. Mol. Cell Biol. 2018, 19, 77–92. [Google Scholar] [CrossRef] [PubMed]
- Bueno, M.; Calyeca, J.; Rojas, M.; Mora, A.L. Mitochondria dysfunction and metabolic reprogramming as drivers of idiopathic pulmonary fibrosis. Redox Biol. 2020, 33, 101509. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhang, W.; Cao, Q.; Wang, Z.; Zhao, M.; Xu, L.; Zhuang, Q. Mitochondrial dysfunction in fibrotic diseases. Cell Death Discov. 2020, 6, 80. [Google Scholar] [CrossRef]
- Zank, D.C.; Bueno, M.; Mora, A.L.; Rojas, M. Idiopathic Pulmonary Fibrosis: Aging, Mitochondrial Dysfunction, and Cellular Bioenergetics. Front. Med. 2018, 5, 10. [Google Scholar] [CrossRef]
- Bargagli, E.; Refini, R.M.; d’Alessandro, M.; Bergantini, L.; Cameli, P.; Vantaggiato, L.; Bini, L.; Landi, C. Metabolic Dysregulation in Idiopathic Pulmonary Fibrosis. Int. J. Mol. Sci. 2020, 21, 5663. [Google Scholar] [CrossRef]
- Mora, A.L.; Bueno, M.; Rojas, M. Mitochondria in the spotlight of aging and idiopathic pulmonary fibrosis. J. Clin. Investig. 2017, 127, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Lightowlers, R.N.; Chrzanowska-Lightowlers, Z.M.; Russell, O.M. Mitochondrial transplantation-a possible therapeutic for mitochondrial dysfunction?: Mitochondrial transfer is a potential cure for many diseases but proof of efficacy and safety is still lacking. EMBO Rep. 2020, 21, e50964. [Google Scholar] [CrossRef] [PubMed]
- McCully, J.D.; Del Nido, P.J.; Emani, S.M. Mitochondrial transplantation for organ rescue. Mitochondrion 2022, 64, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Masuzawa, A.; Black, K.M.; Pacak, C.A.; Ericsson, M.; Barnett, R.J.; Drumm, C.; Seth, P.; Bloch, D.B.; Levitsky, S.; Cowan, D.B.; et al. Transplantation of autologously derived mitochondria protects the heart from ischemia-reperfusion injury. Am. J. Physiol. Heart Circ. Physiol. 2013, 304, H966–H982. [Google Scholar] [CrossRef]
- Weixler, V.; Lapusca, R.; Grangl, G.; Guariento, A.; Saeed, M.Y.; Cowan, D.B.; Del Nido, P.J.; McCully, J.D.; Friehs, I. Autogenous mitochondria transplantation for treatment of right heart failure. J. Thorac. Cardiovasc. Surg. 2021, 162, e111–e121. [Google Scholar] [CrossRef] [PubMed]
- Pabla, N.; Bajwa, A. Role of Mitochondrial Therapy for Ischemic-Reperfusion Injury and Acute Kidney Injury. Nephron 2022, 146, 253–258. [Google Scholar] [CrossRef]
- Lamanilao, G.G.; Dogan, M.; Patel, P.S.; Azim, S.; Patel, D.S.; Bhattacharya, S.K.; Eason, J.D.; Kuscu, C.; Bajwa, A. Key hepatoprotective roles of mitochondria in liver regeneration. Am. J. Physiol. Gastrointest. Liver Physiol. 2023, 324, G207–G218. [Google Scholar] [CrossRef]
- Shi, X.; Zhao, M.; Fu, C.; Fu, A. Intravenous administration of mitochondria for treating experimental Parkinson’s disease. Mitochondrion 2017, 34, 91–100. [Google Scholar] [CrossRef]
- Alexander, J.F.; Seua, A.V.; Arroyo, L.D.; Ray, P.R.; Wangzhou, A.; Heibeta-Luckemann, L.; Schedlowski, M.; Price, T.J.; Kavelaars, A.; Heijnen, C.J. Nasal administration of mitochondria reverses chemotherapy-induced cognitive deficits. Theranostics 2021, 11, 3109–3130. [Google Scholar] [CrossRef]
- Jia, X.; Wang, Q.; Ji, J.; Lu, W.; Liu, Z.; Tian, H.; Guo, L.; Wang, Y. Mitochondrial transplantation ameliorates hippocampal damage following status epilepticus. Anim. Model. Exp. Med. 2023, 6, 41–50. [Google Scholar] [CrossRef]
