
Enhanced CRC Growth in Iron-Rich Environment, Facts and Speculations



Abstract

{kind=link}
{kind=link}
{kind=link}
1. Introduction
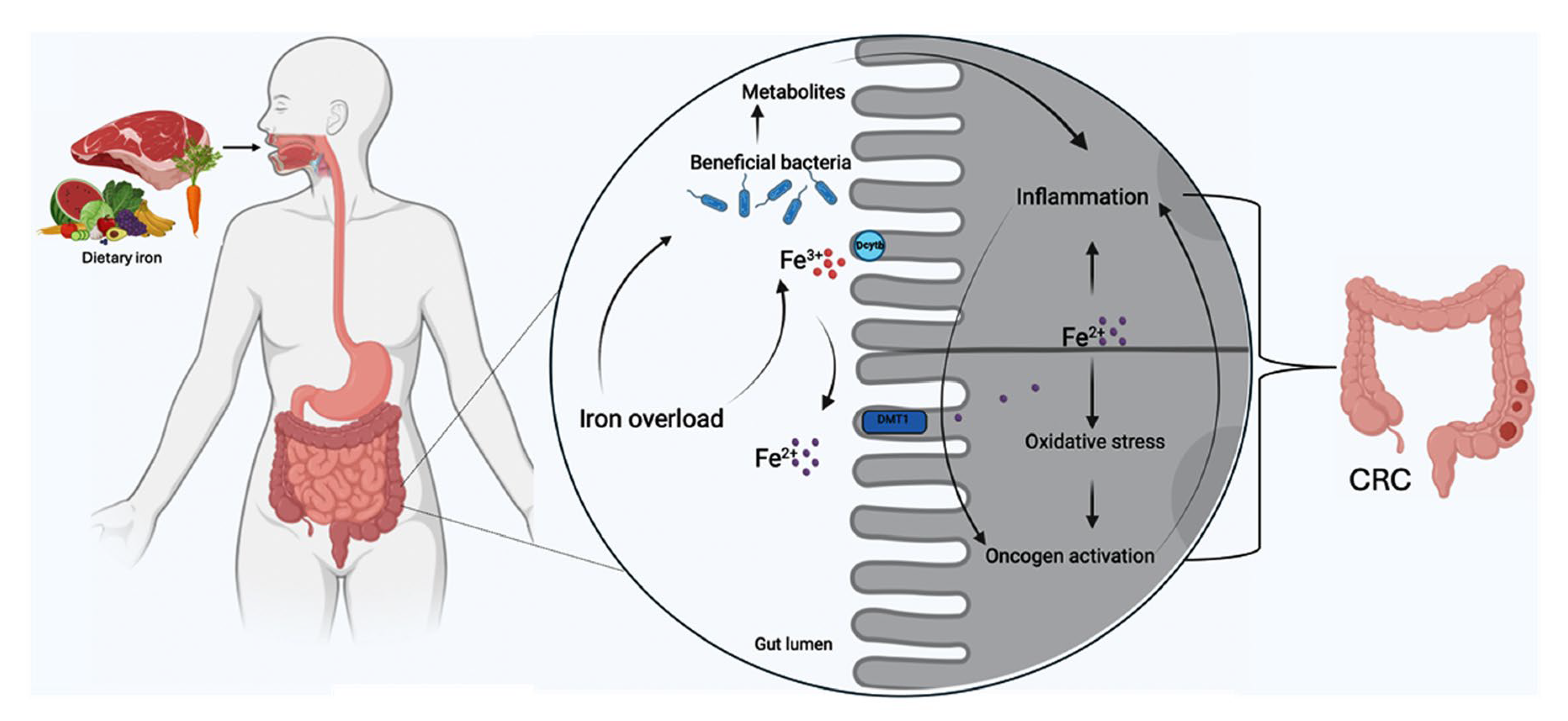
2. The Role of Iron in Colorectal Cancer
3. Iron-Rich Diet and Inflammatory Microbiota
3.1. Intestinal Microbiota and CRC
3.2. Intestinal Microbiota Shift in Response to Iron Availability
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ross, A.C.; Caballero, B.; Cousins, R.J.; Tucker, K.L. Modern Nutrition in Health and Disease; Jones & Bartlett Learning: Burlington, MA, USA, 2020. [Google Scholar]
- Dev, S.; Babitt, J.L. Overview of iron metabolism in health and disease. Hemodial. Int. 2017, 21, S6–S20. [Google Scholar] [CrossRef] [PubMed]
- Bothwell, T.H.; Finch, C.A. Iron Metabolism; Little, Brown & Co.: Boston, MA, USA; Toronto, ON, USA, 1962; p. 339. [Google Scholar]
- Hentze, M.W.; Muckenthaler, M.U.; Galy, B.; Camaschella, C. Two to tango: Regulation of Mammalian iron metabolism. Cell 2010, 142, 24–38. [Google Scholar] [CrossRef] [PubMed]
- Huo, C.; Li, G.; Hu, Y.; Sun, H. The impacts of iron overload and ferroptosis on intestinal mucosal homeostasis and inflammation. Int. J. Mol. Sci. 2022, 23, 14195. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Mori, K.; Li, J.Y.; Barasch, J. Iron, lipocalin, and kidney epithelia. Am. J. Physiol.-Ren. Physiol. 2003, 285, F9–F18. [Google Scholar] [CrossRef]
- Gulec, S.; Anderson, G.J.; Collins, J.F. Mechanistic and regulatory aspects of intestinal iron absorption. Am. J. Physiol. Gastrointest. Liver Physiol. 2014, 307, G397–G409. [Google Scholar] [CrossRef] [PubMed]
- Syed, B.A.; Sargent, P.J.; Farnaud, S.; Evans, R.W. An overview of molecular aspects of iron metabolism. Hemoglobin 2006, 30, 69–80. [Google Scholar] [CrossRef] [PubMed]
- Von Siebenthal, H.K.; Moretti, D.; Zimmermann, M.B.; Stoffel, N.U. Effect of dietary factors and time of day on iron absorption from oral iron supplements in iron deficient women. Am. J. Hematol. 2023, 98, 1356–1363. [Google Scholar] [CrossRef]
- Zijp, I.M.; Korver, O.; Tijburg, L.B.M. Effect of tea and other dietary factors on iron absorption. Crit. Rev. Food Sci. Nutr. 2000, 40, 371–398. [Google Scholar] [CrossRef]
- Scarano, A.; Laddomada, B.; Blando, F.; De Santis, S.; Verna, G.; Chieppa, M.; Santino, A. The chelating ability of plant polyphenols can affect iron homeostasis and gut microbiota. Antioxidants 2023, 12, 630. [Google Scholar] [CrossRef]
- Duck, K.A.; Connor, J.R. Iron uptake and transport across physiological barriers. Biometals 2016, 29, 573–591. [Google Scholar] [CrossRef]