- McCully, J.D.; Del Nido, P.J.; Emani, S.M. Mitochondrial transplantation: The advance to therapeutic application and molecular modulation. Front. Cardiovasc. Med. 2023, 10, 1268814. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.H.; Kim, S.; Kim, Y.; Lee, S.E.; Park, J.H.; Cho, G.; Ha, J.C.; Jung, H.; Lim, S.M.; Han, K.; et al. Human umbilical cord mesenchymal stem cell-derived mitochondria (PN-101) attenuate LPS-induced inflammatory responses by inhibiting NFkappaB signaling pathway. BMB Rep. 2022, 55, 136–141. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.E.; Kang, Y.C.; Kim, Y.; Kim, S.; Yu, S.H.; Park, J.H.; Kim, I.H.; Kim, H.Y.; Han, K.; Lee, H.K.; et al. Preferred Migration of Mitochondria toward Cells and Tissues with Mitochondrial Damage. Int. J. Mol. Sci. 2022, 23, 15734. [Google Scholar] [CrossRef] [PubMed]
- Katrangi, E.; D’Souza, G.; Boddapati, S.V.; Kulawiec, M.; Singh, K.K.; Bigger, B.; Weissig, V. Xenogenic transfer of isolated murine mitochondria into human rho0 cells can improve respiratory function. Rejuvenation Res. 2007, 10, 561–570. [Google Scholar] [CrossRef] [PubMed]
- Kitani, T.; Kami, D.; Kawasaki, T.; Nakata, M.; Matoba, S.; Gojo, S. Direct human mitochondrial transfer: A novel concept based on the endosymbiotic theory. Transplant. Proc. 2014, 46, 1233–1236. [Google Scholar] [CrossRef] [PubMed]
- Kesner, E.E.; Saada-Reich, A.; Lorberboum-Galski, H. Characteristics of Mitochondrial Transformation into Human Cells. Sci. Rep. 2016, 6, 26057. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.J.; Hwang, J.W.; Yun, C.K.; Lee, Y.; Choi, Y.S. Delivery of exogenous mitochondria via centrifugation enhances cellular metabolic function. Sci. Rep. 2018, 8, 3330. [Google Scholar] [CrossRef]
- Tomasek, J.J.; Gabbiani, G.; Hinz, B.; Chaponnier, C.; Brown, R.A. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat. Rev. Mol. Cell Biol. 2002, 3, 349–363. [Google Scholar] [CrossRef]
- Ahmad, T.; Sundar, I.K.; Lerner, C.A.; Gerloff, J.; Tormos, A.M.; Yao, H.; Rahman, I. Impaired mitophagy leads to cigarette smoke stress-induced cellular senescence: Implications for chronic obstructive pulmonary disease. FASEB J. 2015, 29, 2912–2929. [Google Scholar] [CrossRef]
- Fernandez, I.E.; Eickelberg, O. The impact of TGF-beta on lung fibrosis: From targeting to biomarkers. Proc. Am. Thorac. Soc. 2012, 9, 111–116. [Google Scholar] [CrossRef]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef] [PubMed]
- Moeller, A.; Ask, K.; Warburton, D.; Gauldie, J.; Kolb, M. The bleomycin animal model: A useful tool to investigate treatment options for idiopathic pulmonary fibrosis? Int. J. Biochem. Cell Biol. 2008, 40, 362–382. [Google Scholar] [CrossRef] [PubMed]
- Cowley, P.M.; Roberts, C.R.; Baker, A.J. Monitoring the Health Status of Mice with Bleomycin-induced Lung Injury by Using Body Condition Scoring. Comp. Med. 2019, 69, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Jaeger, V.K.; Lebrecht, D.; Nicholson, A.G.; Wells, A.; Bhayani, H.; Gazdhar, A.; Tamm, M.; Venhoff, N.; Geiser, T.; Walker, U.A. Mitochondrial DNA mutations and respiratory chain dysfunction in idiopathic and connective tissue disease-related lung fibrosis. Sci. Rep. 2019, 9, 5500. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, D.; Cardenes, N.; Sellares, J.; Bueno, M.; Corey, C.; Hanumanthu, V.S.; Peng, Y.; D’Cunha, H.; Sembrat, J.; Nouraie, M.; et al. IPF lung fibroblasts have a senescent phenotype. Am. J. Physiol. Lung Cell Mol. Physiol. 2017, 313, L1164–L1173. [Google Scholar] [CrossRef] [PubMed]
- Xie, N.; Tan, Z.; Banerjee, S.; Cui, H.; Ge, J.; Liu, R.M.; Bernard, K.; Thannickal, V.J.; Liu, G. Glycolytic Reprogramming in Myofibroblast Differentiation and Lung Fibrosis. Am. J. Respir. Crit. Care Med. 2015, 192, 1462–1474. [Google Scholar] [CrossRef] [PubMed]
- Sosulski, M.L.; Gongora, R.; Danchuk, S.; Dong, C.; Luo, F.; Sanchez, C.G. Deregulation of selective autophagy during aging and pulmonary fibrosis: The role of TGFbeta1. Aging Cell 2015, 14, 774–783. [Google Scholar] [CrossRef]
- Kim, S.J.; Cheresh, P.; Jablonski, R.P.; Williams, D.B.; Kamp, D.W. The Role of Mitochondrial DNA in Mediating Alveolar Epithelial Cell Apoptosis and Pulmonary Fibrosis. Int. J. Mol. Sci. 2015, 16, 21486–21519. [Google Scholar] [CrossRef] [PubMed]
- Larson-Casey, J.L.; Deshane, J.S.; Ryan, A.J.; Thannickal, V.J.; Carter, A.B. Macrophage Akt1 Kinase-Mediated Mitophagy Modulates Apoptosis Resistance and Pulmonary Fibrosis. Immunity 2016, 44, 582–596. [Google Scholar] [CrossRef]
- Amara, N.; Goven, D.; Prost, F.; Muloway, R.; Crestani, B.; Boczkowski, J. NOX4/NADPH oxidase expression is increased in pulmonary fibroblasts from patients with idiopathic pulmonary fibrosis and mediates TGFbeta1-induced fibroblast differentiation into myofibroblasts. Thorax 2010, 65, 733–738. [Google Scholar] [CrossRef] [PubMed]
- Rangarajan, S.; Bone, N.B.; Zmijewska, A.A.; Jiang, S.; Park, D.W.; Bernard, K.; Locy, M.L.; Ravi, S.; Deshane, J.; Mannon, R.B.; et al. Metformin reverses established lung fibrosis in a bleomycin model. Nat. Med. 2018, 24, 1121–1127. [Google Scholar] [CrossRef] [PubMed]
- Hewitt, R.J.; Maher, T.M. Idiopathic Pulmonary Fibrosis: New and Emerging Treatment Options. Drugs Aging 2019, 36, 485–492. [Google Scholar] [CrossRef]
- Piera-Velazquez, S.; Li, Z.; Jimenez, S.A. Role of endothelial-mesenchymal transition (EndoMT) in the pathogenesis of fibrotic disorders. Am. J. Pathol. 2011, 179, 1074–1080. [Google Scholar] [CrossRef]
- Zeisberg, E.M.; Tarnavski, O.; Zeisberg, M.; Dorfman, A.L.; McMullen, J.R.; Gustafsson, E.; Chandraker, A.; Yuan, X.; Pu, W.T.; Roberts, A.B.; et al. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat. Med. 2007, 13, 952–961. [Google Scholar] [CrossRef]
- Willis, B.C.; duBois, R.M.; Borok, Z. Epithelial origin of myofibroblasts during fibrosis in the lung. Proc. Am. Thorac. Soc. 2006, 3, 377–382. [Google Scholar] [CrossRef] [PubMed]
- Homps-Legrand, M.; Crestani, B.; Mailleux, A.A. Origins of pathological myofibroblasts in lung fibrosis: Insights from lineage tracing mouse models in the single-cell RNA sequencing era. Am. J. Physiol. Lung Cell Mol. Physiol. 2023, 324, L737–L746. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.E.; Kim, I.H.; Kang, Y.C.; Kim, Y.; Yu, S.H.; Yeo, J.S.; Kwon, I.; Lim, J.H.; Kim, J.H.; Han, K.; et al. Mitochondrial transplantation attenuates lipopolysaccharide-induced acute respiratory distress syndrome. BMC Pulm. Med. 2024, 24, 477. [Google Scholar] [CrossRef]
- Cowan, D.B.; Yao, R.; Thedsanamoorthy, J.K.; Zurakowski, D.; Del Nido, P.J.; McCully, J.D. Transit and integration of extracellular mitochondria in human heart cells. Sci. Rep. 2017, 7, 17450. [Google Scholar] [CrossRef]
- Kim, S.; Kim, Y.; Yu, S.H.; Lee, S.E.; Park, J.H.; Cho, G.; Choi, C.; Han, K.; Kim, C.H.; Kang, Y.C. Platelet-derived mitochondria transfer facilitates wound-closure by modulating ROS levels in dermal fibroblasts. Platelets 2022, 34, 2151996. [Google Scholar] [CrossRef]
- Fu, A.; Shi, X.; Zhang, H.; Fu, B. Mitotherapy for Fatty Liver by Intravenous Administration of Exogenous Mitochondria in Male Mice. Front. Pharmacol. 2017, 8, 241. [Google Scholar] [CrossRef]