- Rivella, S.; Crielaard, B.J. Disorders of Iron Metabolism: Iron Deficiency and Iron Overload and Anemia of Chronic Diseases. In Pathobiology of Human Disease; Elsevier: Amsterdam, The Netherlands, 2014. [Google Scholar] [CrossRef]
- Le Blanc, S.; Garrick, M.D.; Arredondo, M. Heme carrier protein 1 transports heme and is involved in heme-Fe metabolism. Am. J. Physiol. Cell Physiol. 2012, 302, C1780–C1785. [Google Scholar] [CrossRef] [PubMed]
- Shayeghi, M.; Latunde-Dada, G.O.; Oakhill, J.S.; Laftah, A.H.; Takeuchi, K.; Halliday, N.; Khan, Y.; Warley, A.; McCann, F.E.; Hider, R.C.; et al. Identification of an intestinal heme transporter. Cell 2005, 122, 789–801. [Google Scholar] [CrossRef] [PubMed]
- McKie, A.T.; Barrow, D.; Latunde-Dada, G.O.; Rolfs, A.; Sager, G.; Mudaly, E.; Mudaly, M.; Richardson, C.; Barlow, D.; Bomford, A.; et al. An iron-regulated ferric reductase associated with the absorption of dietary iron. Science 2001, 291, 1755–1759. [Google Scholar] [CrossRef]
- Gunshin, H.; MacKenzie, B.; Berger, U.V.; Gunshin, Y.; Romero, M.F.; Boron, W.F.; Nussberger, S.; Gollan, J.L.; Hediger, M.A. Cloning and characterization of a mammalian proton-coupled metal-ion transporter. Nature 1997, 388, 482–488. [Google Scholar] [CrossRef]
- Canonne-Hergaux, F.; Gruenheid, S.; Ponka, P.; Gros, P. Cellular and subcellular localization of the Nramp2 iron transporter in the intestinal brush border and regulation by dietary iron. Blood J. Am. Soc. Hematol. 1999, 93, 4406–4417. [Google Scholar]
- Donovan, A.; Lima, C.A.; Pinkus, J.L.; Pinkus, G.S.; Zon, L.I.; Robine, S.; Andrews, N.C. The iron exporter ferroportin/Slc40a1 is essential for iron homeostasis. Cell Metab. 2005, 1, 191–200. [Google Scholar] [CrossRef]
- Han, O.; Kim, E. Colocalization of ferroportin-1 with hephaestin on the basolateral membrane of human intestinal absorptive cells. J. Cell Biochem. 2007, 101, 1000–1010. [Google Scholar] [CrossRef] [PubMed]
- Yeh, K.; Yeh, M.; Mims, L.; Glass, J. Iron feeding induces ferroportin 1 and hephaestin migration and interaction in rat duodenal epithelium. Am. J. Physiol.-Gastrointest. Liver Physiol. 2009, 296, G55–G65. [Google Scholar] [CrossRef] [PubMed]
- Yeh, K.; Yeh, M.; Glass, J. Interactions between ferroportin and hephaestin in rat enterocytes are reduced after iron ingestion. Gastroenterology 2011, 141, 292–299. [Google Scholar] [CrossRef]
- Yang, N.; Zhang, H.; Wang, M.; Hao, Q.; Sun, H. Iron and bismuth bound human serum transferrin reveals a partially-opened conformation in the N-lobe. Sci. Rep. 2012, 2, 999. [Google Scholar] [CrossRef]
- Pantopoulos, K.; Porwal, S.K.; Tartakoff, A.; Devireddy, L. Mechanisms of mammalian iron homeostasis. Biochemistry 2012, 51, 5705–5724. [Google Scholar] [CrossRef] [PubMed]
- Fleming, M.D.; Romano, M.A.; Su, M.A.; Garrick, L.M.; Garrick, M.D.; Andrews, N.C. Nramp 2 is mutated in the anemic Belgrade (b) rat: Evidence of a role for Nramp2 in endosomal iron transport. Proc. Natl. Acad. Sci. USA 1998, 95, 1148–1153. [Google Scholar] [CrossRef]
- Steere, A.N.; Byrne, S.L.; Chasteen, N.D.; Mason, A.B. Kinetics of iron release from transferrin bound to the transferrin receptor at endosomal pH. Biochim. Biophys. Acta (BBA) Gen. Subj. 2012, 1820, 326–333. [Google Scholar] [CrossRef]
- Eckenroth, B.E.; Steere, A.N.; Chasteen, N.D.; Everse, S.J.; Mason, A.B. How the binding of human transferrin primes the transferrin receptor potentiating iron release at endosomal pH. Proc. Natl. Acad. Sci. USA 2011, 108, 13089–13094. [Google Scholar] [CrossRef] [PubMed]
- Andrews, N.C. Iron homeostasis: Insights from genetics and animal models. Nat. Rev. Genet. 2000, 1, 208–217. [Google Scholar] [CrossRef] [PubMed]
- Andrews, N.C. Disorders of iron metabolism. N. Engl. J. Med. 1999, 341, 1986–1995. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, N.; Pantopoulos, K. The IRP/IRE system in vivo: Insights from mouse models. Front. Pharmacol. 2014, 5, 176. [Google Scholar] [CrossRef]
- Rouault, T.A.; Klausner, R.D. Post-transcriptional regulation of genes of iron metabolism in mammalian cells. JBIC J. Biol. Inorg. Chem. 1996, 1, 494–499. [Google Scholar] [CrossRef]
- Vogt, A.-C.S.; Arsiwala, T.; Mohsen, M.; Vogel, M.; Manolova, V.; Bachmann, M.F. On iron metabolism and its regulation. Int. J. Mol. Sci. 2021, 22, 4591. [Google Scholar] [CrossRef]
- De Domenico, I.; Ward, D.M.; Langelier, C.; Vaughn, M.B.; Nemeth, E.; Sundquist, W.I.; Ganz, T.; Musci, G.; Kaplan, J. The molecular mechanism of hepcidin-mediated ferroportin down-regulation. Mol. Biol. Cell 2007, 18, 2569–2578. [Google Scholar] [CrossRef]
- Nemeth, E.; Tuttle, M.S.; Powelson, J.; Vaughn, M.B.; Donovan, A.; Ward, D.M.V.; Ganz, T.; Kaplan, J. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science 2004, 306, 2090–2093. [Google Scholar] [CrossRef]
- Aksan, A.; Farrag, K.; Aksan, S.; Schroeder, O.; Stein, J. Flipside of the coin: Iron deficiency and colorectal cancer. Front. Immunol. 2021, 12, 635899. [Google Scholar] [CrossRef] [PubMed]
- Estêvão, D.; da Cruz-Ribeiro, M.; Cardoso, A.P.; Costa, M.; Oliveira, M.J.; Duarte, T.L.; da Cruz, T.B. Iron metabolism in colorectal cancer: A balancing act. Cell. Oncol. 2023, 46, 1545–1558. [Google Scholar] [CrossRef]
- Zhang, D.-L.; Ghosh, M.C.; Rouault, T.A. The physiological functions of iron regulatory proteins in iron homeostasis-an update. Front. Pharmacol. 2014, 5, 92341. [Google Scholar] [CrossRef]