- Borcherding, N.; Jia, W.; Giwa, R.; Field, R.L.; Moley, J.R.; Kopecky, B.J.; Chan, M.M.; Yang, B.Q.; Sabio, J.M.; Walker, E.C.; et al. Dietary lipids inhibit mitochondria transfer to macrophages to divert adipocyte-derived mitochondria into the blood. Cell Metab. 2022, 34, 1499–1513.e1498. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Li, H.; Yao, Y.; Zhao, T.; Chen, Y.Y.; Shen, Y.L.; Wang, L.L.; Zhu, Y. Stem cell-derived mitochondria transplantation: A novel strategy and the challenges for the treatment of tissue injury. Stem Cell Res. Ther. 2018, 9, 106. [Google Scholar] [CrossRef]
- Acquistapace, A.; Bru, T.; Lesault, P.F.; Figeac, F.; Coudert, A.E.; le Coz, O.; Christov, C.; Baudin, X.; Auber, F.; Yiou, R.; et al. Human mesenchymal stem cells reprogram adult cardiomyocytes toward a progenitor-like state through partial cell fusion and mitochondria transfer. Stem Cells 2011, 29, 812–824. [Google Scholar] [CrossRef] [PubMed]
- Spees, J.L.; Olson, S.D.; Whitney, M.J.; Prockop, D.J. Mitochondrial transfer between cells can rescue aerobic respiration. Proc. Natl. Acad. Sci. USA 2006, 103, 1283–1288. [Google Scholar] [CrossRef] [PubMed]
- Sinclair, K.A.; Yerkovich, S.T.; Hopkins, P.M.; Chambers, D.C. Characterization of intercellular communication and mitochondrial donation by mesenchymal stromal cells derived from the human lung. Stem Cell Res. Ther. 2016, 7, 91. [Google Scholar] [CrossRef]
- Huang, T.; Lin, R.; Su, Y.; Sun, H.; Zheng, X.; Zhang, J.; Lu, X.; Zhao, B.; Jiang, X.; Huang, L.; et al. Efficient intervention for pulmonary fibrosis via mitochondrial transfer promoted by mitochondrial biogenesis. Nat. Commun. 2023, 14, 5781. [Google Scholar] [CrossRef] [PubMed]
- Lancaster, L.H.; de Andrade, J.A.; Zibrak, J.D.; Padilla, M.L.; Albera, C.; Nathan, S.D.; Wijsenbeek, M.S.; Stauffer, J.L.; Kirchgaessler, K.U.; Costabel, U. Pirfenidone safety and adverse event management in idiopathic pulmonary fibrosis. Eur. Respir. Rev. 2017, 26, 146. [Google Scholar] [CrossRef]
- Kim, S.N.; Lee, J.; Yang, H.S.; Cho, J.W.; Kwon, S.; Kim, Y.B.; Her, J.D.; Cho, K.H.; Song, C.W.; Lee, K. Dose-response Effects of Bleomycin on Inflammation and Pulmonary Fibrosis in Mice. Toxicol. Res. 2010, 26, 217–222. [Google Scholar] [CrossRef]
- Ashcroft, T.; Simpson, J.M.; Timbrell, V. Simple method of estimating severity of pulmonary fibrosis on a numerical scale. J. Clin. Pathol. 1988, 41, 467–470. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, S.-E.; Yu, S.-H.; Kim, I.-H.; Kang, Y.C.; Kim, Y.; Yeo, J.S.; Lim, J.H.; Kwon, I.; Kim, J.-H.; Park, S.-W.; et al. Mitochondrial Transplantation Ameliorates Pulmonary Fibrosis by Suppressing Myofibroblast Activation. Int. J. Mol. Sci. 2024, 25, 12783. https://doi.org/10.3390/ijms252312783
Lee S-E, Yu S-H, Kim I-H, Kang YC, Kim Y, Yeo JS, Lim JH, Kwon I, Kim J-H, Park S-W, et al. Mitochondrial Transplantation Ameliorates Pulmonary Fibrosis by Suppressing Myofibroblast Activation. International Journal of Molecular Sciences. 2024; 25(23):12783. https://doi.org/10.3390/ijms252312783
Chicago/Turabian StyleLee, Seo-Eun, Shin-Hye Yu, In-Hyeon Kim, Young Cheol Kang, Yujin Kim, Jeong Seon Yeo, Jun Hyeok Lim, Iksun Kwon, Je-Hein Kim, Se-Woong Park, and et al. 2024. "Mitochondrial Transplantation Ameliorates Pulmonary Fibrosis by Suppressing Myofibroblast Activation" International Journal of Molecular Sciences 25, no. 23: 12783. https://doi.org/10.3390/ijms252312783
APA StyleLee, S.-E., Yu, S.-H., Kim, I.-H., Kang, Y. C., Kim, Y., Yeo, J. S., Lim, J. H., Kwon, I., Kim, J.-H., Park, S.-W., Chang, M.-Y., Han, K., Kim, S.-H., & Kim, C.-H. (2024). Mitochondrial Transplantation Ameliorates Pulmonary Fibrosis by Suppressing Myofibroblast Activation. International Journal of Molecular Sciences, 25(23), 12783. https://doi.org/10.3390/ijms252312783