- Roy, C.N.; Enns, C.A. Iron homeostasis: New tales from the crypt. Blood J. Am. Soc. Hematol. 2000, 96, 4020–4027. [Google Scholar]
- Eisenstein, R.S.; Blemings, K. Iron regulatory proteins, iron responsive elements and iron homeostasis. J. Nutr. 1998, 128, 2295–2298. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L.; Wang, G.L. A nuclear factor induced by hypoxia via de novo protein synthesis binds to the human erythropoietin gene enhancer at a site required for transcriptional activation. Mol. Cell Biol. 1992, 12, 5447–5454. [Google Scholar]
- Semenza, G.L.; Agani, F.; Booth, G.; Forsythe, J.; Iyer, N.; Jiang, B.-H.; Leung, S.; Roe, R.; Wiener, C.; Yu, A. Structural and functional analysis of hypoxia-inducible factor 1. Kidney Int. 1997, 51, 553–555. [Google Scholar] [CrossRef] [PubMed]
- Erez, N.; Milyavsky, M.; Eilam, R.; Shats, I.; Goldfinger, N.; Rotter, V. Expression of prolyl-hydroxylase-1 (PHD1/EGLN2) suppresses hypoxia inducible factor-1α activation and inhibits tumor growth. Cancer Res. 2003, 63, 8777–8783. [Google Scholar]
- Xue, J.; Li, X.; Jiao, S.; Wei, Y.; Wu, G.; Fang, J. Prolyl hydroxylase-3 is down-regulated in colorectal cancer cells and inhibits IKKβ independent of hydroxylase activity. Gastroenterology 2010, 138, 606–615. [Google Scholar] [CrossRef]
- Richardson, N.L.; O’Malley, L.J.; Weissberger, D.; Tumber, A.; Schofield, C.J.; Griffith, R.; Jones, N.M.; Hunter, L. Discovery of neuroprotective agents that inhibit human prolyl hydroxylase PHD2. Bioorg Med. Chem. 2021, 38, 116115. [Google Scholar] [CrossRef]
- Berra, E.; Benizri, E.; Ginouvès, A.; Volmat, V.; Roux, D.; Pouysségur, J. HIF prolyl-hydroxylase 2 is the key oxygen sensor setting low steady-state levels of HIF-1α in normoxia. EMBO J. 2003, 22, 4082–4090. [Google Scholar] [CrossRef] [PubMed]
- Minervini, G.; Quaglia, F.; Tosatto, S.C.E. Insights into the proline hydroxylase (PHD) family, molecular evolution and its impact on human health. Biochimie 2015, 116, 114–124. [Google Scholar] [CrossRef] [PubMed]
- Meneses, A.M.; Wielockx, B. PHD2: From hypoxia regulation to disease progression. Hypoxia 2016, 4, 53–67. [Google Scholar] [PubMed]
- Gaete, D.; Rodriguez, D.; Watts, D.; Sormendi, S.; Chavakis, T.; Wielockx, B. HIF-prolyl hydroxylase domain proteins (PHDs) in cancer—Potential targets for anti-tumor therapy? Cancers 2021, 13, 988. [Google Scholar] [CrossRef]
- Morin, P.J.; Sparks, A.B.; Korinek, V.; Barker, N.; Clevers, H.; Vogelstein, B.; Kinzler, K.W. Activation of β-catenin-Tcf signaling in colon cancer by mutations in β-catenin or APC. Science 1997, 275, 1787–1790. [Google Scholar] [CrossRef]
- Moser, A.R.; Pitot, H.C.; Dove, W.F. A dominant mutation that predisposes to multiple intestinal neoplasia in the mouse. Science 1990, 247, 322–324. [Google Scholar] [CrossRef]
- Ren, J.; Sui, H.; Fang, F.; Li, Q.; Li, B. The application of Apc Min/+ mouse model in colorectal tumor researches. J. Cancer Res. Clin. Oncol. 2019, 145, 1111–1122. [Google Scholar] [CrossRef]
- De Santis, S.; Verna, G.; Serino, G.; Armentano, R.; Cavalcanti, E.; Liso, M.; Dicarlo, M.; Coletta, S.; Mastronardi, M.; Lippolis, A.; et al. Winnie-APCMin/+ mice: A spontaneous model of colitis-associated colorectal cancer combining genetics and inflammation. Int. J. Mol. Sci. 2020, 21, 2972. [Google Scholar] [CrossRef]
- De Santis, S.; Liso, M.; Vacca, M.; Verna, G.; Cavalcanti, E.; Coletta, S.; Calabrese, F.M.; Eri, R.; Lippolis, A.; Armentano, R.; et al. Dysbiosis triggers ACF development in genetically predisposed subjects. Cancers 2021, 13, 283. [Google Scholar] [CrossRef]
- Xue, X.; Taylor, M.; Anderson, E.; Hao, C.; Qu, A.; Greenson, J.K.; Zimmermann, E.M.; Gonzalez, F.J.; Shah, Y.M. Hypoxia-inducible factor-2α activation promotes colorectal cancer progression by dysregulating iron homeostasis. Cancer Res. 2012, 72, 2285–2293. [Google Scholar] [CrossRef] [PubMed]
- Vallée, A.; Lecarpentier, Y.; Vallée, J.-N. The key role of the WNT/β-catenin pathway in metabolic reprogramming in cancers under normoxic conditions. Cancers 2021, 13, 5557. [Google Scholar] [CrossRef]
- Di Grazia, A.; Di Fusco, D.; Franzè, E.; Colella, M.; Strimpakos, G.; Salvatori, S.; Formica, V.; Laudisi, F.; Maresca, C.; Colantoni, A.; et al. Hepcidin Upregulation in Colorectal Cancer Associates with Accumulation of Regulatory Macrophages and Epithelial–Mesenchymal Transition and Correlates with Progression of the Disease. Cancers 2022, 14, 5294. [Google Scholar] [CrossRef]
- Zhao, Y.; Tang, X.; Lei, T.; Fu, D.; Zhang, H. Lipocalin-2 promotes breast cancer brain metastasis by enhancing tumor invasion and modulating brain microenvironment. Front. Oncol. 2024, 14, 1448089. [Google Scholar] [CrossRef] [PubMed]
- Rouault, T.A. Mitochondrial iron overload: Causes and consequences. Curr. Opin. Genet. Dev. 2016, 38, 31–37. [Google Scholar] [CrossRef]
- Chen, X.; Kang, R.; Kroemer, G.; Tang, D. Broadening horizons: The role of ferroptosis in cancer. Nat. Rev. Clin. Oncol. 2021, 18, 280–296. [Google Scholar] [CrossRef]
- Yagoda, N.; Von Rechenberg, M.; Zaganjor, E.; Bauer, A.J.; Yang, W.S.; Fridman, D.J.; Wolpaw, A.J.; Smukste, I.; Peltier, J.M.; Boniface, J.J.; et al. RAS–RAF–MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature 2007, 447, 865–869. [Google Scholar] [CrossRef] [PubMed]
- Ası, J.; Pérez-Martos, A.; Fernández-Silva, P.; Montoya, J.; Andreu, A.L. Iron (II) induces changes in the conformation of mammalian mitochondrial DNA resulting in a reduction of its transcriptional rate. FEBS Lett. 2000, 480, 161–164. [Google Scholar]
- Coffey, R.; Ganz, T. Iron homeostasis: An anthropocentric perspective. J. Biol. Chem. 2017, 292, 12727–12734. [Google Scholar] [CrossRef]
- Lane, D.J.R.; Merlot, A.M.; Huang, M.L.H.; Bae, D.H.; Jansson, P.J.; Sahni, S.; Kalinowski, D.S.; Richardson, D.R. Cellular iron uptake, trafficking and metabolism: Key molecules and mechanisms and their roles in disease. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2015, 1853, 1130–1144. [Google Scholar] [CrossRef]
- Pacella, I.; Grimaldos, A.P.; Rossi, A.; Tucci, G.; Zagaglioni, M.; Potenza, E.; Pinna, V.; Rotella, I.; Cammarata, I.; Cancila, V.; et al. Iron capture through CD71 drives perinatal and tumor-associated Treg expansion. JCI Insight 2024, 9, e167967. [Google Scholar] [CrossRef]
- Vostrejs, M.; Moran, P.L.; Seligman, P.A. Transferrin synthesis by small cell lung cancer cells acts as an autocrine regulator of cellular proliferation. J. Clin. Investig. 1988, 82, 331–339. [Google Scholar] [CrossRef] [PubMed]
- Liang, W.; Ferrara, N. Iron metabolism in the tumor microenvironment: Contributions of innate immune cells. Front. Immunol. 2021, 11, 626812. [Google Scholar] [CrossRef]
- Yadav, A.; Maurya, R.P.; Maurya, A.K. Molecular Pathogenesis of Gut Microbiome Associated Colorectal Cancer: Recent Updates. In Socio-Scientific Interaction in Diabetes and Cancer and Its Management; B P International: Tsim Sha Tsui, Hong Kong, 2023; pp. 162–168. [Google Scholar]
- Yilmaz, B.; Li, H. Gut microbiota and iron: The crucial actors in health and disease. Pharmaceuticals 2018, 11, 98. [Google Scholar] [CrossRef]
- Botta, A.; Barra, N.G.; Lam, N.H.; Chow, S.; Pantopoulos, K.; Schertzer, J.D.; Sweeney, G. Iron reshapes the gut microbiome and host metabolism. J. Lipid Atheroscler. 2021, 10, 160. [Google Scholar] [CrossRef]
- Baas, F.S.; Brusselaers, N.; Nagtegaal, I.D.; Engstrand, L.; Boleij, A. Navigating beyond associations: Opportunities to establish causal relationships between the gut microbiome and colorectal carcinogenesis. Cell Host Microbe 2024, 32, 1235–1247. [Google Scholar] [CrossRef]
- Collins, S.M.; Bercik, P. The Relationship Between Intestinal Microbiota and the Central Nervous System in Normal Gastrointestinal Function and Disease. Gastroenterology 2009, 136, 2003–2014. [Google Scholar] [CrossRef]
- Coleman, O.I.; Haller, D. Microbe–mucus interface in the pathogenesis of colorectal cancer. Cancers 2021, 13, 616. [Google Scholar] [CrossRef] [PubMed]
- Savari, S.; Vinnakota, K.; Zhang, Y.; Sjölander, A. Cysteinyl leukotrienes and their receptors: Bridging inflammation and colorectal cancer. World J. Gastroenterol. WJG 2014, 20, 968. [Google Scholar] [CrossRef]
- Bernstein, H.; Bernstein, C.; Payne, C.M.; Dvorak, K. Bile acids as endogenous etiologic agents in gastrointestinal cancer. World J. Gastroenterol. WJG 2009, 15, 3329. [Google Scholar] [CrossRef] [PubMed]
- Vital, M.; Howe, A.C.; Tiedje, J.M. Revealing the bacterial butyrate synthesis pathways by analyzing (meta) genomic data. mBio 2014, 5, 10–1128. [Google Scholar] [CrossRef]
- Pandey, H.; Tang, D.W.T.; Wong, S.H.; Lal, D. Gut microbiota in colorectal cancer: Biological role and therapeutic opportunities. Cancers 2023, 15, 866. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.; Jin, Y.; Chen, G.; Ma, X.; Zhang, L. Gut microbiota dysbiosis drives the development of colorectal cancer. Digestion 2021, 102, 508–515. [Google Scholar] [CrossRef] [PubMed]
- Yamamura, R.; Inoue, K.Y.; Nishino, K.; Yamasaki, S. Intestinal and fecal pH in human health. Front. Microbiomes 2023, 2, 1192316. [Google Scholar] [CrossRef]
- Macfarlane, G.T.; Gibson, G.R.; Cummings, J.H. Comparison of fermentation reactions in different regions of the human colon. J. Appl. Bacteriol. 1992, 72, 57–64. [Google Scholar]
- Su, L.-K.; Kinzler, K.W.; Vogelstein, B.; Preisinger, A.C.; Moser, A.R.; Luongo, C.; Gould, K.A.; Dove, W.F. Multiple intestinal neoplasia caused by a mutation in the murine homolog of the APC gene. Science 1992, 256, 668–670. [Google Scholar] [CrossRef]
- Singh, A.; Ahmad, N.; Varadarajan, A.; Vikram, N.; Singh, T.; Sharma, S.; Sharma, P. Lactoferrin, a potential iron-chelator as an adjunct treatment for mucormycosis–a comprehensive review. Int. J. Biol. Macromol. 2021, 187, 988–998. [Google Scholar] [CrossRef]
- Ippolito, J.R.; Piccolo, B.D.; Robeson, M.S.; Barney, D.E., Jr.; Ali, J.; Singh, P.; Hennigar, S.R. Iron deficient diets modify the gut microbiome and reduce the severity of enteric infection in a mouse model of S. Typhimurium-induced enterocolitis. J. Nutr. Biochem. 2022, 107, 109065. [Google Scholar] [CrossRef]
- Gu, K.; Wu, A.; Yu, B.; Zhang, T.; Lai, X.; Chen, J.; Yan, H.; Zheng, P.; Luo, Y.; Luo, J.; et al. Iron overload induces colitis by modulating ferroptosis and interfering gut microbiota in mice. Sci. Total Environ. 2023, 905, 167043. [Google Scholar] [CrossRef]
- Liu, Y.; Chen, Y.; Li, Y.; Chen, L.; Jiang, H.; Jiang, L.; Yan, H.; Zhao, M.; Hou, S.; Zhao, C.; et al. Elaborating the mechanism of lead adsorption by biochar: Considering the impacts of water-washing and freeze-drying in preparing biochar. Bioresour. Technol. 2023, 386, 129447. [Google Scholar] [CrossRef]
- Wallert, M.; Ziegler, M.; Wang, X.; Maluenda, A.; Xu, X.; Yap, M.L.; Witt, R.; Giles, C.; Kluge, S.; Hortmann, M.; et al. α-Tocopherol preserves cardiac function by reducing oxidative stress and inflammation in ischemia/reperfusion injury. Redox Biol. 2019, 26, 101292. [Google Scholar] [CrossRef]
- Rescigno, M.; Chieppa, M. Gut-level decisions in peace and war. Nat. Med. 2005, 11, 254–255. [Google Scholar] [CrossRef]
- Bessman, N.J.; Mathieu, J.R.R.; Renassia, C.; Zhou, L.; Fung, T.C.; Fernandez, K.C.; Austin, C.; Moeller, J.B.; Zumerle, S.; Louis, S.; et al. Dendritic cell–derived hepcidin sequesters iron from the microbiota to promote mucosal healing. Science 2020, 368, 186–189. [Google Scholar] [CrossRef] [PubMed]
- Johansson, M.E.V.; Larsson, J.M.H.; Hansson, G.C. The two mucus layers of colon are organized by the MUC2 mucin, whereas the outer layer is a legislator of host–microbial interactions. Proc. Natl. Acad. Sci. USA 2011, 108 (Supp. S1), 4659–4665. [Google Scholar] [CrossRef] [PubMed]
- Hooper, L.V.; Littman, D.R.; Macpherson, A.J. Interactions between the microbiota and the immune system. Science 2012, 336, 1268–1273. [Google Scholar] [CrossRef] [PubMed]
- Vila, A.V.; Imhann, F.; Collij, V.; Jankipersadsing, S.A.; Gurry, T.; Mujagic, Z.; Kurilshikov, A.; Bonder, M.J.; Jiang, X.; Tigchelaar, E.F.; et al. Gut microbiota composition and functional changes in inflammatory bowel disease and irritable bowel syndrome. Sci. Transl. Med. 2018, 10, eaap8914. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chieppa, M.; Kashyrina, M.; Miraglia, A.; Vardanyan, D. Enhanced CRC Growth in Iron-Rich Environment, Facts and Speculations. Int. J. Mol. Sci. 2024, 25, 12389. https://doi.org/10.3390/ijms252212389
Chieppa M, Kashyrina M, Miraglia A, Vardanyan D. Enhanced CRC Growth in Iron-Rich Environment, Facts and Speculations. International Journal of Molecular Sciences. 2024; 25(22):12389. https://doi.org/10.3390/ijms252212389
Chicago/Turabian StyleChieppa, Marcello, Marianna Kashyrina, Alessandro Miraglia, and Diana Vardanyan. 2024. "Enhanced CRC Growth in Iron-Rich Environment, Facts and Speculations" International Journal of Molecular Sciences 25, no. 22: 12389. https://doi.org/10.3390/ijms252212389
APA StyleChieppa, M., Kashyrina, M., Miraglia, A., & Vardanyan, D. (2024). Enhanced CRC Growth in Iron-Rich Environment, Facts and Speculations. International Journal of Molecular Sciences, 25(22), 12389. https://doi.org/10.3390/ijms252212389